R. Lor Randall, MD, FACS
Russell Ward, MD
Bang H. Hoang, MD
Musculoskeletal oncology is a field of medicine that involves the diagnosis and management of neoplastic conditions affecting the musculoskeletal system. This not only entails neoplasia of mesenchymal origin (derived from embryonic mesoderm), but also metastatic carcinoma and a variety of pseudotumorous conditions. Mesenchymal tumors are an extremely heterogeneous group of neoplasms including over 200 benign conditions and 90 types of sarcoma, and so the majority of this chapter is dedicated to them. The relative incidence of benign to malignant disease is 200:1. These tumors are classified histomorphologically based on features of differentiation, but there is considerable overlap. It is favorable to consider these separate conditions as points on a continuum, rather than entirely distinct entities. Classification, nevertheless, is important because it may yield insight to the behavior, treatment response, and overall prognosis. Benign disease, by definition, behaves in a nonaggressive fashion and exhibits little tendency to locally recur or metastasize. Sarcomas (malignant tumors of mesenchymal origin), however, can be rapidly destructive, have metastatic potential, and have a tendency to locally recur.
Neoplastic processes arise in tissues of mesenchymal origin far less frequently than those of ectodermal or endodermal origin. In 2004, soft-tissue and bone sarcomas had an annual incidence in the United States of just more than 8600 and 2400 new cases, respectively. When compared with the overall cancer mortality of 563,000 cases per year in 2004, sarcomas are a small fraction of the problem. However, although they are a relatively uncommon form of cancer, these tumors behave in an aggressive fashion, with currently reported mortality rates in some series of greater than 50%. According to the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) program, approximately 8600 new soft-tissue sarcomas developed in the United States in 2004 with over 3600 sarcoma-related deaths. The associated morbidity is much higher. These tumors inflict a tremendous emotional and financial toll on individuals and society alike. Furthermore, sarcomas preferentially affect older patients, with only 15% occurring in patients younger than 15 years and 40% in patients older than 55 years. Accordingly, as the population ages, the incidence of these conditions will increase.
![]() ETIOLOGY OF MUSCULOSKELETAL TUMORS
ETIOLOGY OF MUSCULOSKELETAL TUMORS
Tumorigenesis is a multifactorial process, which, despite considerable fiscal and intellectual interest, is still poorly understood. There is commonality in genetic mutation that confers to cells the ability to replicate in an unregulated fashion. The development of a colony of abnormally proliferating cells out of normal tissue is referred to as transformation. This process involves acquired mutations in oncogenes, tumor suppressor genes, and other genes that directly or indirectly control proliferation, cell motility, and properties of invasiveness. Such a process may progress beyond the state of benign disease to an aggressive, dedifferentiated state with considerable genomic instability.
To appreciate how bone or soft-tissue tumors develop, one must have a basic understanding of the cell cycle and the regulation thereof. The cell cycle is divided into four distinct phases: G1 (gap 1), S (DNA synthesis), G2 (gap 2), and M (mitosis). After a copy of the entire cellular genome has been synthesized, these duplicate chromosomes are separated, and the cell divides during mitosis. The majority of cell growth occurs during G1. The mature state for mesenchymal cells is normally in a resting, nonproliferative phase designated G0.
Control of the cell cycle is a function of numerous regulatory proteins and checkpoints. These checkpoints allow for the monitoring and repair of the genetic sequence. These proteins are encoded by two basic gene types: oncogenes (stimulatory) and tumor suppressor genes (inhibitory). Oncogenes are genes encoding proteins, which have the inherent ability to transform the host cell to a neoplastic phenotype. Protooncogenes (eg, RAS, WNT, MYC) are wild-type genes that can become oncogenes upon mutation or dysregulation of expression. Tumor suppressor genes (eg, p53, Rb, p21) typically require loss-of-function mutations or mutations in other regulatory genes to impart the neoplastic phenotype. These genes are primarily involved with cell-cycle checkpoints.
Inherited mutations in these genes result in neoplastic predisposition syndromes. In syndromes such as hereditary retinoblastoma and Li-Fraumeni syndrome, one mutated copy of a tumor suppressor gene (Rband p53, respectively) is inherited. Through a variety of causes including deletion, translocation, point mutation, promoter silencing, or other means of loss of heterozygosity, the remaining function of that gene product is lost, and from that cell a tumor is born. Once checkpoint machinery has been disrupted, additional mutations may be accumulated at an increasing rate, allowing genomic instability to spiral out of control. At the extreme, as seen commonly in osteosarcoma, cells contain up to four times the normal number of chromosomes with multiple chromosomal aberrations.
In addition to hereditary factors that predispose one to the development of neoplasia, many well-described environmental factors exist, such as radiation exposure, chemical carcinogens, and certain oncogenic viral infections. More of these factors may surface as meticulous investigation continues in the field of cancer research.
The neoplastic process may arrest in the so-called benign state, with further genomic instability curtailed, or it may progress to a sarcomatous state. For example, if a cell type of origin is a lipoblast, then a lipoma or a liposarcoma may develop. Furthermore, a liposarcoma may progress in its differentiation such that its phenotype, as a high-grade lesion, minimally reflects its lipoblastic origin. This principle is illustrated in Figure 5–1, which shows a myxoid liposarcoma within the substance of a typical intramuscular lipoma. This possibility does not imply, however, that all benign lesions are necessarily at risk for malignant degeneration. It is not a surgical indication to resect a lipoma because of concern for secondary liposarcoma.

![]() Figure 5–1. CT scan of a myxoid liposarcoma within the substance of a long-standing intramuscular lipoma.
Figure 5–1. CT scan of a myxoid liposarcoma within the substance of a long-standing intramuscular lipoma.
Although a plethora of molecular pathways are being studied, understanding of the details of genomic instability and subsequent tumor formation is lacking. There is no single pathway by which all neoplasms arise; instead, multiple genetic targets are altered in a variety of sequences and combinations with the common result of cellular proliferation that is tumorigenesis.
![]() EVALUATION AND STAGING OF TUMORS
EVALUATION AND STAGING OF TUMORS
![]() History and Physical Examination
History and Physical Examination
When evaluating a new patient with a possible tumor, the workup must commence with a thorough history and physical examination. Prior to ordering any diagnostic studies, particular questions must be answered, and the physical characteristics of the mass in question must be assessed. This procedure prevents unnecessary tests and better enables the physician to determine which tests will be most helpful in diagnosing the condition as well as facilitating therapeutic interventions if needed.
The clinical history is of paramount importance (Table 5–1). The age of the patient permits the development of a list of potential diagnoses (Table 5–2), which, when combined with the history, physical examination, and a few additional studies, should permit establishing a diagnosis. The duration and timing of symptoms, rate of growth, presence of pain, and a history of trauma can help elucidate the diagnosis. Specifically, pain or other symptoms occurring at rest or during the night are of particular concern. Additionally, a careful past medical history, family history, and review of systems must not be overlooked.
Table 5–1. Questions that must be asked in the workup of a possible tumor.

Table 5–2. Distribution of bone tumors by age (years).


A thorough physical examination is also critical (Table 5–3). The clinician must assess the location and size of the mass, the quality of the overlying skin, the presence of warmth, any associated swelling, the presence of tenderness, and the firmness of the lesion. For superficial lesions, transillumination and auscultation may also be beneficial. Range of motion of all joints in proximity to the tumor must be recorded, and a complete neurovascular exam must be performed. An assessment of the related lymph node chains and an examination for an enlarged liver or spleen should also be performed.
Table 5–3. Aspects of physical examination that should be documented when evaluating a patient with a mass.

The clinician must also consider pseudotumors in addition to true neoplastic conditions. A history of trauma suggests possible stress fracture of myositis ossificans as a diagnosis. The association of symptoms with physical activity and variations of symptoms with the passage of time are important considerations in establishing a differential diagnosis.
![]() Imaging Studies
Imaging Studies
A. Radiography
Initial evaluation should begin with plain radiography. In every patient with a suspected tumor, orthogonal anteroposterior (AP) and lateral views of the affected area should be obtained. This includes soft-tissue masses as well. Furthermore, in the case of a bone lesion, the entire bone should be imaged. In many cases, radiographic examination is diagnostic, and no further imaging studies are indicated. Although in the case of a more aggressive process the diagnosis may be determined on plain radiographs, further evaluation with advanced studies is usually indicated to determine the extent of disease involvement, as well as the degree of systemic involvement (staging).
The initial radiographic images must be scrutinized. For bone lesions, the location within the bone (eg, diaphyseal, metaphyseal, or epiphyseal; eccentric or central; medullary or surface) facilitates the diagnosis. Epiphyseal tumors are usually benign. Primary sarcomas of bone are usually metaphyseal; however, round cell tumors, such as Ewing sarcoma, multiple myeloma, and lymphoma, are usually medullary diaphyseal lesions. A tumor arising from the surface of a bone may be benign, such as an osteochondroma, or may be malignant, such as a parosteal osteosarcoma.
Terms such as geographic, well-circumscribed, and permeative are useful in describing radiographic abnormalities. Geographic or well-circumscribed implies that the lesion has a distinct, sharply marginated boundary, and suggests benignity (Figure 5–2). Lesions with this feature may exhibit sclerotic borders if the bone itself has reacted to contain the process. A poorly defined, infiltrative process is described as permeative or moth-eaten and reflects a more aggressive process such as malignancy (Figure 5–3), although benign, aggressive processes can exhibit this radiographic feature as well (Figure 5–4). An exception to this rule is multiple myeloma, which frequently demonstrates a punched-out, well-demarcated appearance but in multiple locations.
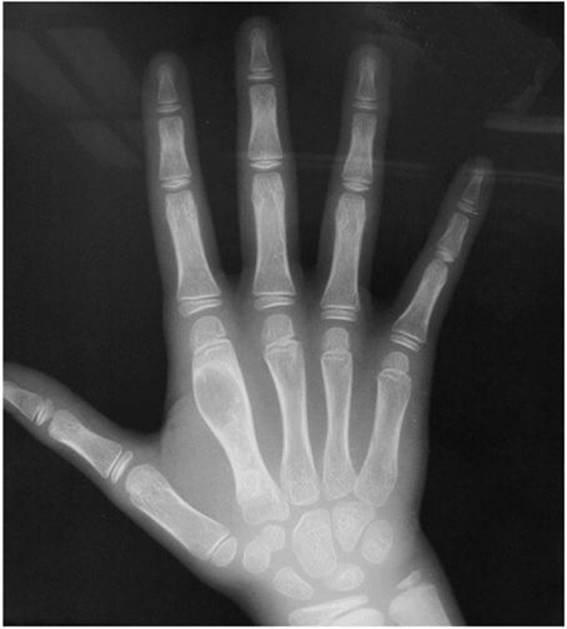
![]() Figure 5–2. Radiograph of an enchondroma of the second metacarpal. Notice its geographic appearance.
Figure 5–2. Radiograph of an enchondroma of the second metacarpal. Notice its geographic appearance.

![]() Figure 5–3. Radiograph of a proximal fibular osteosarcoma demonstrating the destructive, permeative nature of malignant bone tumors.
Figure 5–3. Radiograph of a proximal fibular osteosarcoma demonstrating the destructive, permeative nature of malignant bone tumors.

![]() Figure 5–4. Radiograph of a giant cell tumor of the thumb. This is a typical moth-eaten appearance.
Figure 5–4. Radiograph of a giant cell tumor of the thumb. This is a typical moth-eaten appearance.
Matrix quality is another radiographic feature that aids in diagnosis. Lesions may be entirely radiolucent, sparsely mineralized, or predominantly mineralized, and mineralization may be spiculated, stippled, or composed of rings and arcs. Most osteosarcomas, for example, have a spiculated pattern of mineralization expanding out from the host bone in a sunburst appearance. In contrast, chondroid matrix characteristically forms rings and arcs of mineralization. In soft-tissue lesions, matrix mineralization can be very helpful in diagnosis, as in hemangiomas showing smooth, round mineralized phleboliths, as opposed to synovial sarcoma frequently showing irregular mineralization.
With a careful history, physical examination, and appropriate radiographs, the physician can usually reach a working diagnosis of the lesion. Although some benign and malignant tumors mimic each other, many diagnoses can be ruled out based on information gained without advanced imaging. Factors such as patient age and location as well as radiographic features such as boundary, matrix quality, and reaction of the bone to the lesion can be used to narrow the differential diagnosis. It should be noted that infection can have an extremely variable radiographic appearance, and frequently remains on the differential until ruled out by laboratory studies or biopsy. Tables 5–1 through 5–6 can be used in a step-wise fashion to illustrate the process of focusing the differential diagnosis prior to any advanced imaging and can assist in obtaining the most appropriate studies.
Table 5–4. Skeletal distribution of bone tumors, ranked from most common (1) to least common (5) sites.


Table 5–5. Bone tumors: imaging characteristics, location in a long bone, and beneficial studies, ranked from most common or most beneficial (1) to least common or least beneficial (3).


Table 5–6. Distribution of soft-tissue tumors by age.


B. Isotope Bone Scanning
Technetium-99 radioisotope scans are used to assess the degree of osteoblastic activity of a given lesion (Figure 5–5). In general, they are quite sensitive, with a few exceptions, for active lesions of bone. Accordingly, technetium-99 scans are excellent screening studies for remote lesions (staging). The best indication for a bone scan is suspected multiple bony lesions, such as in metastatic carcinomas and lymphomas of bone. Isotope bone scanning is far simpler to perform, less expensive, and requires less total-body irradiation than skeletal surveys. It is common practice to use serial isotope scans to follow patients with suspected metastatic disease and, at the same time, evaluate the effectiveness of their systemic therapy.

![]() Figure 5–5. Technetium-99 scan demonstrating extensive osteoblastic activity in a patient with meta-static adenocarcinoma.
Figure 5–5. Technetium-99 scan demonstrating extensive osteoblastic activity in a patient with meta-static adenocarcinoma.
Isotope scanning is also used in the staging process of a primary sarcoma of bone such as osteosarcoma to rule out an asymptomatic remote skeletal lesion. Technetium-99 scans are also useful in distinguishing blastic lesions of bone. Given that the study reflects osseous metabolic activity, dormant lesions such as enostoses (bone islands) would not demonstrate significantly increased tracer uptake compared with a blastic prostate metastasis. Inflammatory and traumatic lesions also demonstrate increased activity. It is important to note, however, that multiple myeloma and some metastatic carcinomas (eg, renal cell carcinoma) may not demonstrate increased uptake (ie, false-negative result). Skeletal surveys are preferable for screening for distant involvement in such cases.
C. Computed Tomography and Magnetic Resonance Imaging
Computed tomography (CT) remains a standard imaging procedure for use in well-selected clinical situations. Perhaps the best indication for CT is for smaller lesions that involve cortical structures of bone or spine (Figure 5–6). In such cases, CT is superior to magnetic resonance imaging (MRI) because the resolution of cortical bone using MRI is inferior. CT scan of the chest is the modality of choice for evaluating patients for pulmonary metastasis. Abdominal CT is invaluable in surveying for primary tumor in patients who present with skeletal metastases. For tumors involving the pelvis and sacrum, CT can help elucidate the extent of bone involvement (Figure 5–7). In cases involving a soft-tissue lesion, MRI is far superior to CT unless there is a heavily mineralized process.

![]() Figure 5–6. CT scan of an osteoblastoma arising from the right pedicle of a lumbar vertebra.
Figure 5–6. CT scan of an osteoblastoma arising from the right pedicle of a lumbar vertebra.

![]() Figure 5–7. Pelvic CT demonstrating the bony destruction of the sacrum caused by a giant cell tumor.
Figure 5–7. Pelvic CT demonstrating the bony destruction of the sacrum caused by a giant cell tumor.
MRI is the imaging modality of choice for evaluating bone marrow involvement and noncalcific soft-tissue lesions. The two most common sequences are the T1-weighted and T2-weighted spin echo (Figure 5–8). Short tau inversion recovery (STIR) sequences have been shown to further elucidate the conspicuity of tumors and bone marrow edema against a nonpathologic background. MRI can also demonstrate the normal anatomy of soft structures, including nerves and vessels, thereby eliminating the need for arteriography and myelograms. Dynamic-enhanced MRI, with its ability to estimate tumor blood flow by examining the rate of contrast uptake and clearance, may serve as a predictor of clinical outcome or tumor response to chemotherapy.

![]() Figure 5–8. Synovial sarcoma involving the popliteal fossa. (A) T1-weighted. (B) T2-weighted.
Figure 5–8. Synovial sarcoma involving the popliteal fossa. (A) T1-weighted. (B) T2-weighted.
![]() Laboratory Studies
Laboratory Studies
A. Biopsy
The biopsy is usually the last staging procedure. It is preferred to obtain advanced imaging prior to biopsy to avoid postsurgical artifact on these studies, which may aid in determining a definitive diagnosis. There are three types of biopsy for musculoskeletal neoplasia: excisional, incisional, and needle biopsies. Excisional biopsy is discouraged unless the lesion is particularly small (2–3 cm) or in a location where a cuff of healthy tissue can be resected as a margin without significantly increased morbidity.
Complications relating to biopsy are very common. Accordingly, careful preoperative planning is imperative. Imaging studies aid the surgeon in planning biopsy approach and technique. The highest-quality diagnostic tissue is usually found at the periphery of the lesion, where it interfaces with the normal surrounding tissue. In the case of extracompart-mental primary bone lesions, the periphery of the soft-tissue extension can be sampled without further compromising the structural integrity of the bone. In the case of intramedullary lesions, a round or ovular window should be developed to decrease the risk of fracture, and the defect should be filled with bone wax or cement to avoid unnecessary contamination of the uninvolved soft tissue. There is also evidence that biopsies performed by the treating oncologic surgeon enjoy a lower complication rate than those performed by inexperienced practitioners prior to referral.
The placement of the biopsy site is a major consideration, since resection of the biopsy tract is necessary for many malignant diagnoses. Serious contamination of vital structures such as the popliteal artery or sciatic nerve may result in amputation rather than limb-sparing surgery. Furthermore, transverse incisions should be avoided due to the swath of uninvolved tissue that would need to be resected to incorporate such a biopsy tract into a surgical approach. In many cases, this mandates the use of free or rotational flap coverage that otherwise would not be necessary. Meticulous hemostasis is also mandatory to avoid the formation of a contaminating hematoma. In rare instances, a drain may be helpful, but should exit the skin in line with the biopsy incision.
Obtaining an adequate specimen is critical. A frozen section determines, if viable, diagnostic tissue has been obtained. Definitive diagnosis is rarely possible based on frozen section. In most circumstances, definitive resection should be delayed until final histopathologic diagnosis has been rendered. For the diagnosis of some histologies, special studies may be required such as immunohistochemistry, flow cytometry, fluorescence in situ hybridization, or other cytogenetic studies. Adequate material, handled appropriately, is required for the completion of many of these studies.
Needle biopsies, either core or fine needle, are increasingly being used at experienced tumor centers, especially for presumed lesions that are easily diagnosed, such as round cell tumors or metastatic carcinoma. Because the subtype of tumor frequently determines treatment, architecture of the tumor is generally needed, which requires core rather than fine-needle biopsy. For spine or deep pelvic lesions image-directed needle biopsy is ideal because it avoids excessive multicompartmental contamination. Fine-needle biopsy should be reserved for use in consultation with an experienced cytopathologist. Most current studies show a diagnostic accuracy of 75–85% for needle biopsy compared with more than 95% accuracy for incisional biopsy.
B. Cultures and Special Studies
The damage of biopsy specimens after retrieval can make it impossible to perform special studies such as immunohistochemistry, cytogenetics, flow cytometry, and electron microscopy. For this reason, the surgeon should consult with the pathologist prior to biopsy. For example, formalin preservation of the specimen will preclude many of the above studies. Not only is it important for the operating room personnel to be aware of this, but it is also important for the pathologist to be aware of the clinical history and differential diagnoses, so that the specimen is handled appropriately. It is also wise to culture for bacteria, fungus, and acid-fast bacilli when clinical suspicion warrants.
Molecular diagnostics continue to revolutionize sarcoma diagnostics. This is particularly evident in the decreasing incidence of undifferentiated pleomorphic sarcoma as technologic advances allow better determination of the cell lineage of origin. Specific genomic rearrangements and mutations have been identified in a variety of tumors (Table 5–7). This is not only improving diagnostics, but also treatment. Gastrointestinal stromal tumor (GIST), a malignant mesenchymal neoplasm of the gastrointestinal tract, omentum, and mesentery, expresses a mutant form of c-kit. The KIT gene encodes a tyrosine kinase receptor for which targeted therapy has been developed and has shown significant efficacy. Targeted therapy has also been developed for some subtypes of lymphoma based on the expression pattern of cell surface markers. The status of these cell surface proteins is most often established using flow cytometry.
Table 5–7. Common translocations seen in sarcomas.

![]() Staging Systems
Staging Systems
Staging refers to a critical assessment of the grade and size of a tumor and the extent to which the disease has spread. There are two dominant staging systems for both soft-tissue sarcoma and primary bone sarcoma, which are described in the following sections. The goals of cancer staging include treatment guidance, prognostic stratification, and investigation continuity.
A. System of the American Joint Committee on Cancer
The American Joint Committee on Cancer (AJCC) system (6th edition) is used by most surgical oncologists. This is the four-stage tumor, node, metastasis (TNM) classification system that provides staging criteria for both soft-tissue sarcoma and primary bone sarcoma, as well as all other major malignancies. The distinct staging systems for both soft-tissue sarcoma and primary bone sarcoma entail the size and grade of the primary tumor as well as presence of nodal or distant metastases.
For soft-tissue sarcoma, stage I represents any grade 1 or 2 (out of 4) tumor without nodal or distant metastases. Stage II and III tumors are grade 3 or 4 tumors without metastases. The distinction is in size and depth, where stage III is a tumor of more than 5 cm and deep to fascia (T2b), and stage II is any superficial tumor (T1a and T2a) or less than 5 cm and deep (T1b). Stage IV is any grade, size, or depth tumor with nodal or distant metastases.
For primary bone sarcoma, stage I represents any grade 1 or 2 (out of 4) tumor without noncontiguous or metastatic disease. This is further subdivided by size, with stage IA including tumors of less than 8 cm in largest dimension and stage IB including tumors of 8 cm or greater. It should be noted that this has changed since the AJCC fifth edition, where the distinction was based on intra- versus extracompartmental designation. Stage II represents any grade 3 or 4 tumor without noncontiguous or metastatic disease. This group also is divided into stage IIA and IIB based on 8-cm size. Stage III is any grade or size tumor with noncontiguous disease in the same bone and no distant metastases. Stage IV is any grade or size tumor with distant metastatic disease and is divided into stage IVA and IVB based on isolated pulmonary metastases versus extrapulmonary disease including lymph nodes, respectively.
B. Musculoskeletal Tumor Society System (Surgical Staging System or Enneking System)
Many orthopedic oncologists prefer the Enneking system, which addresses both primary bone and soft-tissue sarcoma, and specifically addresses unique problems associated with sarcoma affecting the extremities. It is a three-stage system with stage I representing low-grade tumors without metastases. Stage II represents high-grade tumors without metastases. The first two groupings are further subdivided into type A and B based on intra- or extracompartmental designation, respectively. Stage III is any tumor with metastatic disease. Although compartmentalization is an important surgical concept, to date, it has not been shown to be a statistically significant prognostic factor.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Meticulous history and physical examination are critical in the diagnosis of bone and soft-tissue tumors.
• Plain radiography should be the initial imaging modality in the evaluation of bone and soft-tissue tumors. Frequently, it is diagnostic, obviating the need for advanced imaging and, at times, biopsy.
• When biopsy is necessary, it should be performed with careful planning by the surgeon prepared to definitively treat all possible diagnoses.
Heck RK, Peabody TD, Simon MA: Staging of primary malignancies of bone. CA Cancer J Clin 2006;56:366. [PMID: 17135693]
Jaffe CC: Response assessment in clinical trials: implications for sarcoma clinical trial design. Oncologist 2008;13:14. [PMID: 18434633]
Kotilingam D, Lev DC, Lazar AJ, et al: Staging soft tissue sarcoma: evolution and change. CA Cancer J Clin 2006;56:282. [PMID: 17005597]
Mankin HJ, Mankin CJ, Simon MA: The hazards of the biopsy, revisited. J Bone Joint Surg Am 1996;78:656. [PMID: 8642021]
Mitsuyoshi G, Naito N, Kawai A, et al: Accurate diagnosis of musculoskeletal lesions by core needle biopsy. J Surg Oncol 2006;94:1. [PMID: 16788939]
Moley JF, Eberlein TJ: Soft-tissue sarcomas. Surg Clin North Am 2000;80:687. [PMID: 10836012]
Oliviera AM, Nascimento AG: Grading in soft tissue tumors: principles and problems. Skeletal Radiol 2001;30:543. [PMID: 11685477]
Ordonez JL, Martins AS, Osuna D, et al: Targeting sarcomas: therapeutic targets and their rational. Semin Diagn Pathol 2008;25:304. [PMID: 19013896]
Simon MA, Finn HA: Diagnostic strategy for bone and soft tissue tumors. J Bone Joint Surg Am 1993;75:622. [PMID: 8478392]
Zahm SH, Fraumeni JF Jr: The epidemiology of soft tissue sarcoma. Semin Oncol 1997;24:504. [PMID: 9344316]
![]() DIAGNOSIS AND TREATMENT OF TUMORS
DIAGNOSIS AND TREATMENT OF TUMORS
BENIGN BONE TUMORS
Benign bone tumors have certain characteristics that favor their diagnosis over malignant conditions. Benign lesions are frequently asymptomatic and, many times, are detected incidentally during workup of an unrelated condition such as minor trauma. The diagnosis can often be made with plain radiography alone. Benign bone tumors are usually well circumscribed, and there is evidence of the host bone successfully reacting to contain the lesion, characterized radiographically by sclerotic margins or a dense osteoblastic reactive zone. In contrast, if the condition is malignant, the patient usually complains of pain, and the radiograph commonly shows a more permeative lesion with lytic destruction and poorly defined margins that suggest rapid progression. Further studies such as MRI or bone scintigraphy are unnecessary for typical benign lesions, such as fibrous dysplasia, enchondroma, or nonossifying fibroma. There is far less cytogenetic information available for benign bone tumors, likely because there is less implication in the treatment. A system of staging exists for benign bone tumors. Stage 1 lesions are considered latent. They are generally asymptomatic, but not always. Although they can progress, they usually resolve. Initially, these lesions should be observed. Stage 2 lesions are considered active. They tend not to resolve spontaneously and are less well demarcated than stage 1 lesions. They frequently require surgical intervention with meticulous attention to complete extirpation because of their propensity for recurrence. Stage 3, or aggressive, lesions demonstrate extensive destruction. Treatment often requires wide en bloc resection.
The more common types of benign bone tumors seen by the practicing orthopedic surgeon are discussed in this section.
![]() Benign Osteoid-Forming Tumors
Benign Osteoid-Forming Tumors
A. Osteoid Osteoma: ICD-9-CM 213.x
The most common benign osteoid-forming tumor is the osteoid osteoma, accounting for 10% of all benign bone tumors. It is more common in males than in females with a peak incidence in the second decade of life. Although it may be present in almost any bone, the proximal femur is the most common location. Dull, aching, nocturnal pain is characteristic, and it is commonly relieved entirely by nonsteroidal anti-inflammatory drugs (NSAIDs) secondary to a high concentration of prostaglandins in the nidus. Osteoid osteoma may have a unique pathogenic nerve supply as well, a unique finding among the bone tumors.
The characteristic radiographic feature of the osteoid osteoma is a central, lytic nidus usually 1 cm or less in diameter. The more common cortically based lesion (Figure 5–9) exhibits extensive reactive sclerosis, creating a fusiform bulge on the bone surface. However, if the nidus is more centrally located in metaphyseal bone, less sclerosis is seen and the radiographic appearance is less diagnostic. If the nidus is close to, or actually in, a joint, as in a femoral neck lesion, the resulting reactive synovitis may mimic a pyarthrosis or rheumatoid arthritis. Technetium bone scans are invariably positive. A CT scan is recommended to better anatomically locate the nidus and confirm the diagnosis.

![]() Figure 5–9. Radiograph (A), isotope bone scan (B), CT scan (C), and photomicrograph (D) of an osteoid osteoma in the femur of a 19-year-old man.
Figure 5–9. Radiograph (A), isotope bone scan (B), CT scan (C), and photomicrograph (D) of an osteoid osteoma in the femur of a 19-year-old man.
In the spine, the typical location for an osteoid osteoma is in the posterior elements. The lumbar spine is most commonly involved followed by the thoracic spine. A secondary scoliosis is usually associated with this presentation with the lesion located at the apex of the concavity. Furthermore, if the nidus is in proximity to a nerve root, radicular pain may develop, which may obscure timely diagnosis.
Histologically, the nidus is seldom larger than 1 cm, and for lesions greater than 2 cm, the term osteoblastoma is reserved, suggesting somewhat more aggressive proliferative features. The nidus is composed of loose, vascular connective tissue and immature, lacy osteoid lined by plump osteoblasts. At the periphery of the nidus, there is bone organized into a tiny trabecular network with centripetally increasing maturity. There is a paucity of cytogenetic data for this entity, which is unlikely to increase rapidly, secondary to the fact that diagnostic tissue is rarely procured in the course of the diagnosis or treatment.
Many cases of osteoid osteoma are stage 1 lesions and can be treated symptomatically with aspirin or NSAIDs until they spontaneously resolve. If the patient fails such treatment, surgical intervention is warranted. If surgery is undertaken, the entire nidus must be eradicated. Resection of surrounding sclerotic bone should not be excessive, because it can severely compromise the structural integrity of the host bone. The so-called burr-down technique is preferred over en bloc resection. The nidus is recognized by the hyperemic, pink hue and is removed by curettage. The burr is then used to advance the margin another 2–3 mm. CT-guided radiofrequency ablation is emerging as an accepted treatment modality. This method employs probes with high-frequency alternating current to induce ionic agitation and frictional heat to induce tumor necrosis. Radiofrequency ablation is used extensively as a less invasive treatment modality with similar success rate as surgical excision.
B. Osteoblastoma: ICD-9-CM 213.x
Osteoblastoma is a large osteoid osteoma that demonstrates a propensity for the posterior elements of the spine. Osteoblastomas are found more commonly in males than in females and occur in the same age group as osteoid osteomas. Osteoblastomas are less common than osteoid osteomas, accounting for 1% of all benign bone tumors. They can occur in the metaphyses of long bones, raising concern of osteosarcoma, and a few are seen in the ankle and wrist. These are usually stage 1–2 lesions.
Radiographically, the osteoblastoma has a more lytic and destructive appearance than the osteoid osteoma. Its nidus, which is greater than 1–2 cm, has a less sclerotic boundary and may take on the appearance of an aneurismal bone cyst. Histologically, however, the nidus is identical to that of an osteoid osteoma. There is rich vascularity in a bed of disorganized, immature osteoid and microtrabeculae lined with a single layer of plump osteoblasts. Multinucleated, osteoclast-like giant cells may be present. Although little cytogenetic data are available, preliminary evidence suggests moderately increased genetic instability over that of osteoid osteoma.
In the spine, the effects of osteoblastoma are similar to osteoid osteoma, although at times more pronounced, including radiating pain and other effects of nerve root or spinal cord impingement (Figure 5–10).

![]() Figure 5–10. Radiograph of an osteoblastoma in the pedicle area of the C3 vertebra of a 14-year-old boy.
Figure 5–10. Radiograph of an osteoblastoma in the pedicle area of the C3 vertebra of a 14-year-old boy.
In patients with osteoblastoma, treatment usually consists of vigorous curettage of the lesion, which may require structural bone grafting if instability results. Radiofrequency ablation may also prove useful in the management of this lesion in certain circumstances.
C. Osteofibrous Dysplasia: ICD-9-CM 213.7
Osteofibrous dysplasia is a rare condition, usually presenting as a stage 1–2 lesion, that is seen almost exclusively in the tibia of children during the first two decades of life. There is a strong male predilection. It commonly affects the anterior cortex resulting in an anterior tibial bow. Osteofibrous dysplasia can be seen in the fibula, and even more rarely, can be seen bilaterally. It is most likely a hamartomatous process and tends to involute spontaneously with skeletal maturity.
In osteofibrous dysplasia (Figure 5–11), lytic changes are seen in the anterior tibial cortex surrounded by sclerotic margins creating a soap-bubble appearance similar to the radiographic picture of fibrous dysplasia and adamantinoma. Histologically, the lytic lesion shows a benign trabecular alphabet soup pattern in a fibrous stroma. Notably, there is prominent osteoblastic rimming of the trabeculae, in contrast with fibrous dysplasia.

![]() Figure 5–11. Radiograph of osteofibrous dysplasia in the tibia of an 8-year-old boy.
Figure 5–11. Radiograph of osteofibrous dysplasia in the tibia of an 8-year-old boy.
As previously eluded to, there is a significant diagnostic dilemma in distinguishing between osteofibrous dysplasia and adamantinoma. If progression is documented or there are other alarming features, diagnostic biopsy is warranted. Osteofibrous dysplasia is vimentin positive but keratin negative, whereas adamantinoma exhibits prominent nests of keratin-positive epithelial cells. When a few scattered keratin-positive cells are seen, it is termed osteofibrous dysplasia–like adamantinoma.
In a report of experience with 35 cases of osteofibrous dysplasia, investigators indicated that early attempts at curettage and grafting of the lesions resulted in a high failure rate because of recurrence. For this reason, they suggested waiting until patients reach the age of 15 years and their disease spontaneously arrests before proceeding with debridement and grafting.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Benign osteoid-forming lesions of bone are typically painful and, especially in the case of osteoid osteoma, may be relieved by aspirin or NSAIDs.
• Osteoblastoma is very similar to osteoid osteoma, but larger, and both show a predilection to the posterior elements of the spine. If there is an associated painful scoliosis, the convexity will point away from the side of the lesion.
• Osteofibrous dysplasia is characterized histologically by the presence of osteoblastic rimming of the immature trabeculae, in contrast to fibrous dysplasia, which has absent osteoblastic rimming.
![]() Benign Chondroid-Forming Tumors
Benign Chondroid-Forming Tumors
A. Enchondroma and the Multiple Enchondromatoses: ICD-9-CM 213.x
Enchondroma refers to a centrally located chondroma of bone. These tumors are relatively common, accounting for greater than 10% of benign bone tumors. In 50% of cases, the tumor is found in the small tubular bones of the hands and feet. It arises in growing bones as a hamartomatous process, but is frequently asymptomatic and may avoid detection until the patient reaches adulthood, at which time it may be discovered in association with a pathologic fracture or as an incidental finding.
Radiographs of enchondromas show geographic lysis with sharp margination and central calcification (Figure 5–12). In the case of an enchondroma of the hand, the cortex is frequently thinned out with slight expansion. In contrast, with involvement of the large long bones, the lesion is centrally located with minimal cortical erosion. Enchondromas are either stage 1 or 2 lesions.

![]() Figure 5–12. Radiograph of an enchondroma of the proximal phalanx of the ring finger.
Figure 5–12. Radiograph of an enchondroma of the proximal phalanx of the ring finger.
Multiple enchondromatosis, or Ollier disease (Figure 5–13), is a rare nonfamilial dysplasia typically seen on half of the body and appears similar to fibrous dysplasia. This condition can be quite extensive with significant involvement of the metaphyses resulting in bowing and shortening of the long bones. Such dramatic changes are not seen in solitary enchondroma. In patients with Maffucci syndrome, multiple enchondromatosis is seen in association with multiple soft-tissue hemangiomas.
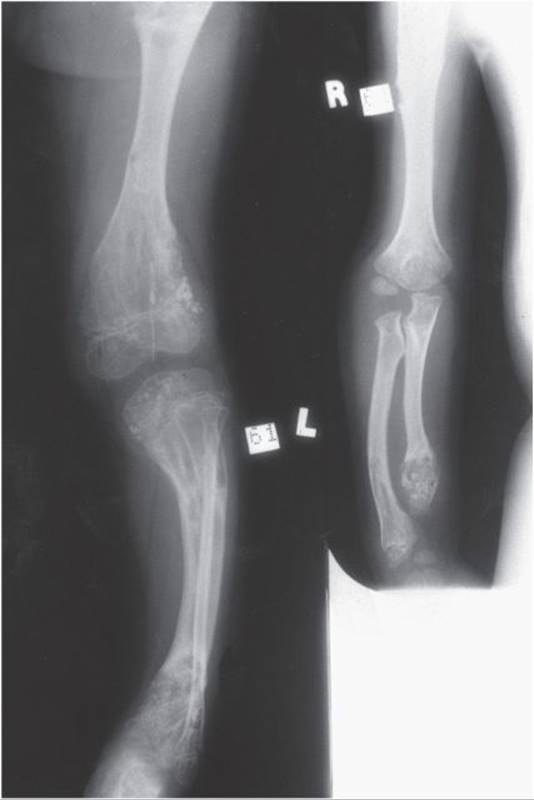
![]() Figure 5–13. Radiograph of Ollier disease of the upper and lower extremities.
Figure 5–13. Radiograph of Ollier disease of the upper and lower extremities.
A large solitary enchondroma converts to low-grade chondrosarcoma in fewer than 5% of cases, and the conversion takes place during adulthood. A solitary enchondroma in the hand rarely converts to chondrosarcoma, although histologically these appear more biologically active. A secondary chondrosarcoma can occur in enchondromatosis up to 20% of the time and may be related to acquired inactivation of certain tumor suppressor genes.
There is no need to treat an asymptomatic patient with a solitary enchondroma, but the patient should be followed radiographically for changes suggesting dedifferentiation. In cases of impending fracture or persistently symptomatic lesions, curettage with margin extension and bone grafting may be performed with a low risk of recurrence. Patients with multiple enchondromatosis must be followed closely because of the increased risk of secondary chondrosarcoma. Patients with Maffucci syndrome are at additional risk for the development of other mesenchymal neoplasia, including hemangiosarcoma and lymphangiosarcoma.
B. Periosteal Chondroma: ICD-9-CM 213.x
A benign chondroma seen on the surface of a bone is called a periosteal chondroma. Patients frequently have more than one lesion, and the most common location is on the proximal humeral metaphysis. Radiographically, the lesions appear to saucerize the underlying cortex (Figure 5–14). These stage 1–2 lesions may grow to a sizable mass, but those larger than 4 cm suggest peripheral chondrosarcoma. Management usually consists of serial imaging to ensure it does not continue growing into adulthood. In concerning cases, simple excision results in low recurrence rates.

![]() Figure 5–14. Radiograph of a periosteal chondroma on the index metacarpal of a 12-year-old boy. Notice the buttress of bone proximally and the characteristic matrix mineralization.
Figure 5–14. Radiograph of a periosteal chondroma on the index metacarpal of a 12-year-old boy. Notice the buttress of bone proximally and the characteristic matrix mineralization.
C. Osteochondroma: ICD-9-CM 213.x
The nonossifying fibroma is the most common benign bone tumor, and the osteochondroma is the second most common. Like the enchondroma, the osteochondroma is a developmental, or hamartomatous, process that arises from a defect in the outer edge of the metaphyseal side of a growth plate, resulting in an exostosis that points away from the joint and moves away from the physis with growth.
Macroscopically, there is a bony base, sharing a medullary communication with the host bone, and a cartilaginous cap (Figures 5–15 and 5–16). They may be pedunculated or sessile. The cartilage cap has a similar columnar organization as a growth plate and synchronously stops growth at skeletal maturity.

![]() Figure 5–15. Radiograph of a solitary osteochondroma on the distal femur of a skeletally immature individual.
Figure 5–15. Radiograph of a solitary osteochondroma on the distal femur of a skeletally immature individual.

![]() Figure 5–16. Typical glistening white appearance of the cartilage cap seen on the same osteochondroma in Figure 5–15.
Figure 5–16. Typical glistening white appearance of the cartilage cap seen on the same osteochondroma in Figure 5–15.
A familial form of osteochondromata, called hereditary multiple exostosis (HME), is an autosomal dominant disorder that is one tenth as common as solitary osteochondroma. Three genetic loci are associated with HME involving the tumor suppressor EXT genes (EXT1, EXT2, and EXT3). This condition exhibits variable penetrance, with the severest forms resulting in severe angular deformities and limb shortening from hundreds of osteochondromata. Forearm involvement can be quite deforming. The metaphyseal portions of the long bones are deformed and widened (Figures 5–17 and 5–18). The histologic findings in the lesions of HME are similar to those in solitary osteochondroma.

![]() Figure 5–17. Radiograph of multiple exostoses involving both hips.
Figure 5–17. Radiograph of multiple exostoses involving both hips.

![]() Figure 5–18. Three-dimensional reconstructed CT scan of the bilateral shoulders and upper thorax of a skeletally immature female with hereditary multiple exostoses.
Figure 5–18. Three-dimensional reconstructed CT scan of the bilateral shoulders and upper thorax of a skeletally immature female with hereditary multiple exostoses.
Conversion to chondrosarcoma is exceedingly rare in solitary osteochondroma and occurs in adulthood. The rate of malignant transformation in HME is approximately 1%, occurring in the cartilaginous cap, usually in the larger proximal lesions.
Osteochondromas are stage 1 lesions. Most children with a solitary osteochondroma are asymptomatic and therefore do not require surgical treatment. In some cases, the lesion may be palpable and irritating. Surgical resection is appropriate in these cases to address the symptoms only and not as a prophylaxis for chondrosarcomatous degeneration. In HME, symptomatic lesions are addressed surgically as needed. Corrective osteotomy is occasionally required for angular deformity. If a previously quiescent lesion begins to enlarge in an adult, it should be removed. The surgical margin should be wide enough to include the entire cartilaginous cap.
D. Chondroblastoma: ICD-9-CM 213.x
The chondroblastoma is a benign cartilage-forming tumor that occurs in the epiphyses or apophyses. When it is diagnosed near or at skeletal maturity, it may expand across the physis or physeal scar. The peak incidence is during the second decade of life with a slight male predominance. The long bones are most often affected, but the patella, talus, and calcaneus are also commonly reported locations. There may be joint involvement presenting with an effusion.
Radiographically, there is sharp demarcation of a radiolucent lesion in the epiphysis with stippled or flocculent calcification. There may be erosion of the subchondral bone with collapse or pathologic fracture (Figure 5–19). There may also be a recognizable aneurismal component. Histologically, there is a background of uniform polyhedral cells with grooved nuclei producing sparse, amorphous chondroid. The cells may be separated by a fine lace of mineralization producing a “chicken wire” appearance. Osteoclast-like giant cells and macrophages are present, especially near areas of hemorrhage or aneurismal conversion.

![]() Figure 5–19. Radiograph of a chondroblastoma in the distal tibia of a 15-year-old boy.
Figure 5–19. Radiograph of a chondroblastoma in the distal tibia of a 15-year-old boy.
Although chondroblastoma presents in a younger age group than giant cell tumor, the two are comparable. Similarities include the location, radiographic appearance, and strikingly similar histologic features. They both typically present as stage 2 or 3 lesions. Also in common is the rare incidence of pulmonary metastasis. When pulmonary metastasis develops, the histology is the same, and they respond well to resection, carrying an excellent prognosis.
Treatment of chondroblastoma usually consists of intralesional curettage with margin extension and bone grafting or structural supplementation with polymethyl methacrylate. The recurrence rate is less that 10% with this form of treatment. When the subchondral bone has been destroyed or the lesion is otherwise more locally aggressive, wide resection with osteoarticular allograft reconstruction has been used with success. Transformation to secondary chondrosarcoma is extremely rare but occurs with increased frequency following radiation therapy.
E. Chondromyxoid Fibroma: ICD-9-CM 213.x
The chondromyxoid fibroma, a very rare tumor, generally affects males in the second or third decade of life. The most common location is the proximal tibial metaphysis, followed by the distal femur and the metatarsals. The tumor is slow growing and accompanied by mild pain and symptoms.
Radiographs of chondromyxoid fibroma show a lytic tumor with sharp sclerotic margins and a pseudoloculated pattern resembling that of a bone cyst. They are eccentric in metaphyseal bone with thinning of the involved cortex (Figure 5–20). Histologic findings include a strange but specific mixture of fibrous, myxomatous, and chondroid tissues, which could mistakenly suggest the diagnosis of chondrosarcoma. There are also frequent osteoclast-like giant cells. The expression pattern of collagens seems to be unique to this entity with predominantly type II, but also types I, III, and VI.

![]() Figure 5–20. Radiograph of a chondromyxoid fibroma in the proximal tibia of an 11-year-old boy.
Figure 5–20. Radiograph of a chondromyxoid fibroma in the proximal tibia of an 11-year-old boy.
Chondromyxoid fibroma usually presents as a stage 2 lesion and has a markedly high propensity for local recurrence. With recurrence rates approaching 25% following simple curettage and bone grafting, aggressive margin extension should be performed. The conversion of chondromyxoid fibroma to secondary chondrosarcoma is extremely rare.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• The matrix of chondroid tumors is characterized by stippled calcification or the presence of rings and arcs of calcification.
• The hallmark of osteochondroma is continuity of the medullary portion of the lesion with the host bone in contrast to the periosteal chondroma, where the host cortex separates the medullary canal of the host bone from the lesion itself.
• Chondromyxoid fibroma is a rare lesion but may be aggressive and have a very high rate of local recurrence.
![]() Benign Fibrous Tumors of Bone
Benign Fibrous Tumors of Bone
A. Fibrous Cortical Defect: ICD-9-CM 213.x
Fibrous cortical defects, or cortical desmoids, are small, hamartomatous fibromas seen almost exclusively in the metaphyseal areas of the lower extremities of growing children. They can be multiple, and as many as 25% of normal children demonstrate these asymptomatic lesions at 5 years of age. The lesions tend to disappear as a result of bone remodeling before skeletal maturity. They may show increased uptake on isotope bone scans.
In the case of fibrous cortical defects, microscopic studies show benign-appearing fibroblasts in a whorled pattern with occasional histiocytes, foam cells, and benign giant cells. The radiographic appearance is so characteristic of this entity (Figure 5–21) that a biopsy is usually not necessary. These are stage 1 lesions and can generally be observed.

![]() Figure 5–21. Radiograph of a metaphyseal fibrous cortical defect in a 15-year-old boy.
Figure 5–21. Radiograph of a metaphyseal fibrous cortical defect in a 15-year-old boy.
B. Nonossifying Fibroma: ICD-9-CM 213.x
Just as the osteoblastoma is considered a larger or more extensive form of osteoid osteoma, the nonossifying fibroma is considered a larger form of the fibrous cortical defect. It is typically seen in the lower extremity of children. Because of its size, it may not entirely resolve by skeletal maturity and can persist into adult life. If the lesion is quite large, approaching 50% of the diameter of the bone, pathologic fracture may ensue. The fracture healing process may facilitate resolution of the lesion. Careful consideration to fracture prophylaxis should be reserved for large lesions in children older than 10 years. Nonossifying fibromas are stage 1 lesions, and neither they nor fibrous cortical defects require biopsy because their radiographic appearance is so characteristic.
With nonossifying fibroma, multiple lesions may take on the appearance of fibrous dysplasia and can be associated with café-au-lait skin defects. Large defects in the tibia can assume the appearance of chondromyxoid fibroma (Figure 5–22). The lesions have a well-defined sclerotic margin with a pseudoloculated lytic center that gives them a soap-bubble radiographic appearance. Histologically, they appear identical to fibrous cortical defects and are characterized by abundant benign fibrous tissue speckled with areas of histiocytes, foam cells, and giant cells. As the lesion involutes in adulthood and the number of giant cells and histiocytes diminishes, large areas of cholesterol deposits become evident, which may suggest the diagnosis of xanthofibroma or xanthoma of bone. Nonossifying fibromas are clearly separated from fibrous dysplasia by the absence of metaplastic osteoid formation in the fibrous stroma.

![]() Figure 5–22. Radiograph of a nonossifying fibroma of the distal tibia.
Figure 5–22. Radiograph of a nonossifying fibroma of the distal tibia.
C. Fibrous Dysplasia: ICD-9-CM 756.54
Fibrous dysplasia can present in a variety of ways: monostotic, polyostotic, and with or without associated syndromes (Figure 5–23). Most cases are diagnosed in the first three decades and have a distinct female predilection. The monostotic presentation is more common than the polyostotic. This condition is a dysplastic anomaly of bone forming mesenchymal tissue with an inability to produce mature lamellar bone. Accordingly, the bone is arrested in an immature woven state with a resultant proliferation of spindled fibroblasts. In the polyostotic form, it tends to be unilateral rather than bilateral. Nevertheless, it can involve any bone in the body. The most common location is the proximal femur where it results in the so-called shepherd’s crook deformity. Other areas frequently involved include the tibia, pelvis, humerus, radius, and ribs.

![]() Figure 5–23. Radiograph of polyostotic fibrous dysplasia of the pelvis.
Figure 5–23. Radiograph of polyostotic fibrous dysplasia of the pelvis.
In addition to bony involvement, patients can demonstrate café-au-lait skin pigmentation. These patches usually have a rough border, in contrast to the smooth border of those seen in neurofibromatosis. Patients with fibrous dysplasia may have associated endocrine problems. For example, 5% of patients with the polyostotic form of fibrous dysplasia also exhibit precocious puberty (McCune-Albright syndrome). Other associated endocrine abnormalities include hyperthyroidism, acromegaly, Cushing disease, and hypophosphatemic osteomalacia. Polyostotic fibrous dysplasia with soft-tissue myxomas is known as Mazabraud syndrome. Fibrous dysplasia can also involve the skull and jaw bones, mimicking ossifying fibroma of jaw bone.
Radiographically, fibrous dysplasia has a ground-glass appearance due to the fine mineralization pattern of the immature woven trabeculae. There is surrounding remodeling of the host bone, which is often expansile. In fibrous dysplasia, microscopic findings include an alphabet soup pattern of metaplastic woven bone scattered through a benign fibrous tissue stroma. The woven trabeculae have a characteristic absence of osteoblastic rimming. Foam cells, giant cells, and cholesterol deposits can be seen. Large cystic areas and even areas of cartilage formation are commonly present.
The molecular basis for fibrous dysplasia is associated with mutations affecting the alpha subunit of G protein. Cells of the osteoblastic lineage are affected, resulting in decreased differentiation and increased proliferation. These mutations cause constitutive elevation of cyclic adenosine monophosphate (cAMP) in fibrous dysplasia and thus alter cAMP target genes such as c-fos, c-jun, IL-6, and IL-11.
Fibrous dysplasia tends to be active during the growing years and then burns out in adult life. Fewer than 1% of lesions convert to osteosarcoma, fibrosarcoma, or even chondrosarcoma. If conversion does occur, it almost always happens during adulthood. Generally, this disease is either stage 1 or 2.
In pediatric patients with active disease, curettage and grafting should be avoided because of high recurrence rates. The goals in treating pediatric patients should be the prevention and treatment of deformity, especially in the lower extremity. Most cases should become quiescent with skeletal maturity. If not, the best surgical treatment in adults consists of rigid fixation with an intramedullary implant with strut grafting as needed. Medical management with bisphosphonates is of benefit in some cases. Irradiation is contraindicated because it may lead to irradiation-induced sarcoma at a later date.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Nonossifying fibromas/fibrous cortical defects may be present in up to one third of the population and are usually detected incidentally.
• If fibrous dysplasia is suspected, a careful examination of the skin should be performed for café-au-lait spots, which are seen in McCune-Albright syndrome.
![]() Cystic Lesions of Bone
Cystic Lesions of Bone
A. Simple Bone Cyst: ICD-9-CM 733.21
Simple bone cysts are a common pseudotumor of bone and the most frequent cause of pathologic fractures in children. Bone cysts usually affect patients between 5 and 15 years of age and occur more often in boys than in girls (2:1) with an incidence of 1 per 10,000 children per year. They are found in the proximal humerus in 50% of cases and in the upper femur in 25%. The calcaneus and pelvis are also uniquely common locations. Patients are asymptomatic until a pathologic fracture occurs. The cystic process continues to grow away from the physis. When it remains in contact with the physis, it is termed “active.” When it separates, it is termed “inactive.”
Radiographs typically show a solitary cyst that is centrally located in the metaphyseal area and has marked thinning of the adjacent cortical bone and a pseudoloculated appearance (Figure 5–24). The bone cyst is filled with a clear serous fluid, and there is increased pressure during the active phase. The fact that this pressure gradually decreases as the cyst becomes inactive suggests a hydrodynamic mechanism. If there is associated fracture, radiographs may show the characteristic “fallen leaf” sign (Figure 5–25).

![]() Figure 5–24. Radiograph of a solitary bone cyst on the proximal humerus of a 13-year-old boy.
Figure 5–24. Radiograph of a solitary bone cyst on the proximal humerus of a 13-year-old boy.

![]() Figure 5–25. Radiographs of a solitary bone cyst with associated pathologic fracture and a “fallen leaf” sign in a 12-year-old girl.
Figure 5–25. Radiographs of a solitary bone cyst with associated pathologic fracture and a “fallen leaf” sign in a 12-year-old girl.
The cyst cavity, lined with a fibrinous membrane that contains giant cells, foam cells, and a slight osteoid formation, is similar to the fibrous tissues seen in other fibrous bone lesions, including fibrous dysplasia. The periosteal covering in the area of a cyst is normal, and thus the pathologic fractures heal normally and in most cases do not require surgery. Unfortunately, the cyst usually persists after fracture union and requires further treatment. Bone-resorbing factors, such as matrix metalloproteinases, prostaglandins, inter-leukin (IL)-1, IL-6, tumor necrosis factor-alpha (TNF-α), and oxygen-free radicals, are demonstrated in the cyst fluid. Nitrate and nitrite levels are also noted to be higher than in serum.
Before the mid-1970s, the standard treatment for a solitary bone cyst was aggressive curettage or even resection followed by bone grafting. In patients with active disease, the recurrence rate was 30–50%, and repeated grafting was frequently necessary. In patients with inactive disease, particularly those older than 15 years, the surgical results were much better and the recurrence rate was lower. Unicameral bone cysts are generally considered stage 1 lesions, but occasionally they may be stage 2. Currently, treatment is a function of location. In weight-bearing bones, such as the proximal femur, lesions should be treated aggressively. Initial management usually involves aspiration/injection with either bone marrow or corticosteroid. The injections are carried out with bone biopsy needles and are repeated three to five times at intervals of 2–3 months, depending on the radiographic response. The best results are when the patient is between 5 and 15 years of age, at which time the disease is active and macrophage activity is greatest in the cyst lining. Curettage and bone grafting may also be an effective modality. Demineralized bone matrix injected in combination with autogenous bone marrow shows encouraging results, with a relatively low recurrence rate and low morbidity.
Physicians should note that sarcomas can take on the radiographic appearance of a solitary bone cyst. For this reason, if needle aspiration does not reveal cystic fluid or if it is impossible to inject contrast material and obtain radiologic confirmation of the diagnosis, an open biopsy is indicated to rule out a sarcoma.
B. Aneurysmal Bone Cyst: ICD-9-CM 733.22
Aneurysmal bone cyst is a hemorrhagic lesion with many characteristics of a giant cell tumor but occurs only half as frequently. Although 75% of the cases of aneurysmal bone cyst occur in patients aged 10–20 years old, giant cell tumor is rare in patients younger than 20 years of age. Both aneurysmal bone cyst and giant cell tumor are more common in females than in males. The femur is the most frequently affected site, followed by the tibia, pelvis, and spine. In the spine, two thirds of aneurysmal bone cysts arise from the posterior elements, and one third arise from the vertebral body.
Initially, the aneurysmal bone cyst appears on radiograph as an aggressive osteolytic lesion with extensive permeative cortical destruction that gives the impression of a malignant process such as Ewing sarcoma or hemorrhagic osteosarcoma. Next, a large aneurysmal bulge occurs outside the bone, with a thin reactive shell of bone forming at the outer edge. Less soap-bubbly pseudoseptation is seen in an aneurysmal bone cyst than in a solitary bone cyst (Figure 5–26).

![]() Figure 5–26. Radiograph of an aneurysmal bone cyst on the proximal femur of a 5-year-old boy.
Figure 5–26. Radiograph of an aneurysmal bone cyst on the proximal femur of a 5-year-old boy.
At the time of biopsy, the aneurysmal bone lesion demonstrates large hemorrhagic cysts, but bleeding is modest. The hemorrhagic cysts are broken up by thick spongy fibrous septae that histologically contain great numbers of large giant cells and have thin osteoid seams. Even if a few mitotic figures are seen, the diagnosis of a benign lesion can remain. A carefully placed biopsy with multiple samples is needed to rule out other well-known skeletal tumors that may demonstrate an aneurysmal component. These include giant cell tumor, chondromyxoid fibroma, and malignant hemorrhagic osteosarcoma. Some authors believe there is no such entity as the aneurysmal bone cyst and that it is merely a morphologic variant of some other underlying neoplastic process. Like the solitary bone cyst, this cyst may have a hydraulic pressure origin that is secondary to hemorrhage and could be traumatically induced. However, abnormal cytogenetic findings were noted in aneurysmal bone cysts, which may suggest a distinct cellular pathogenetic etiology. Specifically, a t(16,17) translocation resulting in a CDH11-USP6 fusion gene product is frequently observed in aneurismal bone cyst. Aneurysmal bone cyst is either a stage 2 or 3 lesion and frequently symptomatic.
If an aneurysmal bone cyst is left untreated, it may involute spontaneously, during which time it develops a heavy shell of reactive bone at the periphery. This involutional process can be hastened by surgical curettage and bone grafting. Radiation is no longer recommended. Another option for treating extremely large lesions is repeated embolization to reduce the rate of hemorrhagic expansion.
C. Epidermoid Cyst: ICD-9-CM 213.x
The least common bone cyst is the epidermoid bone cyst. This lesion is found either in the distal phalanx or in the skull. No other bone is affected. In the case of the phalanx, the cyst is usually the result of nail bed epithelium being driven into the distal phalanx by a crushing blow. The ectopic squamous epithelium produces a keratinized cavity that is filled with clear fluid and creates a surface erosion with a sclerotic reactive base (Figure 5–27). The bulbous cyst seen at the fingertip transilluminates with flashlight examination. Other conditions that might have a similar appearance are the glomus tumor and the enchondroma. The epidermoid cyst is treated with a simple curettage and, in some cases, a bone graft.

![]() Figure 5–27. Radiograph of an epidermoid cyst in the distal phalanx.
Figure 5–27. Radiograph of an epidermoid cyst in the distal phalanx.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• In the treatment of solitary bone cysts, there is a significant rate of recurrence, but the cysts will usually resolve with skeletal maturity.
• If an aneurysmal bone cyst is suspected in the differential diagnosis prior to biopsy, then a telangiectatic osteosarcoma should also be considered.
![]() Giant Cell Tumor of Bone: ICD-9-CM 213.x
Giant Cell Tumor of Bone: ICD-9-CM 213.x
Numerous types of tumors contain giant cells but are not true benign giant cell tumors of bone. Most of the variants are seen in children and include aneurysmal bone cyst, chondroblastoma, simple bone cyst, osteoid osteoma, and osteoblastoma. The giant cell–rich osteosarcoma is the most malignant of the variants, and it is sometimes difficult to distinguish from an aggressive benign giant cell tumor. The giant cell reparative granuloma is a benign variant seen in jaw bones or hand bones and has more spindle cells than a classic giant cell tumor. The brown tumor of hyperparathyroidism is a nonneoplastic variant seen in both primary and secondary hyperparathyroidism. Only after all of the variant conditions are excluded can the diagnosis of benign giant cell tumor be made. Giant cell tumor of bone is now associated with an imbalance in the receptor activator of nuclear factor kappa B/receptor activator of nuclear factor kappa B ligand (RANK/RANKL) system, which is normally associated with osteoclastogenesis.
Between 5 and 10% of all benign bone tumors are true giant cell tumors, occurring most frequently in the third decade of life. They are more frequently found in females than in males. In approximately half of the cases, the tumor is found about the knee. The next most common locations are the distal radius and the sacrum. The tumor is usually painful for several months prior to diagnosis and can cause a pathologic fracture. It can also cause a painful effusion because of its juxtaposition to a major joint. Giant cell tumors may present as either stage 2 or stage 3 disease and less frequently as stage 1. On radiograph, the lesion appears lytic in nature and is located in the epiphyseal-metaphyseal end of a long bone (Figure 5–28). The lesion grows toward the joint surface and frequently comes into contact with articular cartilage but rarely breaks into the joint.

![]() Figure 5–28. Radiograph of a giant cell tumor on the proximal tibia of a 22-year-old woman.
Figure 5–28. Radiograph of a giant cell tumor on the proximal tibia of a 22-year-old woman.
Like the chondroblastoma, the benign giant cell tumor has a 1–2% chance of metastasizing to the lung. Recurrent tumors have up to a 10% chance. Accordingly, pulmonary staging is an important component in the initial evaluation and follow-up of giant cell tumor of bone. The prognosis for survival with this complication is favorable, and the tumors may resolve spontaneously. The benign giant cell tumor can later convert to a malignant condition such as an osteosarcoma or malignant fibrous histiocytoma. It is generally believed that this is secondary to treatment. A conversion rate of 15–20% is reported in patients who were treated previously with more than 3000 cGy of radiation, with conversion occurring 3 or more years after treatment. The conversion rate in patients who do not receive radiation is less than 5%. This finding has come into question with newer radiation therapy modalities.
Until recent years, the standard treatment for giant cell tumor was curettage and bone grafting. The recurrence rate with this treatment was reported to be up to more than 50%. Follow-up treatment consisted of an aggressive resection of the lesion and reconstruction with a large osteoarticular allograft, endoprosthesis, or an excisional arthrodesis. Currently, most surgeons elect an aggressive curettage, followed by high-speed burring and adjuvant phenol, hydrogen peroxide, or liquid nitrogen and by subsequent packing of the defect with bone cement. With this new approach, the recurrence rate is between 10 and 25%. When giant cell tumor infrequently involves an expendable bone, such as the fibula or ilium, it should be primarily resected. En bloc resection continues to be used to treat multiple recurrent tumors, intensive soft-tissue involvement, or massively destructive cases. Embolization may also prove palliative or curative in unresectable cases. For advanced, multiply recurrent, or aggressive metastatic cases, investigators are developing and testing experimental medical protocols, but these remain to be proven. Close follow-up for locally recurrent disease and pulmonary involvement is critical. Surveillance should include a plain chest radiograph every 6–12 months for the first 2–3 years at least.
![]() Hemangioma: ICD-9-CM 213.x
Hemangioma: ICD-9-CM 213.x
Hemangioma of bone is a hamartomatous process that occurs more frequently in females than in males. It is most commonly found in vertebral bodies. It is found only rarely in the diaphysis of a long bone (Figure 5–29). Hemangiomas of bone can be associated with hemangiomas of soft tissue. The spinal lesion is usually discovered as an incidental radiographic finding and demonstrates a characteristic vertically oriented honeycombed or moth-eaten appearance. On rare occasions, a lesion can cause cord compression that may require surgical resection. In such cases, preoperative angiography is critical in evaluating the blood supply to the spinal cord. Alternatively, an attempt at arterial embolization may prove successful and is less aggressive.
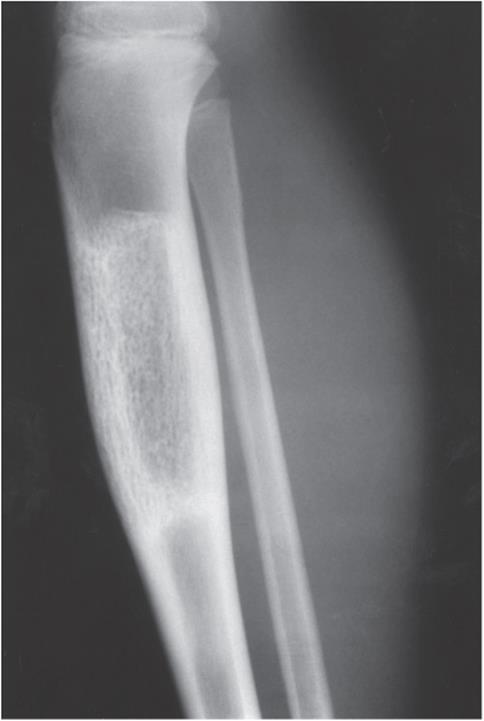
![]() Figure 5–29. Radiograph of a hemangioma of the tibia in a 14-year-old boy.
Figure 5–29. Radiograph of a hemangioma of the tibia in a 14-year-old boy.
Gorham disease, characterized by massive osteolysis in children or young adults, is usually associated with the presence of benign cavernous hemangiomas or lymph-angiomas of bone. This strange condition usually affects a particular area (such as the spine or the hip) but can involve multiple bones of the area and tends to resolve spontaneously (Figure 5–30).

![]() Figure 5–30. Radiograph of a pelvis affected with Gorham disease in a 48-year-old woman.
Figure 5–30. Radiograph of a pelvis affected with Gorham disease in a 48-year-old woman.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Plain radiography is usually diagnostic, and advanced imaging is usually not required except for preoperative planning in benign bone lesions.
• Surgical treatment of most benign bone tumors is reserved for symptomatic lesions unresponsive to conservative measures, those at significant risk of fracture, or for documented enlargement over time.
• Giant cell tumor of bone and chondroblastoma both have an incidence of pulmonary seeding, and chest imaging should be included in the workup and surveillance of these entities.
Balke M, Ahrens H, Streitbuerger A, et al: Treatment options for recurrent giant cell tumors of bone. J Cancer Res Clin Oncol 2009;135:149. [PMID: 18521629]
Balke M, Schremper L, Gebert C, et al: Giant cell tumor of bone: treatment and outcome of 214 cases. J Cancer Res Clin Oncol 2008;134:969. [PMID: 18322700]
Baruffi MR, Neto JB, Barbieri CH, et al: Aneurysmal bone cyst with chromosomal changes involving 7q and 16p. Cancer Genet Cytogenet 2001;129:177. [PMID: 11566352]
Bottner F, Roedl R, Wortler K, et al: Cyclooxygenase-2 inhibitor for pain management in osteoid osteoma. Clin Orthop Relat Res 2001;393:258. [PMID: 11764357]
Bovee JV, van Roggen JF, Cleton-Jansen AM, et al: Malignant progression in multiple enchondromatosis (Ollier’s disease): an autopsy-based molecular genetic study. Hum Pathol 2000;31:1299. [PMID: 11070122]
Cantwell CP, Obyrne J, Eustace S: Current trends in treatment of osteoid osteoma with an emphasis on radiofrequency ablation. Eur Radiol 2004;14:607. [PMID: 14663625]
DiCaprio MR, Enneking WF: Fibrous dysplasia. J Bone Joint Surg Am 2005;87:1848. [PMID: 16085630]
Flemming DJ, Murphey MD, Carmichael BB, et al: Primary tumors of the spine. Semin Musculoskelet Radiol 2000;4:299. [PMID: 11371321]
Harish S, Saifuddin A: Imaging features of spinal osteoid osteoma with emphasis on MRI findings. Eur Radiol 2005;15:2396. [PMID: 15973540]
Kjar RA, Powell GJ, Schilcht SM, et al: Percutaneous radiofrequency ablation for osteoid osteoma: experience with a new treatment. Med J Aust 2006;184:563. [PMID: 16768663]
Knochentumoren A: Local recurrence of giant cell tumor of bone after intralesional treatment with and without adjuvant therapy. J Bone Joint Surg Am 2008;90:1060. [PMID: 18451399]
Oliveira AM, Hsi BL, Weremowicz S, et al: USP6 (Tre2) fusion oncogenes in aneurysmal bone cyst. Cancer Res 2004;64:1920. [PMID: 15026324]
Parekh SG, Donthineni-Rao R, Ricchetti E, et al: Fibrous dysplasia. J Am Acad Orthop Surg 2004;12:305. [PMID: 15469225]
Radhakrishnan K, Rockson SG: Gorham’s disease: an osseous disease of lymphangiogenesis. Ann N Y Acad Sci 2008;1131:203. [PMID: 18519972]
Randall RL, Nork SE, James PJ: Aggressive aneurysmal bone cyst of the proximal humerus. Clin Orthop Relat Res 2000;370:212. [PMID: 10660716]
Robinson P, White LM, Sundaram M, et al: Periosteal chondroid tumors: radiologic evaluation with pathologic correlation. Am J Roentgenol 2001;177:1183. [PMID: 11641198]
Romeo S, Oosting J, Rozeman LB, et al: The role of noncartilage-specific molecules in differentiation of cartilaginous tumors: lessons from chondroblastoma and chondromyxoid fibroma. Cancer2007;110:385. [PMID: 17559135]
Rougraff BT, Kling TJ: Treatment of active unicameral bone cysts with percutaneous injection of demineralized bone matrix and autogenous bone marrow. J Bone Joint Surg Am 2002;84-A:921. [PMID: 12063325]
Salerno M, Avnet S, Alberghini M, et al: Histogenic characterization of giant cell tumor. Clin Orthop Relat Res 2008;466:2081. [PMID: 18543051]
Staals EL, Bacchini P, Mercuri M, et al: Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am 2007;89:987. [PMID: 17473135]
Suneja R, Grimer RJ, Belthur M, et al: Chondroblastoma of bone: long-term results and functional outcome after intralesional curettage. J Bone Joint Surg Br 2005;87:974. [PMID: 15972914]
Sung AD, Anderson ME, Zurakowski D, et al: Unicameral bone cyst: a retrospective study of three surgical treatments. Clin Orthop Relat Res 2008;466:2519. [PMID: 18679761]
MALIGNANT BONE TUMORS
Malignant bone tumors are primarily treated with wide resection followed by limb salvage surgery in current treatment regimens. Depending on the histology, this is augmented with the use of adjuvant chemotherapy or radiation therapy or both. Limb salvage surgery has been advanced significantly in the past two decades with improvements in megaprostheses and techniques associated with the use of allografts. Recent series report survival of megaprostheses about the knee of 80–90% and 60–80% at 5 and 10 years, respectively. Newer methods of fixation of megaprostheses are showing promising results for long-term survival and ease of revision surgery.
Allograft reconstruction continues to be useful but is used less frequently in the reconstruction of a major weight-bearing joint and is now primarily used in metadiaphyseal reconstructions.
![]() Osteoid-Forming Sarcomas
Osteoid-Forming Sarcomas
Aside from multiple myeloma, osteosarcoma of bone is the most common primary malignant tumor of bone, constituting 20% of all primary malignancies of bone. In the United States, between 500 and 1000 new cases are diagnosed each year. The global incidence is felt to be between 1 and 3 per million people annually. There are currently many subtypes of osteoid-forming sarcomas, ranging from the extremely low-grade variants, such as parosteal osteosarcoma, to the extremely high-grade variants, such as osteosarcoma secondary to Paget disease.
The molecular pathobiology is a subject of intense investigation. Several gene families were investigated as potential biomarkers of disease progression. Among these are genes involved with angiogenesis (eg, vascular endothelial growth factor [VEGF]), growth factors and their receptors (eg, transforming growth factor beta, Wnt receptor LRP5, HER2), cytoskeletal protein (eg, ezrin), and cellular senescent protein (ie, telomerase).
This discussion begins with the more common, central form of sarcoma that is seen in children and known as classic osteosarcoma.
A. Classic Osteosarcoma: ICD-9-CM 170.x
The classic form of osteosarcoma is typically seen in patients in their second or third decade, with a peak in the adolescent growth spurt. It occurs more frequently in males than in females and is found in the metaphyseal areas of long bones, with 50% of lesions about the knee joint (Figures 5–31 and 5–32). The distal femur is the most common site, followed by the proximal tibia and then the proximal humerus. It is rare to see osteosarcoma in the small bones of the feet or hands or in the spine. When seen in the foot, it occurs in the larger bones of the hindfoot. The prognosis is more favorable for a tumor in a small bone than for one in a large bone.

![]() Figure 5–31. Osteosarcoma of the distal femur of a 15-year-old female patient. Notice the sunburst appearance.
Figure 5–31. Osteosarcoma of the distal femur of a 15-year-old female patient. Notice the sunburst appearance.
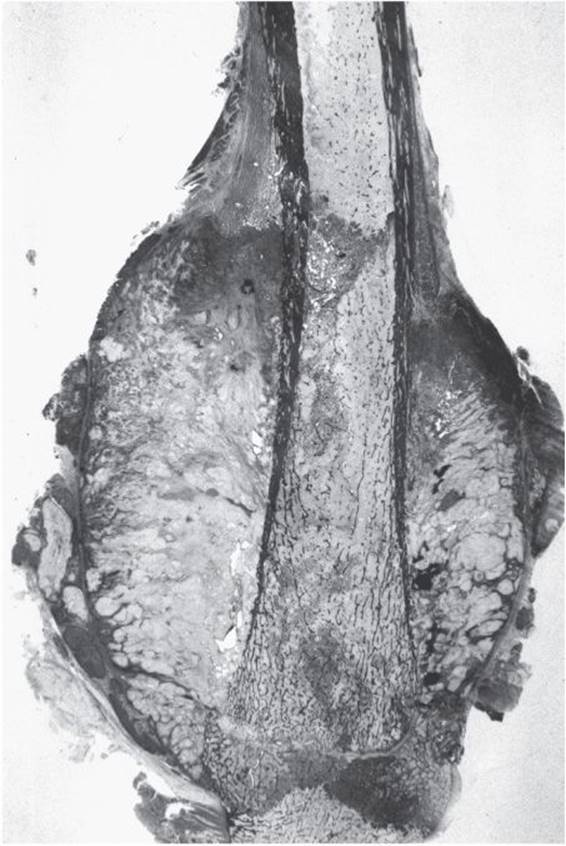
![]() Figure 5–32. Gross surgical specimen from Figure 5–31. Notice the sharp upper medullary margin located about the same level as the extracortical mass. The tumor has not invaded the growth plate.
Figure 5–32. Gross surgical specimen from Figure 5–31. Notice the sharp upper medullary margin located about the same level as the extracortical mass. The tumor has not invaded the growth plate.
Most patients with classic osteosarcoma have symptoms of pain before a tumor is noticeable. A mass near a major joint may exist for several weeks or even months before a diagnosis is made. Dilated veins may be seen in the overlying skin. The radiographic findings include permeated lytic destruction of metaphyseal bone, with eventual cortical breakthrough into the subperiosteal space and subsequent formation of a Codman triangle at the diaphyseal end of the tumor (Figure 5–33). As the tumor continues to push its way into the extracortical soft tissue, a typical sunburst pattern of neoplastic bone may be seen outside the involved bone.
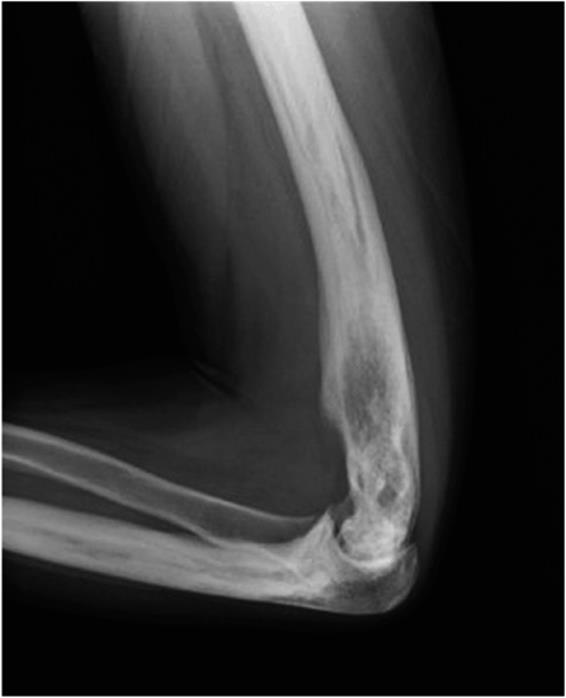
![]() Figure 5–33. Radiograph of the elbow of a 16-year-old patient who carries diagnoses of hereditary retinoblastoma and melorheostosis. Increasing elbow pain and the anterior diaphyseal Codman triangle were the presentation of an underlying osteosarcoma.
Figure 5–33. Radiograph of the elbow of a 16-year-old patient who carries diagnoses of hereditary retinoblastoma and melorheostosis. Increasing elbow pain and the anterior diaphyseal Codman triangle were the presentation of an underlying osteosarcoma.
In fewer than 2–25% of cases, an additional lesion may be found at a higher level in the femur. Such a so-called skip lesion may portend a worse probability of survival and should be considered a true metastatic focus (stage III [Enneking], stage III [AJCC]). Approximately 50% of osteosarcomas are of the more typical osteoblastic type, followed by chondroblastic, with a small percentage of them fibroblastic. Whether the subtype portends a better or worse prognosis is controversial. Confounding variables such as multidrug resistance (P-glycoprotein expression) may be differentially expressed in different subtypes. P-glycoprotein overexpression itself bears a substantial relationship to clinical outcome. More recently, higher serum VEGF levels, as a presumed surrogate marker for higher levels of tumor VEGF production, have been associated with poorer survival.
Staging of osteosarcoma must include an MRI of the entire involved bone (Figure 5–34). This technique offers excellent contrast of the extracortical portion of the tumor and at the same time gives good intramedullary contrast of the high-signal tumor next to a low-signal fatty marrow. The periphery of the tumor can readily be appreciated and represents the most anaplastic and rapidly growing part of the tumor. This region is the best tissue for a biopsy because it is easy to reach, soft enough for a diagnostic frozen section, and representative of the most aggressive portion of the tumor. Furthermore, the MRI provides the necessary anatomic data to determine the level of transection through the host bone for a safe margin and to determine whether a limb-sparing procedure is feasible.

![]() Figure 5–34. Short tau inversion recovery sequence MRI of osteosarcoma of the femur of a 19-year-old woman.
Figure 5–34. Short tau inversion recovery sequence MRI of osteosarcoma of the femur of a 19-year-old woman.
Before the advent of adjuvant multidrug chemotherapy, the treatment of osteosarcoma was radical amputation. Eighty percent of these patients proceeded to die from disseminated pulmonary disease. Today, with the combination of chemotherapy and surgical treatment, the prognosis for 5-year survival approaches 70%.
The drugs commonly used today include high-dose methotrexate, doxorubicin, cisplatin, and ifosfamide. The use of interferon in poor responders and patients suffering from disseminated disease is currently under investigation. They are administered intravenously in cyclic intervals of 3–4 weeks for approximately 11–15 weeks prior to surgery. Surveillance imaging studies are performed during this period to assess possible reduction in tumor volume. Tumor necrosis secondary to neoadjuvant chemotherapy, determined at the time of tumor resection, is an important prognostic factor. Patients with greater than 90% tumor necrosis have a significantly improved 5-year survival rate, approaching 85%. Approximately half of patients demonstrate this response to current chemotherapy regimens. Furthermore, the postoperative drug regimen can be adjusted based on this evaluation.
In extremity osteosarcoma, limb-sparing surgery, with wide resection of the tumor, is the standard approach. Amputation is reserved for the exceptional or recurrent case. In fewer than 10% of cases, amputation is performed at a level approximately 5 cm above the upper pole of the tumor. Limb salvage techniques continue to evolve with reconstruction options including large prostheses, structural allografts, and composite reconstructions. Endoprosthetics are composed of modular components in various lengths, linked together with taper fittings (Figures 5–35 and 5–36). The intramedullary stems are of various diameters and lengths and are usually cemented. The immediate functional results are excellent, with minimal early complications. However, subsequent loosening at 5–10 years occurs in as many as 15–30% of cases. Newer methods of fixation of megaendoprosthetics are showing promising results. Another limb-sparing procedure consists of the use of an osteoarticular allograft alone or in combination with a prosthesis. The major drawback with large bone allografts is a 10–15% chance of infection, nonunion, or stress fracture, especially in the immunosuppressed patient receiving chemotherapy. The use of an excisional arthrodesis was more popular in the past but is rarely elected today because patients have better function with a mobile joint.

![]() Figure 5–35. (A) Two examples of distal femoral replacement systems. (B) Modularity of system allows different-size intercalary body segments.
Figure 5–35. (A) Two examples of distal femoral replacement systems. (B) Modularity of system allows different-size intercalary body segments.

![]() Figure 5–36. Lateral radiograph of distal femoral replacement system in skeletally immature patient. Expansion with larger intercalary body segment is possible.
Figure 5–36. Lateral radiograph of distal femoral replacement system in skeletally immature patient. Expansion with larger intercalary body segment is possible.
Prior to the introduction of chemotherapy, the finding of pulmonary metastasis portended a very poor prognosis. Today, however, in larger tumor centers where aggressive surgical approaches with multiple thoracotomies and continued chemotherapy are used, the 5-year survival rate is approximately 30%. Patients with skeletal metastases, or so-called metachronous osteosarcoma, however, have a significantly worse prognosis, unless it is a solitary, resectable metastasis.
Molecular oncologic evaluation of osteosarcoma specimens is beginning to elucidate factors involved in its pathogenesis. The p53 suppressor genes have an increased mutation rate in osteosarcoma. Osteosarcomas with a mutation in the p53 gene have a significantly higher rate of genomic instability, including multiple duplicate chromosomes and frequent hyperdiploid state. However, wild-type TP53 and MDM2 in and of themselves are not of prognostic value. Loss of heterozygosity of the Rb gene is a predictive feature of osteosarcoma. The F33 isoform also demonstrates a strong correlation with osteosarcoma disease progression. ErbB-2 (HER-2/neu), a protooncogene, and transforming growth factor-beta, isoform 3 expression is also correlated with a worse prognosis in osteosarcoma patients. Controversy surrounds the significance of cytoplasmic versus membranous staining in HER-2/neu expression as it relates to prognosis in osteosarcoma.
B. Hemorrhagic or Telangiectatic Osteosarcoma: ICD-9-CM 170.x
Telangiectatic osteosarcoma, an extremely lytic and destructive variant of classic osteosarcoma, is seen in the same age group and location. Its radiographic appearance is similar to that of an aneurysmal bone cyst, thereby making the diagnosis difficult (Figure 5–37). The pathologic specimen is hemorrhagic, with microscopic evaluation demonstrating the presence of malignant-appearing stromal cells with giant cells.

![]() Figure 5–37. Radiograph of hemorrhagic osteosarcoma in a 6-year-old girl.
Figure 5–37. Radiograph of hemorrhagic osteosarcoma in a 6-year-old girl.
Because hemorrhagic osteosarcoma is a high-grade, purely lytic tumor, the incidence of pathologic fracture in the early course of the disease is high. If significant contamination of the adjacent neurovascular structures results, a pathologic fracture may necessitate amputation rather than limb salvage (Figure 5–38). This situation must be carefully evaluated with a preoperative MRI. Accordingly, in cases with significant risk for fracture during the preoperative treatment regimen, it may be appropriate to immobilize the involved extremity or proceed with limb-sparing surgery earlier than usual. Prior to the advent of aggressive multidrug chemotherapy, the prognosis for patients with hemorrhagic osteosarcoma was extremely poor. At present, however, it is the same as the prognosis for patients who have classic osteosarcoma and is treated with similar protocols.

![]() Figure 5–38. Clinical photograph of patient who sustained pathologic fracture through a distal femoral osteosarcoma contaminating the neurovascular structures precluding limb salvage.
Figure 5–38. Clinical photograph of patient who sustained pathologic fracture through a distal femoral osteosarcoma contaminating the neurovascular structures precluding limb salvage.
C. Parosteal Osteosarcoma: ICD-9-CM 170.x
Parosteal osteosarcoma is a low-grade variant arising in an exophytic pattern from the cortical surface of bone. There is no medullary involvement. It is low grade, with a 5-year survival rate in excess of 90% and a 10-year survival rate of 80%. It accounts for 3–4% of all osteosarcomas.
The tumor is composed of a spindle cell fibroblastic component with well-developed bone trabeculae. There also may be areas of cartilage present. Osteoblasts are well differentiated, and few mitotic figures are present.
Parosteal osteosarcoma is more common in females than in males and affects a slightly older age group than classic osteosarcoma (see Table 5–2). It is a slow-growing tumor with minimal symptoms initially. It is metaphyseal in origin, with the vast majority of cases involving the posterior aspect of the distal femur (Figure 5–39).

![]() Figure 5–39. Radiograph (A) and CT scan (B) of a parosteal osteosarcoma of the distal femur in a 21-year-old woman.
Figure 5–39. Radiograph (A) and CT scan (B) of a parosteal osteosarcoma of the distal femur in a 21-year-old woman.
Because the parosteal osteosarcoma is low grade, it does not respond well to either chemotherapy or radiation therapy. Therefore, the only treatment is wide surgical resection. This usually requires distal femur removal, but in smaller cases, side resection of the posterior cortex and tumor only may be feasible, sparing the knee joint. Nevertheless, a negative tumor margin is imperative. Otherwise, recurrence is likely. Recurrence may occur as late as 5–10 years because of the tumor’s slow growth.
On occasion, low-grade parosteal osteosarcoma can dedifferentiate into a high-grade sarcoma. Such a lesion carries a similar prognosis to classic osteosarcoma.
D. Periosteal Osteosarcoma: ICD-9-CM 170.x
Periosteal osteosarcoma is another surface osteosarcoma of low to intermediate grade. This lesion represents less than 2% of all osteosarcomas. It arises beneath the periosteum, elevating it and inducing vigorous neoosteogenesis with a predominant chondroblastic differentiation. It is slightly more common in females, with a peak incidence in the second decade of life. It almost exclusively arises in the long bone. The lesion can mimic an aneurysmal bone cyst or periosteal chondroma radiographically (Figure 5–40).

![]() Figure 5–40. Radiograph of a periosteal osteosarcoma of the distal tibia in a 15-year-old boy.
Figure 5–40. Radiograph of a periosteal osteosarcoma of the distal tibia in a 15-year-old boy.
Because of its low to intermediate grade, periosteal osteosarcoma is generally not treated with chemotherapy but may be in more advanced cases. Wide surgical resection is the modality of choice. It also carries a better prognosis than classic osteosarcoma. Approximately 25% of patients succumb to metastatic disease within 2–3 years. The surgical treatment is usually a limb-sparing procedure, and because the tumor is more diaphyseal in location, the adjacent joints may often be spared.
E. Secondary Osteosarcoma: ICD-9-CM 170.x
Osteosarcoma can arise from benign disease through a process that may involve a second mutation and usually occurs at a later age (see Table 5–2). Among the benign conditions that can result in secondary osteosarcoma are Paget disease, osteoblastoma, fibrous dysplasia, benign giant cell tumor, bone infarction, and chronic osteomyelitis.
The classic example of a secondary osteosarcoma is seen in a small percentage of patients with Paget disease. Pagetic osteosarcomas, which represent approximately 3% of all osteosarcomas, are the most common osteosarcomas in the older (>65 years) age group. The most frequent location for pagetic osteosarcoma is the humerus, followed next by the pelvis and femur. The typical patient has a long history (15–25 years) of dull, aching pain associated with the inflammation of Paget disease before a new acute pain arises in an area of recent lytic destruction and the diagnosis of pagetic osteosarcoma is established (Figure 5–41). The prognosis for patients with pagetic osteosarcoma is extremely poor (5-year survival rate of approximately 8%). Because of the older age group involved, chemotherapy is usually not an option secondary to intolerance.

![]() Figure 5–41. Radiograph of a pagetic osteosarcoma of the tibia.
Figure 5–41. Radiograph of a pagetic osteosarcoma of the tibia.
F. Low-Grade Intramedullary Osteosarcoma: ICD-9-CM 170.x
Another rare and low-grade osseofibrous variant of osteosarcoma is the central or intramedullary form. Although this variant has a microscopic appearance similar to that of parosteal osteosarcoma, it is usually located in metaphyseal bone about the knee joint in adults between 15 and 65 years of age. Males and females are equally affected. Radiographically, intramedullary osteosarcoma creates a sclerotic density in metaphyseal bone (Figure 5–42). Like the parosteal osteosarcoma, the low-grade intramedullary osteosarcoma carries an excellent prognosis and can be treated with local surgery alone.

![]() Figure 5–42. Radiograph (A) and CT scan (B) of a low-grade intramedullary osteosarcoma in the distal femur of a 65-year-old man.
Figure 5–42. Radiograph (A) and CT scan (B) of a low-grade intramedullary osteosarcoma in the distal femur of a 65-year-old man.
G. Irradiation-Induced Osteosarcoma: ICD-9-CM 170.x
Radiation-induced osteosarcoma may arise after any form of significant radiation exposure (in excess of 30 Gy) (Figure 5–43). Onset is usually delayed an average of 15 years (range, 3–55 years). Other irradiation-induced sarcomas, besides the osteosarcoma type, include irradiation-induced fibrosarcoma and malignant fibrous histiocytoma. All of these secondary sarcomas are invariably high grade and carry a poor prognosis for survival, with a very high rate of metastasis.

![]() Figure 5–43. Radiograph of irradiation-induced osteosarcoma of the peritrochanteric area in a 35-year-old woman.
Figure 5–43. Radiograph of irradiation-induced osteosarcoma of the peritrochanteric area in a 35-year-old woman.
H. Multicentric Osteosarcoma: ICD-9-CM 170.x, or 199.0
Multicentric osteosarcoma has two clinical presentations: (1) synchronous, occurring in childhood and adolescence, and (2) metachronous, occurring in adults. The synchronous type is a high-grade sclerosing intramedullary type, which is lethal. The adult form is less aggressive, with a lower-grade histo-logic appearance, but prognosis remains grim (Figure 5–44).

![]() Figure 5–44. Isotope bone scan of multicentric osteosarcoma in an 8-year-old girl.
Figure 5–44. Isotope bone scan of multicentric osteosarcoma in an 8-year-old girl.
I. Soft-Tissue Osteosarcoma: ICD-9-CM 171.x
Osteosarcoma can occur in muscle tissue outside bone and accounts for approximately 4% of all osteosarcomas (Figure 5–45). Soft-tissue osteosarcoma is rarely seen in patients younger than 40 years. The number of cases is equal in females and males, and the tumor is usually seen in large muscle groups of the pelvis and thigh areas.

![]() Figure 5–45. Radiograph of a soft-tissue osteosarcoma in the calf area of a 67-year-old woman.
Figure 5–45. Radiograph of a soft-tissue osteosarcoma in the calf area of a 67-year-old woman.
Soft-tissue osteosarcoma must be differentiated from the more common myositis ossificans. Although soft-tissue osteosarcoma shows heavy mineralization in the central area (see Figure 5–45), myositis ossificans has a zonal pattern of ossification, with the mature, dense ossification concentrating at the periphery of the lesion.
The treatment of soft-tissue osteosarcoma is the same as for the high-grade osseous form and includes a wide resection and adjuvant chemotherapy. The prognosis is worse with the soft-tissue form of osteosarcoma, with a high rate of chemotherapy resistance.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Osteosarcoma is best confirmed by biopsy of the leading edge of the soft-tissue component if possible.
• Parosteal osteosarcoma usually occurs on the posterior aspect of the distal femur, exhibiting a “stuck-on” appearance.
![]() Chondroid-Forming Sarcomas
Chondroid-Forming Sarcomas
Chondroid-forming sarcomas are a heterogeneous group of neoplasms consisting of a cartilage-based histology. A cornerstone to the diagnosis of chondrosarcoma is the absence of osteoid formation. If any osteoid is present with a malignant stroma, the tumor is considered an osteosarcoma with chondroblastic features. It is important to make the distinction because chondrosarcomas behave differently from osteosarcomas. However, this can be a difficult task. The surgeon must consider the age of the patient and carefully assess the radiographic and histologic features to confirm the diagnosis.
A. Primary or Central Conventional Chondrosarcoma: ICD-9-CM 170.x
The typical primary chondrosarcoma is a low-grade tumor seen in adults between 30 and 60 years of age. The tumor is found more frequently in men than in women. Minimal symptoms of pain may occur over a period of several years before a radiograph is obtained. The pelvis and femur are the most common locations, followed by the rib cage, proximal humerus, scapula, and upper tibia. Primary chondrosarcoma is extremely rare in small bones, including the hand and foot. The metaphysis is the most common location in a long bone; however, a diaphyseal location is not unusual.
Approximately 85% of central chondrosarcomas are low-grade lesions with a typical matrix calcification that can be described as flocculated with multiple rings and arcs. Radiographic criteria are more useful in distinguishing between enchondroma and low-grade chondrosarcoma than are histologic ones. Frequently cited radiographic criteria suggestive of more aggressive biologic potential include endosteal scalloping greater than 50% of the cortical width, change over time, and adjacent radiolucency near an area of typical chondroid matrix calcification (Figure 5–46). The high-grade lesions are rare, and radiographically, they lose their typical lobulated and calcific pattern and take on the appearance of a more permeative high-grade tumor, such as a malignant fibrous histiocytoma. At the same time, histologically, the high-grade chondrosarcomas lose their chondroid matrix pattern, which is replaced with that of a more aggressive spindle cell tumor.

![]() Figure 5–46. Radiograph of a low-grade primary chondrosarcoma in the distal femur of an 83-year-old man.
Figure 5–46. Radiograph of a low-grade primary chondrosarcoma in the distal femur of an 83-year-old man.
Because of the weakened cortex, the patient usually complains of local pain not experienced with an enchondroma. Because most chondrosarcomas are low grade, they do not respond well to adjuvant irradiation or chemotherapy. Therefore, aggressive surgical management is imperative. However, optimal surgical management is controversial. Although wide en bloc resection is ideal from a margin standpoint, it can often produce considerable morbidity. On the contrary, aggressive intralesional resection (curettage) and margin expansion with adjuvant therapy (eg, phenol or liquid nitrogen) can reduce morbidity and may provide equal local control. In fact, some authors found that for grade 1 chondrosarcoma the margin of resection is not significant in terms of local recurrence or disease progression.
In general, the prognosis for low-grade central chondrosarcoma is very good, with a low rate of pulmonary metastasis if the primary lesion is widely resected. Nevertheless, recurrences can occur late, even over 15 years later. For any intermediate- or high-grade chondrosarcomas, wide en bloc resection is mandatory (Figure 5–47).


![]() Figure 5–47. Preoperative radiograph of a large central chondrosarcoma in the proximal humerus of a 52-year-old woman (A), placement of a Neer prosthesis (B), and postoperative radiograph (C).
Figure 5–47. Preoperative radiograph of a large central chondrosarcoma in the proximal humerus of a 52-year-old woman (A), placement of a Neer prosthesis (B), and postoperative radiograph (C).
B. Secondary Chondrosarcoma: ICD-9-CM 170.x
The vast majority of secondary chondrosarcomas arise from osteochondromas in patients afflicted with HME. Patients with solitary osteochondromas do not generally form secondary chondrosarcomas in their lesions, making prophylactic removal unnecessary and unwarranted unless the solitary lesion is otherwise symptomatic. Even in patients with HME, the rate of malignant degeneration is less than 1% and generally does not occur in patients prior to skeletal maturity. However, patients with secondary chondrosarcoma tend to be younger than those with primary chondrosarcomas (see Table 5–2). The lesions tend to be slow growing with minimal to mild symptoms. The most common site is the pelvis, followed by the proximal femur, proximal humerus, and ribs. Plain radiographs demonstrate a flocculated calcific pattern (Figure 5–48). An osteochondroma with a cartilage cap thicker than 1–2 cm should raise suspicion of a secondary chondrosarcoma. The overall prognosis for patients with secondary or peripheral chondrosarcoma is even better than that for patients with primary or central chondrosarcoma. Surgical removal, without violation of the cartilage cap, is the only effective treatment modality.

![]() Figure 5–48. CT scan of a secondary peripheral chondrosarcoma in the ilium of a 56-year-old man with hereditary multiple exostoses.
Figure 5–48. CT scan of a secondary peripheral chondrosarcoma in the ilium of a 56-year-old man with hereditary multiple exostoses.
C. Dedifferentiated Chondrosarcoma: ICD-9-CM 170.x
Dedifferentiated chondrosarcoma is the most malignant variant of chondrosarcoma, accounting for between 5 and 10% of all chondrosarcomas. It is heralded by the transformation of areas of conventional chondrosarcoma into malignant fibrous histiocytoma or osteosarcoma. Histologically, it is characterized by two distinct but neighboring areas of low- to intermediate-grade malignant chondroid tumor and heterogeneous high-grade sarcoma. Dedifferentiated chondrosarcoma occurs in older patients, usually between 50 and 70 years of age. It is found in the same areas affected by central primary chondrosarcomas, including the pelvis, femur, and proximal humerus (Figure 5–49). Radiographs show areas of rarefaction within the tumor with cortical attenuation. Pathologic fracture is not uncommon.

![]() Figure 5–49. Radiograph of dedifferentiated chondrosarcoma in the distal femur of a 73-year-old woman.
Figure 5–49. Radiograph of dedifferentiated chondrosarcoma in the distal femur of a 73-year-old woman.
The prognosis in dedifferentiated chondrosarcoma is bleak, with the majority of patients developing and dying of metastatic disease within 1 year (historically, 1-year survival rate approached 10%). Chemotherapy and radiation therapy are less effective than in malignant fibrous histiocytoma or osteosarcoma that arose de novo. Surgical resection remains the mainstay of treatment, with adjuvant modalities employed in younger patients.
D. Clear Cell Chondrosarcoma: ICD-9-CM 170.x
Clear cell chondrosarcoma is a rare low-grade variant of chondrosarcoma. Clear cell lesions occur more often in males than in females and are usually seen in patients between 20 and 50 years of age. The vast majority of lesions are found in the femoral head (Figure 5–50). The radiographic appearance is one of a lytic tumor with sharp margination and a central matrix calcification, creating the appearance of a chondroblastoma. Although microscopic examination reveals the presence of some giant cells, as seen in chondroblastoma, areas of low-grade chondrosarcoma are also evident in which no giant cells are seen. Even on gross examination, the clear cell chondrosarcoma does not look like a chondrosarcoma, which explains why it is frequently mistaken for a chondroblastoma in younger adult patients. The tumor cells have abundant glycogen, giving them their characteristic clear cell phenotype. Although no significant genetic alteration is found in clear cell chondrosarcoma, newer findings show that alkaline phosphatase activity may correlate with prognosis.
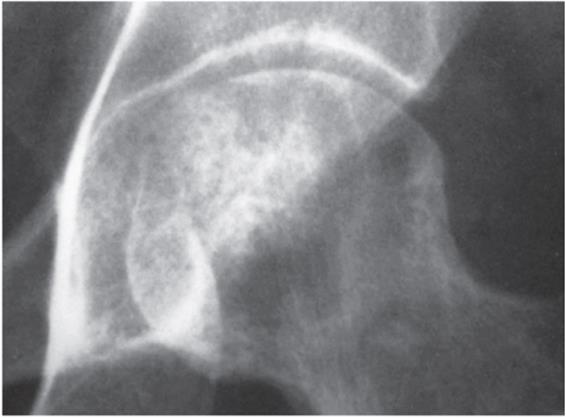
![]() Figure 5–50. Radiograph of clear cell chondrosarcoma of the femoral head in a 25-year-old man.
Figure 5–50. Radiograph of clear cell chondrosarcoma of the femoral head in a 25-year-old man.
The treatment for clear cell chondrosarcoma is a wide excision and reconstruction. The prognosis with this type of treatment is good. In contrast, when lesions are mistaken for chondroblastomas and treatment consists of simple curettage and bone grafting, the prognosis is poor and the recurrence rate is high.
E. Mesenchymal Chondrosarcoma: ICD-9-CM 170.x, or 171.x
Another rare variant of chondrosarcoma is the mesenchymal chondrosarcoma. It is a highly cellular tumor composed of primitive mesenchymal cells with foci of cartilage differentiation. This tumor involves the soft tissue in a third of cases, occurs more frequently in females than in males, and is seen in young adults (see Table 5–2). The jaw is the most common location, followed by the spine and ribs, with few cases noted in long bones.
Mesenchymal chondrosarcoma is a high-grade tumor with histologic features of low-grade chondrosarcoma. Heavily calcified areas, mixed with areas of malignant round cells, may give it the appearance of Ewing sarcoma or solitary fibrous tumor.
Treatment consists of resection, with a wide margin if possible, and adjuvant chemotherapy and radiation therapy. Despite aggressive treatment, the prognosis is very poor, with a high incidence of pulmonary metastasis.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Typical chondroid-forming sarcomas are not sensitive to radiation therapy or chemotherapy and are treated with surgery alone.
• Pain, endosteal scalloping greater than 50% of the cortical width, or radiolucency adjacent to an otherwise typical-appearing enchondroma should alert the practitioner to the possibility of chondrosarcoma.
![]() Round Cell Tumors
Round Cell Tumors
This so-called group of tumors is composed of distinct tumors that, other than their similar microscopic appearance using hematoxylin-eosin stain, are quite different. They behave and are treated in a variety of ways, given that each arises from a different cell type.
A. The Ewing Sarcoma Family of Tumors
1. Ewing sarcoma: ICD-9-CM 170.x—Ewing sarcoma is a well-known clinical entity originally described by James Ewing as a diffuse endothelioma of bone. Since the time of his description, many theories have evolved regarding the tumor’s true histogenesis. Based on electron microscopic and immunohistochemical findings, experts currently believe the tumor represents an undifferentiated member of the family of neural tumors distinct from neuroblastoma. The Ewing sarcoma family of tumors (ESFT) also includes the less common primitive neuroectodermal tumor (PNET) and Askin tumors. ESFTs have been shown to express chimeric transcription factors that result from reciprocal translocations involving chromosome 22, containing the Ewing sarcoma gene (EWS). Ninety percent of the time t(11:22) is seen, resulting in a EWS/FLI-1 chimera. Less frequently, t(21:22) and t(7:22) are seen. The resulting transcription factors are put under the control of the EWS promoter. Recently, investigations have elucidated a handful of downstream targets, including VEGF (encoding vascular endothelial growth factor) and CAV1 (encoding caveolin-1), which are thought to be necessary for tumorigenesis. Furthermore, they are being investigated as possible targets for directed therapy.
In 90% of cases, Ewing sarcoma is found in patients between 5 and 25 years of age. If the patient is younger than 5 years, the most likely diagnosis is metastatic neuroblastoma. Males are affected more frequently than females and carry a worse prognosis. The pelvis is the most common location, followed by the femur, tibia, humerus, and scapula. However, because Ewing sarcoma is a myelogenous tumor, it can be found in any bone in the body (Figure 5–51).
![]() Figure 5–51. MRI of the hindfoot of a 9-year-old girl with calcaneal Ewing sarcoma.
Figure 5–51. MRI of the hindfoot of a 9-year-old girl with calcaneal Ewing sarcoma.
Ewing sarcoma appears radiographically as a central lytic tumor of the diaphyseal-metaphyseal bone. It creates extensive permeative destruction of cortical bone, and as it breaks through under the periosteum, it takes on a typical onionskin, multilaminated appearance. Another radiographic feature is the reactive hair-on-end appearance created by bone forming along the periosteal vessels that run perpendicularly between the cortex and the elevated periosteum (Figure 5–52).

![]() Figure 5–52. Radiograph of periosteal response in Ewing sarcoma of the femur in a 15-year-old boy.
Figure 5–52. Radiograph of periosteal response in Ewing sarcoma of the femur in a 15-year-old boy.
Ewing sarcoma can frequently masquerade as osteomyelitis because it is a high-grade lesion with resultant areas of necrosis; liquefaction may occur that may be mistaken for pus. Furthermore, patients frequently present with systemic symptoms of low-grade intermittent fever and elevated white cell count and erythrocyte sedimentation rate (ESR). Microscopically, small roundlike cells predominate in densely packed sheets. Formation of pseudorosettes may also be seen (in <20%). The rosette-like patterns are more frequently seen in PNET.
Ewing sarcoma is an aggressive malignancy with high local recurrence and metastatic rates. Patients with locally resectable disease treated with multidrug chemotherapy have a 5-year survival rate of approximately 70% (Figure 5–53). Currently, most chemotherapy is driven by large cooperative trials including induction and adjuvant multidrug chemotherapy with drugs such as vincristine, doxorubicin, cyclophosphamide, dactinomycin, and ifosfamide. Unfortunately, 15–25% of patients present with nonlocalized disease. For the patient who presents with advanced metastatic disease, the 5-year survival rate is 30%. Resection of lung metastasis, if possible, does improve survival.

![]() Figure 5–53. Pretreatment MRI (A) and MRI following 10 weeks of neoadjuvant chemotherapy (B) of Ewing sarcoma of the fibula in a 14-year-old boy.
Figure 5–53. Pretreatment MRI (A) and MRI following 10 weeks of neoadjuvant chemotherapy (B) of Ewing sarcoma of the fibula in a 14-year-old boy.
Ewing sarcoma is a radiosensitive tumor. Historically, this was a modality of choice, employing 45–50 Gy over 5 weeks to treat local disease. Because of the not insignificant risk of secondary sarcomas, surgery was investigated as the primary modality for local control. If the margins are contaminated, local irradiation must still be used postoperatively. Postoperative radiotherapy is also generally accepted after complete resection of a tumor showing a poor response to neoadjuvant chemotherapy, with improved local control rates having been illustrated. Preoperative radiotherapy, although not generally used, may have a role in lesions that are on the border of resectability.
2. Primitive neuroectodermal tumor: ICD-9-CM 170.x, or 171.x—PNET is the less common relative to Ewing sarcoma. Like Ewing, this tumor demonstrates expression of neural markers by immunocytochemistry. PNET also exhibits the t(11:22) translocation with the resulting EWS/FLI-1 fusion protein. In fact, because of such similarities, it is generally agreed that PNET and Ewing sarcoma represent ends of a spectrum of disease.
By strict criteria, PNET is a rare tumor, representing approximately 10% of Ewing-like tumors. The demographics are identical to those of Ewing-like tumors. Treatment of PNET is similar to that of Ewing sarcoma; however, the survival rate is slightly less. Accordingly, some authors feel it should be distinguished from Ewing sarcoma.
B. Lymphoma: ICD-9-CM 200.x
Lymphoblastic tumors are considered systemic neoplasms of the lymphatic organs, including the bone marrow, and they account for 7% of all malignant bone tumors. They can be roughly divided into Hodgkin lymphomas and non-Hodgkin lymphomas, both of which can affect bone. Of the two groups, the lymphomas associated with Hodgkin disease carry a much better prognosis. When they are found in bone, they tend to be localized and have a considerable blastic response, especially when involving the vertebra.
There are two main types of non-Hodgkin lymphomas. The type emphasized in this section is the primary lymphoma of bone, in which a localized lytic destruction occurs in a single bone, and the results of staging studies (including an isotope bone scan, a CT scan of the chest and abdomen, and marrow aspiration) all prove negative for other areas of involvement. The other type is the more generalized or systemic form of lymphoma, in which many lymphoid organs are involved, including the lymph nodes, liver, spleen, and bone. The prognosis is better for an isolated primary lymphoma of bone, but years later involvement may become generalized or systemic and carry a worse prognosis. This is similar to the case with plasma cell tumors, in which the findings in a patient can change from that of a solitary plasmacytoma with an excellent prognosis to that of the multiple myeloma form of the disease with a poor prognosis.
Primary lymphoma of bone, which was formerly called reticulum cell sarcoma of bone, accounts for approximately half of all lymphomas. To meet the criteria of being a primary bone lymphoma, there must be a 4- to 6-month interval from the onset of skeletal manifestations to the development of systemic disease. It occurs more frequently in males than in females, is usually found in patients older than 25 years, and affects the spine or pelvis in more than 50% of the cases. In the extremities, the femur is the most commonly involved area, followed next by the humerus and the tibia. Polyostotic involvement occurs in 10–40% of cases.
Radiographic findings in primary lymphoma include extensive lytic permeation of cortical bone, with minimal sclerotic response in diaphyseal, metaphyseal, and epiphyseal locations (Figure 5–54). MRI studies demonstrate that the actual marrow involvement is frequently more extensive than the cortical disruption seen on simple radiographs suggests.

![]() Figure 5–54. Radiograph of a lymphoma in the proximal humerus of a 64-year-old woman.
Figure 5–54. Radiograph of a lymphoma in the proximal humerus of a 64-year-old woman.
The most common histologic types of lymphoma of bone are the large cell or mixed small and large cell types. The cells tend to demonstrate little cytoplasmic structure. However, the nuclear pattern shows indented and folded nuclear patterns and a prominent pink-staining nucleolus, which may help to distinguish it histologically from Ewing sarcoma. Immunohistochemical staining is often necessary to differentiate Ewing sarcoma from the B-cell and T-cell subtypes of lymphoma. In the case of lymphomas, the glycogen stain is usually negative, but the reticulum stain is often positive.
In primary lymphoma of bone, as in Ewing sarcoma, multidrug chemotherapy has greatly improved the 5-year survival rate, which now is approximately 70% for patients with either of these tumors. Like Ewing sarcoma, primary lymphoma of bone is highly sensitive to local irradiation. If the primary lymphoma is localized, a wide resection and limb-salvage reconstruction may be carried out, thereby avoiding the need for local irradiation, and possibly effect a cure. However, if the involvement is more extensive, as is commonly the case, it is necessary to use intralesional techniques such as cemented intramedullary nails or a long-stem prosthesis and subsequently use whole bone irradiation, similar to the management of metastatic carcinoma with pathologic fractures. In cases of extensive systemic involvement, bone marrow transplantation can be used.
C. Plasma Cell Tumor
A bone tumor composed of malignant monoclonal plasma cells is referred to as a myeloma or plasmacytoma. It is rare for a patient to have a solitary myeloma or plasmacytoma. Tumors are almost always found on multiple bony sites, in which case the term multiple myeloma is used.
1. Myeloma: ICD-9-CM 203.x—Multiple myeloma, which is the most common primary tumor of bone, accounts for 45% of all malignant bone tumors. It is the second most common hematopoietic malignancy. An estimated 90% of cases are in patients older than 40 years. It accounts for 1% of all malignancies in Caucasians and 2% in African Americans.
The disease is characterized by a triad of osteolytic punched-out lesions (multifocal) (Figure 5–55), neoplastic proliferation of atypical plasma cells, and a monoclonal gammopathy. Diagnostic criteria are established for myeloma. Major criteria include plasmacytosis on biopsy of a lesion, marrow plasmacytosis, and an abnormal serum protein electrophoresis and light (Bence Jones) proteinuria. It causes bony destruction similar to that caused by lymphomas, with most lesions occurring in the trunk, hip, and shoulder areas. Knowledge of the biology of multiple myeloma continues to increase at an astounding rate, and investigation toward targeted therapy grows proportionally. Targeted therapy trials have included inhibitors of cell surface receptors such as VEGF-R and IGF-1R, inhibitors of cell signaling pathways such as the MAPK pathway and the MTOR pathway, janus kinase inhibitors, histone deacetylase inhibitors, and many others.

![]() Figure 5–55. Radiograph of multiple myeloma in the femoral shaft of a 72-year-old man.
Figure 5–55. Radiograph of multiple myeloma in the femoral shaft of a 72-year-old man.
Lesions are rarely found distal to the knee or elbow. Approximately 3% of patients with myeloma have a sclerotic form of the disease, which appears to carry a better prognosis and is associated with peripheral neuropathy. The serum protein electrophoresis shows an elevated monoclonal immunoglobulin on either the a or y spike. Bence Jones proteinuria is secondary to light-chain immunoglobulin spillover. Occasionally, electrophoresis of a urine sample yields positive results, whereas that of a serum sample yields negative results. In aggressive forms of myeloma, the extensive bone breakdown causes hypercalcemia, which can lead to a semicomatose state and, over a long period, results in nephrocalcinosis. Renal damage also results from protein plugging of the renal tubules, and renal failure may ensue.
A marrow aspirate usually demonstrates the abnormal plasma cells. These cells show an eccentrically placed nucleus in a well-structured eosinophilic cytoplasm. Although normal B-cell–derived plasma cells produce antibodies, the abnormal B-cell–derived plasma cells produce immunoglobulin that is ineffective, which helps explain the increased infection rate in patients with myelomas. Patients may also demonstrate extraosseous infiltrates, with the majority seen in the upper airway and oral cavity. Amyloidosis may be seen concurrently in 10–15% of cases. A quarter of these have extensive cardiac involvement. In such cases, the median survival is 4 months.
Plain radiographs show myeloma lesions to be sharply demarcated lytic lesions with minimal periostitis. Pathologic fixation is frequent. Bone scans have a high false-negative rate thought to be caused by almost exclusive osteoclast activity. For this reason, a skeletal survey instead of a bone scan is important in the staging of this disease.
Fewer than 2% of myeloma cases demonstrate the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-component spike, skin changes, scleroses of bone).
Although treatment and prognosis have improved, myeloma remains a fatal disease, with more than 90% of patients dying within 2–3 years. Chemotherapy such as melphalan and cortisone may induce a transient remission in 50–70% of cases. Bisphosphonates have been introduced to the treatment algorithm with results showing reduced skeletal-related events, pain, and increased time to progression.
Local treatment of myeloma is similar to that of meta-static disease, with cemented intramedullary nails and prosthetic devices used after an intralesional debridement. The amount of bleeding at the surgical site is usually extensive, similar to that encountered with surgery for metastatic renal cell carcinoma and certain thyroid metastases. After surgery, the entire bone should be irradiated with 5500 cGy. Spinal lesions should be handled just like metastatic tumors, as discussed in a later section.
2. Solitary myeloma: ICD-9-CM 203.8—Solitary lesions are rare (Figure 5–56). By definition, there must be no marrow involvement. Seventy-five percent of these cases have an entirely normal serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP). The remaining 25% may have mild abnormalities. Vertebral involvement is the most common site. Patients also tend to be younger (<50 years old). Unfortunately, 70% of these solitary cases develop multiple myeloma within 3 years. Until this happens, the treatment is only local, with a wide resection if possible or intralesional debridement and reconstruction followed by radiation therapy.

![]() Figure 5–56. Radiograph of a solitary plasmacytoma in the proximal femur of a 46-year-old man.
Figure 5–56. Radiograph of a solitary plasmacytoma in the proximal femur of a 46-year-old man.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Ewing sarcoma is characterized by a chimeric transcription factor that is a product, most often, of the t(11:22), resulting in an altered expression pattern in the neoplastic lineage.
• Because of the high level of sensitivity of lymphoma to chemotherapy and radiation therapy, wide resection is infrequently performed.
• Because of the propensity for myeloma to remain cold on isotope bone scanning, a skeletal survey is the study of choice for skeletal staging.
![]() Fibrous Sarcomas of Bone
Fibrous Sarcomas of Bone
Malignant fibrous tumors of bone are clinically similar to osteosarcoma, but they affect an older (<20 years) age group of patients and show a complete absence of tumor osteoid formation. The two major tumors in this category are fibrosarcoma and malignant fibrous histiocytoma.
A. Fibrosarcoma of Bone: ICD-9-CM 170.x
Fibrosarcoma of bone is a malignant spindle cell tumor seen in an older patient population with a peak incidence in the fourth decade. It is 10 times less frequent than osteosarcoma but tends to involve similar locations. The most common site of fibrosarcoma is the distal femur, followed next in order by the proximal tibia, pelvis, proximal femur, and proximal humerus. It is rarely seen in the spine, hand, or foot.
On radiograph, fibrosarcomas appear to be almost purely osteolytic and permeative, similar to lymphomas. For this reason, they are painful and can lead to a pathologic fracture. Microscopically, myofibroblastic differentiation with osteoid formation or histiocytes permits distinction from fibroblastic osteosarcoma and malignant fibrous histiocytoma (MFH) of bone. The low-grade form is characterized by malignant-appearing fibroblasts that form a large amount of collagen fiber, giving the appearance of an aggressive desmoplastic fibroma. The high-grade form is characterized by a more anaplastic fibroblast with a higher index of mitotic activity and less collagen fiber formation. It is common to see a basket-woven or storiform pattern in the microscopic picture.
The prognosis and treatment are directly related to the histologic grade of the tumor. Low-grade fibrosarcoma has a better prognosis than osteosarcoma does, but it must be treated by means of an aggressive and wide resection to avoid local recurrence. Because the low-grade form has a low mitotic index, adjuvant chemotherapy and radiation therapy are of little help. High-grade fibrosarcoma has a prognosis and a rate of metastasis that are similar to those of osteosarcoma, and it is usually treated in a similar manner with a combination of surgery and, if the patient is young enough to tolerate the systemic toxicity, adjuvant chemotherapy.
B. Malignant Fibrous Histiocytoma of Bone: ICD-9-CM 170.x
Prior to 1970, MFH was rarely diagnosed in bone but was commonly found in soft tissue. Now MFH is more common in bone than fibrosarcoma, but the two types of tumor run a similar clinical course. MFH of bone is seen in middle-age and older adults (see Table 5–2), is more common in males than in females, and affects the same bony sites as fibrosarcoma and osteosarcoma.
MFH is a purely lytic tumor that shows aggressive permeation of metaphyseal-diaphyseal bone, similar to the findings in lymphoma (Figure 5–57). Lytic destruction is diffuse, with no evidence of a periosteal response of blastic repair. Microscopic analysis of MFH usually shows the tumor to be high grade and have highly anaplastic fibroblasts mixed with malignant histiocytes and a few giant cells in a typical storiform pattern.

![]() Figure 5–57. Radiograph (A) and T1-weighted MRI (B) of malignant fibrous histiocytoma in the distal femur of a 50-year-old woman.
Figure 5–57. Radiograph (A) and T1-weighted MRI (B) of malignant fibrous histiocytoma in the distal femur of a 50-year-old woman.
Because MFH is closely related to the high-grade fibrosarcoma, it carries a poor prognosis, with high rates of local recurrence and metastasis. The treatment program is therefore similar to that for high-grade fibrosarcoma and osteosarcoma, and it includes an aggressive wide resection and the use of adjuvant chemotherapy.
![]() Adamantinoma of Bone: ICD-9-CM 170.x
Adamantinoma of Bone: ICD-9-CM 170.x
Adamantinomas account for only 0.33% of all malignant bone tumors; occur with equal frequency in males and females, usually during the second and third decades of life; are found in the tibia in 90% of cases; and are usually diaphyseal in location, frequently starting in the anterior cortex. The cause of adamantinoma remains unknown, although angioblastic synovial cells and epithelial cells were considered in the past. Newer investigations, including immunohistochemistry and electron microscopic studies, lend support to the hypothesis of an epithelial origin, which goes along with the histologic appearance of a basal cell carcinoma and might explain the common site of origin subcutaneously in the anterior tibial cortex. The name adamantinoma was given to the tibial lesion because its histologic appearance is similar to that of the adamantinoma of jaw bone (ameloblastoma), but the two entities have no other relationship clinically.
In patients with adamantinoma, the radiograph shows a benign tumor with a lytic central core that is surrounded by reactive sclerotic bone that typically bulges the anterior cortex and thus takes on the appearance of either fibrous dysplasia or osteofibrous dysplasia (Figure 5–58). One consideration in the differential diagnosis is that osteofibrous dysplasia is painless, whereas pain is a frequent symptom in adamantinoma. Another is that benign fibrous lesions of bone stop growing at bone maturity, whereas the adamantinoma continues on into adult life, at which point a biopsy of the progressive lytic portion of the disease should be performed. There have been cases of osteofibrous dysplasia combined with small areas of adamantinoma scattered in the benign osseofibrous tissue. In fact, more recently, one variant has been termed osteofibrous dysplasia–like adamantinoma and appears to have less overall biologic potential. Adamantinoma is also occasionally found in both the tibia and fibula, so the physician should look for multiple sites.




![]() Figure 5–58. Adamantinoma of the tibia. Initial anteroposterior radiograph (A), lateral radiograph (B), bone scan (C), and MRI (D). Immediate postoperative radiographs after resection with intercalary allograft reconstruction and vascularized fibula transport are shown in E and F. Anteroposterior and lateral radiographs from 3 years postoperatively are shown in G and H. Clinical photographs from 3 years postoperatively are shown in I–K.
Figure 5–58. Adamantinoma of the tibia. Initial anteroposterior radiograph (A), lateral radiograph (B), bone scan (C), and MRI (D). Immediate postoperative radiographs after resection with intercalary allograft reconstruction and vascularized fibula transport are shown in E and F. Anteroposterior and lateral radiographs from 3 years postoperatively are shown in G and H. Clinical photographs from 3 years postoperatively are shown in I–K.
Microscopic findings include nests or cords of epithelial or angioid tissue growing in a fibrous tissue stroma, which can give adamantinoma the appearance of a low-grade angiosarcoma or a metastatic carcinoma. Cytologically, chromosomal abnormalities are frequent, especially gains of chromosomes 7, 8, 12, and 19.
Adamantinoma grows extremely slowly, over many years, but on occasion metastasizes to regional lymph nodes and the lung. For this reason, it should be treated by a wide resection, which in most cases is a segmental diaphyseal resection followed by an allograft reconstruction over an intramedullary nail. Because of the low-grade nature of this tumor, adjuvant irradiation or chemotherapy is rarely indicated. Even if pulmonary metastases occur, they can be resected, and there is a fairly good prognosis for survival.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Adamantinoma is on a continuum with osteofibrous dysplasia and should be distinguished by increasing pain and radiographic progression.
![]() Vascular Sarcomas of Bone
Vascular Sarcomas of Bone
Vascular sarcomas are relatively rare. They include the hemangioendothelioma, angiosarcoma, and hemangiopericytoma of bone. The terms hemangioendothelioma and angiosarcoma are frequently used synonymously; however, the first term refers to a low-grade tumor, and the second term usually suggests a higher grade lesion with a poorer prognosis.
A. Hemangioendothelioma: ICD-9-CM 170.x
The hemangioendothelioma, also known as angiosarcoma, which is more common in males than in females, is seen in a wide range of ages between the second and seventh decades. The femur, pelvis, spine, and ribs are the usual sites of origin, and the diaphyses and metaphyses of the long bones are also involved. One third of cases are multicentric, usually in the same bone or limb.
Radiographically, the lesion appears purely lytic. The more anaplastic the disease process is, the less reactive bone is. The clinical picture varies widely, depending on the histologic grade of the tumor. The low-grade lesions look like benign hemangiomas, are slow growing, and carry an excellent prognosis. The high-grade lesions are fast-growing lytic lesions with a poor prognosis.
Treatment depends on the histologic grade. The low-grade lesions do well with simple curettage and bone graft, but the high-grade lesions require a more aggressive wide resection and reconstruction. Adjuvant chemotherapy and radiation therapy can be considered for high-grade lesions, especially in patients with multifocal disease.
![]() Chordomas: ICD-9-CM 170.x
Chordomas: ICD-9-CM 170.x
Chordoma of bone is rare and accounts for 4% of malignant bone tumors. It takes its origin from the primitive notochord and has the clinical appearance of a chondrosarcoma. Chordomas affect males more frequently than females and are seen in patients between 30 and 80 years of age. Although 50% of the tumors are sacrococcygeal in origin, 37% arise in the sphenoccipital area, and the remainder of tumors arise from vertebral bodies of the cervical or lumbar spine. The cranial lesions are seen in a younger age group and carry a poor prognosis because of the dangerous location next to the brain, where surgical removal is difficult.
On radiograph, the chordoma appears as a centrally located lytic process that has minimal sclerotic response at the periphery and may show slight matrix calcification, as in a chondrosarcoma. By definition, chordoma is a midline lesion. If the sacrum is involved, the lesion is seen usually in the lower three sacral segments and presents as an extracortical lobulated mass both in front and behind the sacrum. Because of the slow tumor growth, pain may not occur early, but constipation can be an early symptom that results from pressure on the rectum. Because the true anatomic borders are not readily defined by routine radiography, it is best to image this tumor with CT or MRI (Figure 5–59).

![]() Figure 5–59. Sacral chordoma in middle-aged woman: T2 sagittal image (A) and T2 transverse image (B).
Figure 5–59. Sacral chordoma in middle-aged woman: T2 sagittal image (A) and T2 transverse image (B).
Microscopically, nests or cords of cells, sprinkled in a sea of mucinous tissue, give an appearance similar to low-grade chondrosarcoma. In most cases, large vacuolated cells appear like a signet ring and are referred to as physaliferous cells.
Treatment for the sacral lesions is an aggressive wide resection, which can be difficult because of excessive bleeding. Significant neurogenic bowel and bladder deficits can result. At present, it is common to use adjuvant radiation therapy to help reduce the chance of postoperative recurrence. Newer studies recommend using up to 5000 cGy preoperatively, followed by a boost of 1500 cGy postoperatively. If the surgeon is successful in obtaining clean margins, the local recurrence rate is approximately 30%. With contaminated margins, the recurrence rate climbs to 65%. Recurrence 10–15 years following surgery is common. Because of the low-grade characteristics of the chordoma, it is rare to see a pulmonary metastasis, even after a local recurrence following an inadequate local surgical resection.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Information provided in the history and physical examination is critical in determining malignant versus benign characteristics of bone tumors.
• Survival in Ewing sarcoma and osteosarcoma has been dramatically improved with refined neoadjuvant chemotherapy, and the timing and duration of that treatment modality can affect outcome.
• Biopsy of bone lesions with suspected malignant potential should include a portion of the periphery of the lesion, particularly when there is an associated soft-tissue component, as this is where the most diagnostic tissue is found.
Anthouli-Anagnostopoulou FA, Hatziolou E, Papachristou G, et al: Juxtacortical osteosarcoma. A distinct malignant bone neoplasm. Adv Clin Pathol 2004;3:127. [PMID: 11080792]
Avnet S, Longhi A, Salerno M, et al: Increased osteoclast activity is associated with aggressiveness of osteosarcoma. Int J Oncol 2008;33:1231. [PMID: 19020756]
Bacci G, Balladelli A, Palmerini E, et al: Neoadjuvant chemotherapy for osteosarcoma of the extremities in preadolescent patients: the Rizzoli Institute experience. J Pediatr Hematol Oncol 2008;30:908. [PMID: 19131777]
Bacci G, Ferrari S, Bertoni F, et al: Histologic response of high-grade nonmetastatic osteosarcoma of the extremity to chemotherapy. Clin Orthop Relat Res 2001;386:186. [PMID: 11347833]
Bacci G, Forni C, Longhi A, et al: Local recurrence and local control of non-metastatic osteosarcoma of the extremities: a 27-year experience in a single institution. J Surg Oncol 2007;96:118. [PMID: 17577221]
Barrille-Nion S, Barlogie B, Bataille R, et al: Advances in biology and therapy of multiple myeloma. Hematology (Am Soc Hematol Educ Program) 2003;248. [PMID: 14633785]
Bruns J, Elbracht M, Niggemeyer O: Chondrosarcoma of bone: an oncological and functional follow-up study. Ann Oncol 2001;12:859. [PMID: 11484965]
Burger R, Le Gouill S, Tai YT, et al: Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol Cancer Ther 2009;8:26. [PMID: 19139110]
Cesari M, Bertoni F, Bacchini P, et al: Mesenchymal chondrosarcoma. An analysis of patients treated at a single institution. Tumori 2007;93:423. [PMID: 18038872]
Crapanzano JP, Ali SZ, Ginsberg MS, et al: Chordoma: a cytologic study with histologic and radiologic correlation. Cancer 2001;93:40. [PMID: 11241265]
Desai SS, Jambhekar N, Agarwal M, et al: Adamantinoma of tibia: a study of 12 cases. J Surg Oncol 2006;93:429. [PMID: 16550582]
Donati D, Yin J, Di Bella C, et al: Local and distant control in non-metastatic pelvic Ewing’s sarcoma patients. J Surg Oncol 2007;96:19. [PMID: 17345611]
Ewing J: Diffuse endothelioma of bone. Proc NY Pathol Soc 1921;21:17. [No PMID]
Gelderblom H, Hogendoorn PC, Dijkstra SD, et al: The clinical approach towards chondrosarcoma. Oncologist 2008;13:320. [PMID: 18378543]
Han I, Oh JH, Na YG, et al: Clinical outcome of parosteal osteosarcoma. J Surg Oncol 2008;97:146. [PMID: 18050289]
Hoang BH, Kubo T, Healey JH, et al: Expression of LDL receptor-related protein 5 (LRP5) as a novel marker for disease progression in high-grade osteosarcoma. Int J Cancer 2004;109:106. [PMID: 14735475]
Kanamori M, Antonescu CR, Scott M, et al: Extra copies of chromosomes 7, 8, 12, 19, and 21 are recurrent in adamantinoma. J Mol Diagn 2001;3:16. [PMID: 11227067]
Khanna C, Wan X, Bose S, et al: The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med 2004;10:182. [PMID: 14704791]
Kilpatrick SE, Geisinger KR, King TS, et al: Clinicopathologic analysis of HER-2/neu immunoexpression among various histologic subtypes and grades of osteosarcoma. Mod Pathol 2001;14:1277. [PMID: 11743051]
Mandahl N, Gustafson P, Mertens F, et al: Cytogenetic aberrations and their prognostic impact in chondrosarcoma. Genes Chromosomes Cancer 2002;33:188. [PMID: 11793445]
Ocio EM, Mateos MV, Maiso P, et al: New drugs in multiple myeloma: mechanisms of action and phase I/II clinical findings. Lancet Oncol 2008;9:1157. [PMID: 19038762]
Ogose A, Hotta T, Kawashima H, et al: Elevation of serum alkaline phosphatase in clear cell chondrosarcoma of bone. Anticancer Res 2001;21:649. [PMID: 11299821]
Overholtzer M, Rao PH, Favis R, et al: The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci USA 2003;100:11547. [PMID: 12972634]
Pring ME, Weber KL, Unni KK, et al: Chondrosarcoma of the pelvis. A review of sixty-four cases. J Bone Joint Surg Am 2001;83-A:1630. [PMID: 11701784]
Rizzo M, Ghert MA, Harrelson JM, et al: Chondrosarcoma of bone: analysis of 108 cases and evaluation for predictors of outcome. Clin Orthop Relat Res 2001;391:224. [PMID: 11603673]
Roland Durr H, Wegener B, Krödel A, et al: Multiple myeloma: surgery of the spine: retrospective analysis of 27 patients. Spine (Phila Pa 1976) 2002;27:320. [PMID: 11805699]
Schwab JH, Antonescu CR, Athanasian EA, et al: A comparison of intramedullary and juxtacortical low-grade osteogenic sarcoma. Clin Orthop Relat Res 2008;466:1318. [PMID: 18425560]
Scully SP, Ghert MA, Zurakowski D, et al: Pathologic fracture in osteosarcoma: prognostic importance and treatment implications. J Bone Joint Surg Am 2002;84-A:49. [PMID: 11792779]
Streitburger A, Ahrens H, Balke M, et al: Grade I chondrosarcoma of bone: the Munster experience. J Cancer Res Clin Oncol 2009;135:543. [PMID: 18855011]
Tallini G, Dorfman H, Brys P, et al: Correlation between clinicopathological features and karyotype in 100 cartilaginous and chordoid tumours. A report from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. J Pathol 2002;196:194. [PMID: 11793371]
Tian E, Zhan F, Walker R, et al: The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 2003;349:2483. [PMID: 14695408]
Weiss A, Khoury JD, Hoffer FA, et al: Telangiectatic osteosarcoma: the St. Jude Children’s Research Hospital’s experience. Cancer 2007;109:1627. [PMID: 17351949]
BENIGN SOFT-TISSUE TUMORS
Soft tissue can be defined as nonepithelial, extraskeletal mesenchymal tissue exclusive of the reticuloendothelial system and glia. This definition would include fat, fibrous tissue, muscle, and the relating neurovascular structures.
Benign soft-tissue tumors, by definition, represent a differentiated neoplastic process with a limited capacity for autonomous growth. They generally demonstrate a marginal capacity to invade locally with infrequent local recurrence. Because of the extensive numbers of benign soft-tissue tumors, discussion is limited here to the more common entities.
![]() Lipomas
Lipomas
The lipoma is by far the most common soft-tissue tumor, accounting for approximately 50% of all soft-tissue tumors. Lipoma outnumbers liposarcoma by a ratio of 100:1. Cytogenetic abnormalities have been reported in 50–80% of lipomas. There are a large number of variants including the superficial subcutaneous lipoma; the intramuscular lipoma; the spindle cell lipoma; the angiolipoma; the benign lipoblastoma; and the lipomas of tendon sheaths, nerves, synovium, periosteum, and lumbosacral area.
A. Superficial Subcutaneous Lipoma: ICD-9-CM 214.x
The most frequently seen type of lipoma is the superficial subcutaneous type, which can be solitary or multiple. Subcutaneous lipomas occur with equal frequency in men and women and seem to arise spontaneously during the fifth and sixth decade of life. The most common locations are the back, shoulder, and neck.
On palpation, this tumor is soft and ballotable. Although it is found more commonly in obese patients, the size of the lipoma does not correlate with the weight of the patient. Lipomas do not reduce in volume with weight loss. They generally grow to a limited size, and sarcomatous degeneration does not occur. Surgical treatment is usually cosmetic in nature, and the recurrence rate is less than 5%.
B. Intramuscular Lipoma: ICD-9-CM 214.8
The deep intramuscular lipoma is seen in adults between 30 and 60 years of age, affects men more frequently than women, and is commonly found in the large muscles of the extremities. The lesions are slow growing and painless. The intramuscular lipoma has a characteristic radiolucency that contrasts with the surrounding muscle (Figure 5–60). On MRI, this tumor demonstrates a uniform high-signal image on the T1-weighted spin-echo sequence. On gross examination, the tumor can appear quite infiltrative in surrounding muscle and has a faint yellow color on sectioning. Histologic studies show that the intramuscular lipoma, like the subcutaneous lipoma, is composed of benign lipocytes with small pyknotic nuclei that are difficult to see on the surface of the large fat-laden cell. When samples are taken for biopsy purposes, the pathologist must take care to rule out a low-grade, well-differentiated liposarcoma that can coexist with a benign lipoma. On rare occasions, a lipoma can have chondroid or osseous hamartomatous elements that have caused it to be classified as a mesenchymoma in the past. In other cases, evidence of hemorrhage or necrosis can be found in a lipoma and creates low-signal changes on the MRI that are similar to the changes seen in liposarcoma.

![]() Figure 5–60. Radiograph (A) and coronal view T1-weighted MRI (B) of an intramuscular lipoma in the quadriceps muscle of a 72-year-old man.
Figure 5–60. Radiograph (A) and coronal view T1-weighted MRI (B) of an intramuscular lipoma in the quadriceps muscle of a 72-year-old man.
A marginal surgical excision is indicated for treatment of intramuscular lipoma. Local recurrence rates of 15–60% are reported.
C. Spindle Cell Lipoma: ICD-9-CM 214.x
The spindle cell lipoma is seen typically in the posterior neck and shoulder area in men between 45 and 64 years of age. On gross examination, the spindle cell lipoma has the appearance of an ordinary lipoma but with areas of gray-white gelatinous foci streaking through it. Microscopic examination of these areas reveals the presence of benign fibroblasts. Thus, with imaging studies, dense areas are scattered throughout the normal radiolucent areas of a lipoma. On MRI, findings generally consist of a low-signal streaking through the typical high-signal pattern of a benign lipoma.
The treatment for this lesion is a simple marginal resection. The chance for local recurrence is minimal.
D. Angiolipoma: ICD-9-CM 214.x
The angiolipoma (Figure 5–61) is a subcutaneous lesion seen in young adults (see Table 5–6), usually on the forearm. Multiple lesions are frequently present and usually painful because of their vascularity. Grossly, the lobular lipoma demonstrates vascular channels. Treatment of angiolipoma consists of marginal excision.

![]() Figure 5–61. Radiograph (A) and T1-weighted MRI (B) of a soft-tissue angiolipoma in the volar aspect of the forearm of a 27-year-old woman.
Figure 5–61. Radiograph (A) and T1-weighted MRI (B) of a soft-tissue angiolipoma in the volar aspect of the forearm of a 27-year-old woman.
E. Diffuse Lipomatosis: ICD-9-CM 214.9
An extremely rare variant of the lipoma is diffuse lipomatosis, characterized by the presence of multiple superficial and deep lipomas that involve one entire extremity or the trunk and usually have their onset during the first 2 years of life. Histologically, an individual lesion in a patient with diffuse lipomatosis looks no different from a typical solitary lipoma. When lipomatosis of a nerve occurs, the involved limb or digit may become massive in size, sometimes making it impossible to remove the fatty tumors surgically. If this is the case, amputation may be indicated.
F. Lumbosacral Lipoma: ICD-9-CM 214.8
The lumbosacral lipoma occurs in the lumbosacral area posterior to a spina bifida defect. It is frequently associated with both intradural and extradural lipomas and thus can result in neurologic deficits. Although lumbosacral lipoma is generally considered a pediatric tumor, it can be seen in adults (Figure 5–62).

![]() Figure 5–62. T1-weighted MRI of a lumbosacral lipoma.
Figure 5–62. T1-weighted MRI of a lumbosacral lipoma.
Surgical treatment consists of a marginal resection of the entire lipoma, including the portion arising from the vertebral canal and lumbosacral roots.
G. Benign Lipoblastoma and Diffuse Lipoblastomatosis: ICD-9-CM 215.x
The benign and diffuse types of lipoblastoma are seen in the extremities or trunk of infants. The lesions, solitary or multiple, can be superficial or deep in muscle tissue. They demonstrate cellular immaturity, with lipoblasts similar to the myxoid form of liposarcoma. Even with the cellular aggressiveness of the lesions, the prognosis is excellent following simple surgical resection.
H. Hibernoma: ICD-9-CM 215.x
Hibernoma, a rare lipoma usually seen in young adults (see Table 5–6), commonly occurs in the scapular and interscapular regions, is painless and slow growing, and ranges between 10 and 15 cm in diameter. The hibernoma is composed of finely granular or vacuolated cells characteristic of brown fat and contains a considerable amount of glycogen. The treatment is marginal surgical resection with a low potential for recurrence.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• In the histologic characterization of lipomatous lesions, biopsy is subject to considerable sampling error. Therefore, if incisional biopsy is indicated, the most suspicious portion of the lesion should be obtained.
• The differential diagnosis of macrodactyly should include lipomatosis of nerve.
• MRI findings of a lesion that shares the same signal intensity as subcutaneous fat on all sequences are diagnostic of a benign to low-grade lipomatous lesion.
![]() Benign Vascular Tumors
Benign Vascular Tumors
Benign vascular proliferative tumors are the second most common benign tumor after lipomas. Three types of vascular tumors are discussed here: hemangiomas, lymphangiomas, and glomus tumors.
Like lipomas, angiomas occur in a wide variety of clinical conditions seen more often in females than in males. The most common type of angioma is the hemangioma, which can be a superficial cutaneous lesion or a deep, intramuscular one. The lymphatic counterpart of the hemangioma is known as the lymphangioma or hygroma. In most cases, the lesion is solitary or localized. If it is extensive and involves an entire limb, the term angiomatosis is used. Because most hemangiomas and lymphangiomas are congenital, the term hamartomatous or arteriovenous malformation is applied in their classification. Hemangiomas and lymphangiomas arise from developmental dysplasias of the endothelial tube, whereas glomus tumors arise from pericytes, which are cells that lie outside the endothelial tube. Most vascular anomalies arise sporadically, but some familial, autosomal dominant inheritance patterns are also described. Genetic analysis of these families identified specific gene mutations supporting the genomic role in the regulation of angiogenesis.
A. Hemangioma: ICD-9-CM 228.x
Hemangiomas are the most frequently seen tumors of childhood and account for 7% of all benign tumors.
1. Capillary hemangioma: ICD-9-CM 228.x—The most common type of hemangioma is the solitary capillary type, also referred to as the port-wine stain, which appears as an elevated red to purple cutaneous lesion on the head or neck. The lesion occurs during the first few weeks after birth, grows rapidly over a period of several months, and regresses over a 7-year period in 75–90% of cases.
Because of the spontaneous regression, no treatment is needed in most cases. In the past, treatment consisted of cryosurgery, sclerotherapy, or irradiation, but frequently this treatment was worse than the disease itself. Today, when treatment is required, the treatment of choice involves the use of selective laser coagulation.
2. Cavernous hemangioma: ICD-9-CM 228.x—The cavernous hemangioma is larger and less common than the capillary hemangioma. The enlarged vascular spaces of the cavernous lesion give it the appearance of a cluster of purple grapes. It lies deep in the extremity, with common involvement of muscles and even the synovial membrane of the joints (synovial hemangioma).
Imaging may be characteristic (Figure 5–63). In some patients with deep intramuscular forms of hemangioma, the skin shows no abnormalities and no phleboliths are apparent on radiograph. With MRI, deep intramuscular hemangiomas can be easily detected by the characteristic mixed-signal serpiginous pattern seen in the T1-weighted image.

![]() Figure 5–63. Clinical appearance (A) and radiographic appearance (B) of a cavernous hemangioma in the foot of one patient, and T1-weighted and T2-weighted MRIs (C) of a cavernous hemangioma of another patient.
Figure 5–63. Clinical appearance (A) and radiographic appearance (B) of a cavernous hemangioma in the foot of one patient, and T1-weighted and T2-weighted MRIs (C) of a cavernous hemangioma of another patient.
The muscle lesions are usually asymptomatic until intralesional hemorrhage occurs either spontaneously or after a minor injury. The pain symptoms are usually short lived but recur infrequently. In some patients, the pain is more severe and associated with muscle contracture and joint deformity. These patients may require surgical resection of the scarred-down lesion to allow for better joint function and to reduce the pain. In rare cases of multiple hemangiomas involving the entire limb, amputation may be indicated. Vascular embolization of the feeder vessels can be attempted but may lead to a significant compartment syndrome, with severe contractures or with loss of muscle strength and limitation of joint movement.
3. Arteriovenous hemangioma: ICD-9-CM 228.x—The arteriovenous hemangioma is seen in young patients (see Table 5–6), usually in the head, neck, or lower extremity. It is associated with significant arteriovenous shunting in the tumor, which creates increased perfusion. This results in increased local temperature, pain, and continuous thrill or bruit over the mass. In the extremity, it also results in an overgrowth of the limb (Klipel-Trenaunay syndrome).
If shunting is excessive, surgical removal of the hemangioma may be necessary to prevent consumptive coagulopathy and high-output cardiac failure (Kasabach-Merritt syndrome). Arteriograms are helpful in determining the degree of shunting prior to treatment. Embolization or surgical ligation of feeder vessels is frequently not a successful form of treatment.
4. Epithelioid hemangioma (Kimura disease): ICD-9-CM 228.x—This cutaneous hemangioma is found on the head or neck in women between 20 and 40 years old. It is associated with inflammatory changes and eosinophilia, and it sometime ulcerates. Its name is derived from the epithelial appearance of the endothelia-lined capillary structures.
5. Pyogenic granuloma: ICD-9-CM 228.x—The pyogenic granuloma is a polypoid capillary hemangioma that affects the skin or mucosal surfaces of males and females in all age groups. It may be associated with trauma and is found about the mouth, gingivae, or fingers. The lesions have a purplered color, bleed easily, and ulcerate.
B. Lymphangioma: ICD-9-CM 228.x
The lymphangioma is nothing more than an angioma composed of lymphatic endothelial tubes filled with lymphatic fluid, rather than being filled with blood, as the hemangioma is. Lymphangiomas can be localized, which occurs with the cystic hygroma, and they are usually seen about the head, neck, or axilla of young boys and girls (see Table 5–6). As with hemangiomas, the larger lymphomas are cavernous lesions seen in older patients with deeper involvement. In both lymphangioma and hemangioma, because of increased regional perfusion, bony overgrowth can occur (Figure 5–64).

![]() Figure 5–64. Radiograph of a lymphangioma in the forearm and hand of a 23-year-old woman.
Figure 5–64. Radiograph of a lymphangioma in the forearm and hand of a 23-year-old woman.
C. Glomus Tumor: ICD-9-CM 215.x
The glomus tumor arises from the hemangiopericyte, which is a cell seen at the periphery of the capillary vascular network and normally involved with the regulation of blood flow through the capillary system. Microscopic examination of the tumor reveals large vascular spaces surrounded by a homogeneous field of round epithelioid hemangiopericytes, with no evidence of mitotic activity.
The glomus tumor is a pink lesion that usually measures less than 1 cm in diameter. It represents 1.6% of all soft-tissue tumors and occurs with equal frequency in men and women, usually between 20 and 40 years of age. Although the tumor is found most commonly in the subungual area of a digit, where it is readily visible, it also occurs subcutaneously on the hand, wrist, forearm, or foot, where it may be invisible and thus difficult to diagnose until localized lancinating pain leads to a surgical exploration. Glomus tumors have sporadically been reported in deep soft tissues, viscera, and intraosseous locations as well. After the lesion is surgically removed, the pain subsides and recurrence occurs in fewer than 10% of cases.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Radiographic findings of phleboliths are diagnostic of hemangioma.
• Glomus tumors are usually diagnosed at a very small size due to characteristic location in the fingertip and extreme cold sensitivity.
![]() Extraabdominal Desmoid Tumors (Aggressive Fibromatosis): ICD-9-CM 215.x
Extraabdominal Desmoid Tumors (Aggressive Fibromatosis): ICD-9-CM 215.x
In comparison with the infantile fibrous lesions mentioned earlier, the desmoid tumor is seen in older children and young adults up through 40 years of age. Whereas abdominal desmoids are seen in the abdominal wall of women following pregnancy, the extraabdominal desmoids usually occur in men and are more common in proximal areas about the shoulder and buttock, followed next by the posterior thigh, popliteal area, arm, and forearm. In most cases, it presents as a solitary tumor. Multicentric involvement is seen at times, however, and can be associated with Gardner syndrome, which is characterized by polyposis of the large bowel and by craniofacial osteomas. In patients with familial adenomatous polyposis (FAP), an inherited disease caused by mutations in the APC gene, desmoids are a significant source of morbidity and mortality. The APC gene, located on chromosome 5, encodes for a 300-kDa protein, in which a germline mutation is an early event in tumor formation.
Desmoids are deep-seated tumors that arise from muscle fascial planes and infiltrate extensively into adjacent muscle tissue, tendons, joint capsules, and even bone. Compared with malignant fibrosarcomas, desmoids are poorly marginated and thus difficult to resect surgically. Desmoids can engulf surrounding vessels and nerves, whereas fibrosarcomas usually push these structures aside. A desmoid may cause local pain and grow quite rapidly, suggesting a malignant tumor. The desmoid tends to grow more longitudinally along muscle planes to a considerable size, frequently resulting in restricted joint motion about the shoulder, hip, or knee. Because the local aggressiveness of desmoids is so similar to that of malignant fibrosarcomas or MFHs, some experts believe the desmoid may be a low-grade fibrosarcoma that has lost its potential to metastasize; however, molecular analyses may suggest otherwise.
On gross examination, a desmoid tumor is firm and heavily collagenized. Microscopically, it has a low mitotic index, similar to that of a plantar or palmar fibromatosis. Radiographically, a desmoid is noncalcified and appears dense in comparison with normal muscle. It is easily seen in soft window CT scanning. More exact presurgical imaging can be obtained with MRI (Figure 5–65). As with an abdominal desmoid, an extraabdominal desmoid physical injury may play a role in the activation of a preexisting oncogene located in the damaged fibroblast.

![]() Figure 5–65. T1-weighted MRI of a desmoid tumor in the gluteal area of a 45-year-old woman.
Figure 5–65. T1-weighted MRI of a desmoid tumor in the gluteal area of a 45-year-old woman.
Desmoids are usually treated surgically with an aggressive wide resection similar to that used in treating a primary sarcoma. Even following a margin-negative resection of the desmoid, the recurrence rate may approach 50%. For this reason, it is common to administer 50 Gy of radiation to the surgical site starting 2 weeks postoperatively. With radiation therapy, the recurrence rate decreases to 15%. In rare cases an amputation may be necessary after multiple recurrences. A few cases of spontaneous involution of desmoid tumors are reported after 40 years of age.
Based on clinical and experimental evidence, estrogen may play a role in the development of desmoid tumors. Accordingly, agents such as tamoxifen are being used in some centers because of their antiestrogen effects. NSAIDs were also implemented in attempts to treat aggressive cases. Cytotoxic chemotherapy has also been instituted in selected unresectable cases, especially associated with familial adenomatous polyposis, with some success.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Desmoids are benign but may be extremely locally aggressive, with up to 50% recurring locally after margin-negative resection alone.
![]() Benign Tumors of Peripheral Nerves
Benign Tumors of Peripheral Nerves
Benign tumors of peripheral nerve sheaths are common and take their origin from Schwann cells, which normally produce myelin and collagen fiber.
A. Neurilemoma: ICD-9-CM 215.x
The neurilemoma (neurinoma or benign schwannoma) is the least common of the benign tumors of peripheral nerve sheaths. It usually affects individuals between 20 and 50 years of age and occurs with equal frequency in men and women. It has a predilection for spinal roots and for superficial nerves on the flexor surfaces of both upper and lower extremities. In most cases, the lesion is solitary, but multiple lesions are occasionally seen in von Recklinghausen disease. The neurilemmoma is slow growing and rarely causes pain or a neurologic deficit.
Unlike the neurofibroma, which has a fusiform appearance, the neurilemoma is round (Figure 5–66). Microscopic studies reveal the presence of a characteristic Verocay body, which consists of palisading Schwann cells and is found in the fibrotic Antoni A substance of the tumor. Other areas reveal a more mucinous Antoni B substance. Neurilemomas may occur in an axial fashion involving spinal roots, often presenting as a dumbbell-shaped extradural defect (Figure 5–67). In comparison with the less restricted peripheral lesions, the nerve root lesions are more apt to cause pain associated with neurologic deficiency because of their bony constriction.

![]() Figure 5–66. T1-weighted MRI of a neurilemoma of the ulnar nerve in a 69-year-old man.
Figure 5–66. T1-weighted MRI of a neurilemoma of the ulnar nerve in a 69-year-old man.

![]() Figure 5–67. Myelogram of a neurilemoma in the cervical spine.
Figure 5–67. Myelogram of a neurilemoma in the cervical spine.
In some cases, simple excision of the neurilemoma is clinically indicated, which often can be performed without serious damage to the nerve. If the patient is asymptomatic, observation is appropriate because there is little chance for malignant degeneration.
B. Solitary Neurofibroma: ICD-9-CM 215.x
The solitary neurofibroma is a fusiform fibrotic tumor arising centrally from a smaller peripheral nerve (Figure 5–68). The tumor is seen with equal frequency in men and women, usually between 20 and 30 years of age. It is 10 times more common than the multiple form seen in von Recklinghausen disease, is usually smaller, and carries less chance of malignant degeneration. Microscopic examination of the solitary neurofibroma shows interlacing bundles of elongated spindle cells with benign-appearing nuclei and occasionally with areas resembling the Antoni A tissue seen in the neurilemoma.

![]() Figure 5–68. Photographic appearance of a solitary neurofibroma.
Figure 5–68. Photographic appearance of a solitary neurofibroma.
Treatment of the solitary neurofibroma consists of simple excision. Iatrogenic damage to the nerve fascicles is more likely than with resection of the neurilemoma, due to the intertwined growth of the neurofibroma.
C. Neurofibromatosis (von Recklinghausen Disease): ICD-9-CM 237.7
von Recklinghausen disease is a familial dysplasia, inherited as an autosomal dominant trait, with an incidence of approximately 1 in every 3000 live births. The disease usually begins during the first few years of life with the emergence of small café-au-lait spots. Over time, these lesions grow in number and size. Unlike the lesions seen in fibrous dysplasia, the lesions in von Recklinghausen disease do not have rough edges. If a patient has more than six lesions that have smooth edges and are greater than 1.5 cm in diameter, the diagnosis of von Recklinghausen disease is certain.
Later in life, the patient develops multiple neurofibromas, each of which appears as a soft cutaneous nodule (Figure 5–69). This pedunculated skin lesion, which is called fibroma molluscum, can be large and pendulous. More pathognomonic of the disease is the plexiform neurofibroma, occurring in 25% of patients, which appears in larger nerves and can involve an entire extremity (see Figure 5–69). When the overlying skin of an extremity is loose and hyperpigmented, the condition is called elephantiasis neuromatosa, or “elephant man syndrome.” (It is now thought that John Merrick, the so-called elephant man, was actually affected by Proteus syndrome.) Among the bony changes seen in von Recklinghausen disease are scoliosis in up to 20%, bowing and/or pseudarthrosis of the tibia in 5%, spinal meningocele, scalloping of the vertebra, and osteolytic lesions in bone.

![]() Figure 5–69. Cutaneous manifestations of neurofibromatosis.
Figure 5–69. Cutaneous manifestations of neurofibromatosis.
A major threat to the patient’s life is that a malignant sarcoma will develop from one of the large and deep neurofibromas. This occurs at a later age in 3–5% of patients.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Neurilemomas are usually easily dissected off the associated nerve, whereas neurofibromas generally are far more intertwined in the nerve fascicles, making nerve-sparing resection more difficult.
• Patients with neurofibromatosis have a lifetime risk of secondary sarcoma of up to 5%.
![]() Intramuscular Myxomas: ICD-9-CM 215.x
Intramuscular Myxomas: ICD-9-CM 215.x
The intramuscular myxoma is a rare tumor seen in patients older than 40 years and affecting the large muscles about the thighs, shoulders, buttocks, and arms. It is a slow-growing, well-marginated tumor that has the gelatinous physical quality of a ganglion cyst or myxoid liposarcoma. The intramuscular myxoma causes no pain and can grow to greater than 15 cm in diameter. Although it appears radiolucent on CT scan, MRI demonstrates an intermediate signal on the T1-weighted image and an extremely high signal on the T2-weighted image. Multiple myxomas are associated with polyostotic fibrous dysplasia in Mazabraud syndrome.
The intramuscular myxoma can be resected marginally. After this procedure, the recurrence rate is extremely low.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Many soft-tissue lesions have very characteristic findings on imaging studies, but those with nonspecific findings should be diagnosed with needle or incisional biopsy rather than excisional biopsy.
Blei F: Basic science and clinical aspects of vascular anomalies. Curr Opin Pediatr 2005;17:501. [PMID: 16012263]
Blei F: Congenital lymphatic malformations. Ann N Y Acad Sci 2008;1131:185. [PMID: 18519970]
Crawford AH, Schorry EK: Neurofibromatosis update. J Pediatr Orthop 2006;26:413. [PMID: 16670560]
Faurschou A, Togsverd-Bo K, Zachariae C, et al: Pulsed dye laser vs. intense pulsed light for port-wine stains: a randomized side-by-side trial with blinded response evaluation. Br J Dermatol 2009;160:359. [PMID: 19120324]
Gega M, Yanagi H, Yoshikawa R, et al: Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol 2006;24:102. [PMID:16382119]
Gombos Z, Zhang PJ: Glomus tumor. Arch Pathol Lab Med 2008;132:1448. [PMID: 18788860]
Kang HJ, Shin SJ, Kang ES: Schwannomas of the upper extremity. J Hand Surg Br 2000;25:604. [PMID: 11106529]
Lev D, Kotilingam D, Wei C, et al: Optimizing treatment of desmoid tumors. J Clin Oncol 2007;25:1785. [PMID: 17470870]
Marler JJ, Mulliken JB: Current management of hemangiomas and vascular malformations. Clin Plast Surg 2005;32:99. [PMID: 15636768]
Murphey MD, Carroll JF, Flemming DJ, et al: From the archives of the AFIP: benign musculoskeletal lipomatous lesions. Radiographics 2004;24:1433. [PMID: 15371618]
Nielsen GP, O’Connell JX, Rosenberg AE: Intramuscular myxoma: a clinicopathologic study of 51 cases with emphasis on hypercellular and hypervascular variants. Am J Surg Pathol 1998;22:1222. [PMID: 9777984]
Shields CJ, Winter DC, Kirwan WO, et al: Desmoid tumours. Eur J Surg Oncol 2001;27:701. [PMID: 11735163]
Signoroni S, Frattini M, Negri T, et al: Cyclooxygenase-2 and platelet-derived growth factor receptors as potential targets in treating aggressive fibromatosis. Clin Cancer Res 2007;13:5034. [PMID: 17785554]
Skapek SX, Frattini M, Negri T, et al: Vinblastine and methotrexate for desmoid fibromatosis in children: results of a Pediatric Oncology Group Phase II Trial. J Clin Oncol 2007;25:501. [PMID: 17290057]
Sorensen SA, Mulvihill JJ, Nielsen A: Long-term follow-up of von Recklinghausen neurofibromatosis: survival and malignant neoplasms. N Engl J Med 1986;314:1010. [PMID: 3083258]
MALIGNANT SOFT-TISSUE TUMORS
Sarcomas are capable of invasive, locally destructive growth with a tendency to recur and to metastasize. All sarcomas do not behave the same, however. Some sarcomas, such as dermatofibrosarcoma protuberans, rarely metastasize. Undifferentiated pleomorphic sarcoma, in contrast, does so with alacrity.
A. Fibrohistiocytic Tumors: ICD-9-CM 171.x
Until recently, MFH was the most common soft-tissue sarcoma seen in adults (Figure 5–70). Strangely, although more frequently encountered than other adult soft-tissue sarcomas, the cell type(s) of origin remain unclear. However, ongoing developments in molecular diagnostics have allowed the determination of the origin of an increasing number of cases. The latest World Health Organization classification for sarcomas no longer includes MFH as a distinct entity. The current nomenclature for the majority of MFH is undifferentiated pleomorphic sarcoma.

![]() Figure 5–70. Clinical appearance (A), T1-weighted MRI (B), T2-weighted MRI (C), and resected surgical specimen (D) of a large pleomorphic malignant fibrous histiocytoma in the posterior thigh of a 55-year-old man.
Figure 5–70. Clinical appearance (A), T1-weighted MRI (B), T2-weighted MRI (C), and resected surgical specimen (D) of a large pleomorphic malignant fibrous histiocytoma in the posterior thigh of a 55-year-old man.
1. Pleomorphic malignant fibrous histiocytoma/undifferentiated high-grade pleomorphic sarcoma: ICD-9-CM 171.x—Undifferentiated high-grade pleomorphic sarcoma occurs more frequently in men than in women by a ratio of 1.2:1, primarily affecting individuals between 50 and 70 years of age. Usually it is a deep lesion found in the large muscles about the thigh, hip, and retroperitoneal areas. The tumor may be asymptomatic.
On gross examination, the tumor appears multinodular and may demonstrate several separate satellite lesions in the same muscle belly, especially at the superior and interior poles. It may be necrotic and ranges in color from dirty gray to a reddish tan. Microscopy demonstrates that it is composed of malignant fibroblasts mixed with anaplastic and pleomorphic histiocytes.
The prognosis and treatment vary, depending on the size and location of the tumor. The overall local recurrence potential is 45%, with a 40% incidence of metastasis to the lung and with a 10% incidence of regional lymph node involvement. Tumors smaller than 5 cm in diameter and found in a subcutaneous location in the distal body parts carry a good prognosis, with a 5-year survival rate of 80%, whereas tumors that are 5 cm or more in diameter and located deep in a more proximal muscle group carry a poor prognosis, with a 5-year survival rate of only 55%.
Although the treatment depends on the clinical situation, it generally consists of an aggressive wide resection after careful preoperative staging, including an MRI of the primary tumor and CT scan of the chest. Amputation is rare, with limb salvage possible in the majority of cases.
The use of adjuvant radiation therapy is important in reducing the local recurrence rate. Many clinicians administer 50–55 Gy to a wide area, followed by a boost of 60–66 Gy aimed at the surgical site. An attempt is made to leave a longitudinal strip of tissue out of the field of radiation to reduce the chance of postirradiation edema distal to the treatment site. Some centers advocate preoperative and postoperative radiation with 50 Gy given before resection and approximately 15 Gy given postoperatively. Some institutions employ preoperative radiation exclusively. Local recurrence rates are generally between 5 and 25%.
The use of adjuvant chemotherapy is more controversial. Because limited data suggest that chemotherapy results in a significant improvement in survival and because most patients are older individuals who cannot tolerate the high-dose protocols, medical oncologists are divided on whether to advocate the use of chemotherapeutic agents in the treatment of undifferentiated pleomorphic sarcoma.
2. Giant cell malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma with giant cells: ICD-9-CM 171.x—The giant cell type of MFH also affects older patients and is seen in large muscle groups. Histologically, multiple osteoclastic giant cells are seen, and there may be areas of hemorrhage. It carries a similar prognosis for pulmonary metastasis, local recurrence, and overall survival.
3. Inflammatory malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma with prominent inflammation: ICD-9-CM 171.x—The inflammatory type of MFH affects the older age groups and is more common in the retroperitoneal areas. Histologically, it has prominent benign-appearing xanthomatous cells and mixed inflammatory cells including neutrophils, eosinophils, and occasional lymphocytes and plasma cells. There is some evidence that this entity may be a form of dedifferentiated liposarcoma. Although it has a similar rate of pulmonary metastasis, review of the literature suggests less favorable overall survival with an increased rate of disease-related mortality. This is probably related to the more frequent retroperitoneal location.
B. Fibrosarcoma: ICD-9-CM 171.x
Fifty years ago, fibrosarcoma was considered the most common of the soft-tissue sarcomas, secondary to imprecise pathologic classification of MFH, certain liposarcomas, rhabdomyosarcoma, leiomyosarcomas, and malignant peripheral nerve sheath tumors. Currently, fibrosarcoma is considered one of the least common soft-tissue sarcomas. The diagnosis is reserved for those tumors in which the histology demonstrates a uniform fasciculated growth pattern of spindle cells (malignant fibroblasts). It is clinically similar to MFH, occurs with nearly equal frequency in men and women, is found in patients between 30 and 55 years of age, is sometimes slow growing and painless, and tends to affect deep fascial structures of muscle about the knee and thigh, followed next by the forearm and leg.
On gross examination, fibrosarcoma appears as a firm and lobulated lesion that has a yellowish white to tan color. The lesion may demonstrate a few calcific or osseous deposits on radiographic exam. Microscopy reveals spindle, uniformly shaped fibroblasts oriented in a herringbone pattern. Cells show varying degrees of mitotic activity. Fibrosarcomas contain no malignant histiocytes.
The treatment and prognosis depend on the grade of tumor in a particular patient. Low-grade fibrosarcoma is nearly the same tumor as a benign desmoid tumor and has an extremely low rate of metastasis. However, high-grade fibrosarcoma requires an aggressive wide surgical resection, along with radiation therapy, and has a pulmonary metastasis rate of 50–60%. Lymph node involvement is rare. The use of chemotherapy is considered controversial in patients with fibrosarcoma, as it is in patients with MFH.
C. Myxofibrosarcoma: ICD-9-CM 171.x
Also known as myxoid MFH, myxofibrosarcoma is a relatively common sarcoma in the elderly, seen in the 50- to 80-year age group. The thigh is the most common location followed by the arm and shoulder girdle. Most are large at presentation and may be low grade, with little meta-static potential, or high-grade, with metastases occurring in 20–35% of cases. Larger tumors with increased necrosis exhibit an increased rate of metastasis. Local recurrence is seen in approximately one half of cases after resection, and it is well described that lesions have the propensity to recur at a higher grade, having accumulated chromosomal aberrations. This property, combined with the fact that no specific cytogenetic abnormalities have been identified, highlights the role of genomic instability in the malignant degeneration of this entity. Largely because of the peak age group, chemotherapy is infrequently recommended; however, radiotherapy is routinely delivered either preoperatively or postoperatively.
D. Dermatofibrosarcoma Protuberans: ICD-9-CM 171.x
Dermatofibrosarcoma protuberans, a low- to intermediate-grade fibrohistiocytic tumor, is unique because of its nodular cutaneous location. It is seen more commonly in males than females and occurs in young or middle-age (20–40 years) adults. It is typically located about the trunk and proximal extremities. Antecedent trauma is recorded in 10–20% of cases. Dermatofibrosarcoma protuberans begins as a painless subcutaneous nodule or nodules and slowly develops into an elevated multinodular plaque (Figure 5–71). Microscopic examination of the lesion reveals the same storiform or basket-weave pattern of a benign or malignant fibrous histiocytoma but with a very low mitotic index. The pattern tends to infiltrate extensively into surrounding subcutaneous fat and skin, which accounts for the high local recurrence rate, sometimes reported to approach 50%.

![]() Figure 5–71. Clinical appearance of dermatofibrosarcoma protuberans on the bottom of the heel of a 30-year-old man.
Figure 5–71. Clinical appearance of dermatofibrosarcoma protuberans on the bottom of the heel of a 30-year-old man.
Characteristic cytogenetic abnormalities are described with characteristic features such as reciprocal t(17;22) (q22;q13) or, more commonly, supernumerary ring chromosomes containing sequences from chromosomes 17 and 22. The specific cytogenetic rearrangement may not be as critical as the resulting fusion product of collagen 1 alpha 1 and platelet-derived growth factor (COL1A1-PDGFB), which is detected in the vast majority of cases.
Surgical treatment, consisting of an aggressive resection, is associated with a lower recurrence rate of 20%. Because of the low mitotic index, radiation therapy is not usually indicated, and the chance of pulmonary metastasis is only 1%.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Malignant fibrous histiocytoma has recently been reclassified largely due to increased ability to determine cell lineage with modern molecular techniques.
E. Liposarcomas: ICD-9-CM 171.x
Liposarcoma is the second most common soft-tissue sarcoma after undifferentiated pleomorphic sarcoma. Like MFH, liposarcoma is a tumor of older (40–60 years) patients and can be large and deep seated. Four types of liposarcoma are discussed in the following sections. The well-differentiated type and the myxoid type are associated with a low chance for lung metastasis, whereas the round cell and the pleomorphic types tend to behave more aggressively.
1. Well-differentiated liposarcoma: ICD-9-CM 171.x—This very low-grade tumor affects individuals who are 40–60 years of age and occurs more frequently in men than in women. It grows extremely slowly and reaches a large size without causing pain. The deep-seated tumor is found in the retroperitoneum, buttock, or thigh. In some cases of well-differentiated liposarcoma, findings include inflammation and sclerosis.
On gross examination, this tumor has a fatty lobulated appearance similar to a benign lipoma. Even under the microscope, many large areas of the tumor appear benign. However, with proper sampling, the pathologist will find a few areas of lipoblast activity to suggest the diagnosis of a liposarcoma. MRI findings are sometimes difficult to distinguish from a large deep lipoma (Figure 5–72).

![]() Figure 5–72. T1-weighted MRI of a well-differentiated liposarcoma in the thigh of a 63-year-old man.
Figure 5–72. T1-weighted MRI of a well-differentiated liposarcoma in the thigh of a 63-year-old man.
In cases of well-differentiated liposarcoma, a conservative wide resection is performed to avoid local recurrence. Adjuvant radiation therapy is not helpful, and chemotherapy is never used. The chance of metastatic disease is very low, and the prognosis for survival is excellent.
2. Myxoid liposarcoma: ICD-9-CM 171.x—Myxoid liposarcoma is the most common fat sarcoma, accounting for 40–50% of all liposarcomas. The myxoid type is low to intermediate grade and seen in older patients (see Table 5–6). The clinical presentation is similar to the well-differentiated liposarcoma.
Gross examination of a myxoid liposarcoma reveals a lobulated pattern with some areas that appear similar to those of a lipoma but with other myxomatous areas. Microscopic examination shows myxoid tissue with areas of signet ring lipoblasts. It is common to find a delicate pattern of capillaries running through the myxoid areas. MRI frequently demonstrates a heterogeneous high- and low-signal pattern typical of myxoid liposarcoma but not present in cases of benign lipoma (Figure 5–73).

![]() Figure 5–73. Sagittal view T1-weighted MRI of a myxoid liposarcoma in the thigh of a 32-year-old man.
Figure 5–73. Sagittal view T1-weighted MRI of a myxoid liposarcoma in the thigh of a 32-year-old man.
Characteristic translocations are also seen in myxoid liposarcoma. The predominant type is t(12;16)(q13;p11); however, t(12;22)(q13;q12) is also described. Multifocal myxoid liposarcoma is also described. Consideration for additional advanced axial imaging should be entertained with this histologic subtype. Although myxoid liposarcoma carries a very good prognosis, the tumor should be removed with wide margins. There is current debate over the use of neoadjuvant chemotherapy versus neoadjuvant radiotherapy, with this entity showing responsiveness to both. Adjuvant radiotherapy is still widely used today.
3. Round cell and pleomorphic liposarcoma: ICD-9-CM 171.x—These high-grade liposarcomas are seen in the same locations and age group as the well-differentiated and myxoid subtypes. But unlike the latter, the round cell and pleomorphic types are fast-growing tumors that may be painful.
In cases of round cell or pleomorphic liposarcoma, the lesion does not have a fatty appearance on gross examination but instead looks more like an MFH or a fibrosarcoma. Moreover, on MRI, the lesion appears more like an MFH, with a low-signal pattern in the T1-weighted image and a high-signal pattern in the T2-weighted image. Microscopically, the round cell type of liposarcoma shows areas of uniformly shaped round cells similar to those found in Ewing sarcoma or lymphoma and also shows areas of myxoid tissue. In the pleomorphic type of liposarcoma, large and bizarre giant cells occur similar to those found in undifferentiated pleomorphic sarcoma and rhabdomyosarcoma.
In round cell and pleomorphic liposarcoma, there is an early and high rate of pulmonary metastasis. Accordingly, the prognosis for survival is poor. Thus, the treatment should include aggressive resection, adjuvant radiation therapy as necessary, and chemotherapy in selected patients.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• CT scan of the abdomen and pelvis should be included in the staging and surveillance of liposarcomas due to the incidence of associated retroperitoneal tumors.
• Liposarcoma is particularly subject to sampling error with biopsy, so every effort should be made to sample the most aggressive-appearing portion of the tumor.
F. Rhabdomyosarcomas: ICD-9-CM 171.x
Rhabdomyosarcomas account for 20% of all soft-tissue sarcomas. The embryonal and alveolar types of rhabdomyosarcoma affect pediatric patients, and the rarer pleomorphic type affects adults.
1. Embryonal rhabdomyosarcoma: ICD-9-CM 171.x—The embryonal type is seen in patients from birth to 15 years of age and encountered more frequently in boys than in girls. It is most common in the head and neck area. The so-called botryoid form is seen as a cluster of grapes under mucous membranes in the vagina, bladder, or retroperitoneal area. Histologically, it is a round cell tumor-like Ewing sarcoma, but some rhabdomyoblasts with cross striations are present in a few areas. The presence of anaplasia, or areas of cells containing enlarged, bizarre, hyperchromatic nuclei, is associated with a more aggressive phenotype.
Embryonal rhabdomyosarcoma is treated with local surgical resection plus preoperative and postoperative chemotherapy consisting of vincristine, dactinomycin, cyclophosphamide, and doxorubicin given in cyclic courses during a 2-year span. If the surgical margins are contaminated, local radiation therapy is used. With this program, the 5-year survival rate is 80%. Prior to the advent of chemotherapy, it was only 10%.
2. Alveolar rhabdomyosarcoma: ICD-9-CM 171.x—This type of rhabdomyosarcoma affects individuals between 10 and 25 years of age and is found more commonly in males than in females. Besides affecting the head and neck, it can be seen in the extremities, especially the thigh and calf. Microscopic examination of the lesion reveals a typical alveolar pattern of round cells, with fewer rhabdomyoblasts seen in this type of rhabdomyosarcoma than in the embryonal type. This type of rhabdomyosarcoma is associated with the fusion genes PAX3-FKHR or PAX7-FKHR. Although not definitive, the presence of the translocation t(2;13)/PAX3-FKHR may be an adverse prognostic factor, with molecular screening being implemented in the future. Currently, the treatment is the same as for the embryonal type, but the prognosis is a bit worse.
3. Pleomorphic rhabdomyosarcoma: ICD-9-CM 171.x—In the 1940s, pleomorphic rhabdomyosarcoma was a popular histologic diagnosis, and MFH was a rare one. Based on today’s criteria, most of the old cases classified as pleomorphic rhabdomyosarcoma would now be classified as undifferentiated pleomorphic sarcoma. Currently, the pleomorphic type of rhabdomyosarcoma is the rarest type.
Pleomorphic rhabdomyosarcoma is a high-grade tumor that affects middle-age and older adults and is seen most commonly in the large muscle groups of the proximal extremities, usually the lower extremities. Microscopic examination of the tumor reveals large atypical giant cells, along with racket- or tadpole-shaped malignant rhabdomyoblasts that stain positive for glycogen, actin, and myosin. The tumor carries a poor prognosis and is associated with a high rate of metastasis to the lung. The treatment for pleomorphic rhabdomyosarcoma is similar to that for undifferentiated pleomorphic sarcoma and consists of a wide local resection and adjuvant radiation therapy. Chemotherapy is rarely indicated.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Alveolar rhabdomyosarcoma is associated with characteristic translocations t(1;13) or t(2;13) resulting in the PAX7-FKHR or PAX3-FKHR fusion products, respectively. This affects prognosis and may ultimately affect treatment protocols as well.
G. Leiomyosarcoma: ICD-9-CM 171.x
Leiomyosarcoma is a rare soft-tissue tumor whose cell type of origin is smooth muscle. It is seen in the middle-age adult (see Table 5–6) and is much more common in women than in men. Its usual locations, in order of frequency, are retroperitoneal, intraabdominal, cutaneous, and subcutaneous. In some cases, the lesion has a venous wall origin and is found in the vena cava or large vessels of the leg. On microscopic examination, leiomyosarcoma can demonstrate a palisading, orderly fascicular pattern similar to a malignant schwannoma. A specific immunohistochemical staining for actin may be helpful in the differential diagnosis.
The prognosis and treatment for leiomyosarcoma are similar to those for fibrosarcoma. However, leiomyosarcomas of venous wall origin have a worse prognosis because they are difficult to resect and have a high rate of pulmonary metastasis.
H. Synovial Sarcomas: ICD-9-CM 171.x
Synovial sarcoma (Figure 5–74) is the fourth most common soft-tissue sarcoma. It is seen in young adults between 15 and 35 years of age and affects males slightly more than females. The name of this tumor suggests a synovial cell origin, but only 10% of synovial sarcomas are found in a major joint. Nevertheless, they frequently arise from juxtaarticular structures, especially around the knee, and they can also arise from tendon sheaths, bursal sacs, fascial planes, and deep muscles. Synovial sarcomas can be seen about the shoulder, arm, elbow, and wrist and are the most common soft-tissue sarcoma in the foot.

![]() Figure 5–74. Radiograph (A) and microscopic appearance (B) of a synovial sarcoma in the shoulder of a 20-year-old woman.
Figure 5–74. Radiograph (A) and microscopic appearance (B) of a synovial sarcoma in the shoulder of a 20-year-old woman.
Synovial sarcomas initially grow slowly and cause pain in approximately half of affected patients. The tumors may appear after an injury, and because dystrophic calcification or even heterotopic bone formation is seen in half of the cases, the tumors are assumed to be a benign process for 2–4 years before a diagnostic biopsy is performed.
Microscopic examination of the tumor shows a typical biphasic pattern composed of epithelium-like cells that form nests, clefts, or tubular structures surrounded by malignant fibroblastic spindle cells. The epithelium-like cells produce a mucinous material that suggests a synovial cell origin, although this origin is unlikely. A monophasic form of synovial sarcoma is described and reported to consist of a dominant fibroblastic or epithelial cell pattern. If the lesion shows no biphasic component, however, it is difficult to confirm the diagnosis of synovial sarcoma.
Molecular characterization of this tumor reveals a particular translocation, t(X;18), representing the fusion of SYT (at 18q11) with either SSX1 or SSX2 (both at Xp11). Both SYT and SSX are transcription factors whose fusion product is seen in the majority of synovial sarcomas.
Despite the slow growth of synovial sarcoma, the 5- and 10-year survival rates are only 50 and 25%, respectively. In cases in which the tumors are heavily calcified, the 5-year survival rate is 80%. Because of the poor prognosis, the treatment plan should include aggressive wide resection, along with both radiation therapy and chemotherapy. Recent evidence shows that ifosfamide-based regimens are associated with improved patient outcomes. Lymph node involvement is seen in 20% of affected patients and may require a surgical excision followed by local radiation therapy.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Synovial sarcomas can present as small, calcified soft-tissue tumors in up to 50% of cases.
I. Malignant Peripheral Nerve Sheath Tumor: ICD-9-CM 171.x
A malignant peripheral nerve sheath tumor can arise from a preexisting benign solitary neurofibroma but more frequently arises from the multiple lesions of neurofibromatosis type 1. In both cases, the tumor mass is usually larger than 5 cm in diameter and may arise from a large deep neurogenic structure such as the sciatic nerve (Figure 5–75) or one of the spinal roots. Smaller nerves, even cutaneous branches, however, can give rise to these sarcomas. Malignant degeneration from a solitary neurofibroma usually occurs after 40 years of age with a 5-year survival rate of 75%. In contrast, patients whose tumor arose from the lesions of neurofibromatosis type 1 are generally younger and have a 5-year survival rate of 30%. Surgical treatment consists of a wide resection if possible. Adjuvant radiation and chemotherapy are used in selected cases.

![]() Figure 5–75. Clinical appearance of a café-au-lait defect in the skin overlying a malignant schwannoma in the buttock area of a 42-year-old man (A), and gross appearance of the tumor in the resected sciatic nerve (B).
Figure 5–75. Clinical appearance of a café-au-lait defect in the skin overlying a malignant schwannoma in the buttock area of a 42-year-old man (A), and gross appearance of the tumor in the resected sciatic nerve (B).
J. Malignant Vascular Tumors
1. Kaposi sarcoma: ICD-9-CM 176.x—Of the malignant vascular tumors, Kaposi sarcoma is the most common with four specific subtypes: (1) chronic, (2) lymphadenopathic, (3) transplant associated, (4) acquired immunodeficiency syndrome (AIDS) related. Its pathogenesis is related to infection with Kaposi sarcoma–associated herpesvirus while in the immunocompromised state. It is found directly beneath the skin, generally in the lower extremity of adults, is seen more often in men than in women, and is endemic in central Africa. The cutaneous lesions seen frequently in the foot and ankle area are purplish in color and are nodular (Figure 5–76). Microscopic examination of Kaposi sarcoma shows an aggressive vascular pattern with rare mitosis. However, over a period of many years, the tumor progresses into a full-blown angiosarcoma or fibrosarcoma. Cytotoxic chemotherapeutic alternatives are limited due to the immunocompromised status of the host. With continued investigation in the field, antiviral medications will likely become a mainstay in the treatment of this disease. Although the behavior of Kaposi sarcoma is a function of the immunologic status of the patient and other variables, the overall mortality rate is 10–20%.

![]() Figure 5–76. Clinical appearance of Kaposi sarcoma of the foot.
Figure 5–76. Clinical appearance of Kaposi sarcoma of the foot.
2. Angiosarcoma: ICD-9-CM 171.x—Soft-tissue angiosarcoma is rare, accounting for less than 1% of all sarcomas. Although angiosarcomas are usually cutaneous lesions and tend to affect men more than women, they sometimes take the form of a deep tumor, and they are typically seen in the upper extremities of women who have chronic lymphedema following radical breast surgery and radiation therapy. Histologic examination of angiosarcoma shows anaplastic endothelial cells surrounded by reticulum fiber. Prognosis for the older patient is poor. Smaller lesions in younger (<50 years) patients have a distinctively better outcome. The treatment is wide resection, sometimes with radiation therapy.
3. Solitary fibrous tumor/hemangiopericytoma: ICD-9-CM 171.x—The diagnosis of solitary fibrous tumor versus hemangiopericytoma is a topic of debate. It is now thought that many lesions previously characterized as hemangiopericytomas are actually extrapleural solitary fibrous tumors. Both may range from very low-grade to high-grade sarcoma. These rare perivascular tumors are thought to arise from pericytes. Pericytes are highly arborized perivascular cells that line capillaries and venules. The lesion, which affects male and female adults with equal frequency, is usually found deep in muscle bellies and generally located in the thigh or retroperitoneal area of the pelvis. Microscopic examination of the malignant solitary fibrous tumor reveals tightly packed cells with round nuclei with moderate amounts of cytoplasm with poorly defined borders. Bifurcating sinusoidal vessels that have a typical staghorn appearance are characteristic of the classic hemangiopericytoma. Cytogenetic analysis reveals multiple chromosome translocations including t(12:19) and t(13:22). Treatment consists of a wide surgical resection, followed by local radiation therapy. Some authors recommend preoperative embolization or afferent vessel ligation (or both) intraoperatively.
MISCELLANEOUS SOFT-TISSUE SARCOMAS
The remaining soft-tissue sarcomas are rare and only a brief description of their clinical patterns is summarized.
A. Soft-Tissue Chondrosarcoma: ICD-9-CM 171.x
There are three types of soft-tissue chondrosarcomas.
1. Myxoid chondrosarcoma: ICD-9-CM 171.x—The myxoid chondrosarcoma is sometimes referred to as a chordoid sarcoma because it looks like a chordoma. It is a slow-growing tumor seen in adults, usually in deep structure of the leg. It has a myxoid appearance, does not calcify, and is low grade. Like the chordoma, the myxoid chondrosarcoma responds only to surgical removal.
2. Mesenchymal chondrosarcoma: ICD-9-CM 171.x—This tumor affects individuals between 15 and 40 years of age, is found deep in the lower extremity and neck areas, is fast growing, and carries a poor prognosis because of the high risk of pulmonary metastasis. Calcification may be seen on radiograph, and microscopic examination reveals round cells scattered in a chondroid matrix. Treatment consists of a wide resection in conjunction with chemotherapy and radiation therapy.
3. Synovial chondrosarcoma: ICD-9-CM 171.x—The conversion of a synovial chondromatosis to a malignant synovial chondrosarcoma is an extremely rare phenomenon. It can occur with lesions of the hip or knee region in older (>60 years) adults.
B. Ewing Sarcoma: ICD-9-CM 171.x
Extraskeletal Ewing sarcoma can be found in individuals between 10 and 30 years of age and is usually located in the paravertebral area, thorax, or deep muscle area of the lower extremity. It is a fast-growing tumor with minimal pain symptoms. It carries the same prognosis as its counterpart in bone and is treated with the same combination of surgery, chemotherapy, and radiation therapy.
C. Alveolar Soft Part Sarcoma: ICD-9-CM 171.x
This round cell sarcoma affects more females than males, is usually found in patients between 15 and 35 years of age, and arises in the deep muscle tissue of the lower extremity, usually the thigh. Alveolar soft part sarcoma is a slow-growing tumor but carries a poor prognosis because of early pulmonary metastasis. The tumor has increased vascularity and is thought to originate from a neurogenic stem cell. It derives its name from its alveolar pattern, which is seen on microscopic examination and can cause this tumor to be mistaken for an alveolar form of rhabdomyosarcoma. A cytogenetic, unbalanced abnormality, t(x;17)(p11.2;q25), is described. Treatment of alveolar soft part sarcoma consists of a wide surgical resection plus radiation therapy and chemotherapy.
D. Epithelioid Sarcoma: ICD-9-CM 171.x
Although this superficial skin lesion is seen most commonly in the palm of the hand, it can also be found on the dorsum of the forearm or on the plantar aspect of the foot. It is a slow-growing tumor that affects patients between 20 and 30 years of age, causes minimal pain symptoms, and is associated with ulceration.
Because epithelioid sarcoma has a whitish color that under the microscope demonstrates cords of epithelium-like cells, it can be mistaken for a synovial sarcoma. Moreover, because of its firm multilobulated presentation, the epithelioid sarcoma may be mistaken for a plantar of palmar fibromatosis (Figure 5–77).

![]() Figure 5–77. Clinical appearance of epithelioid sarcoma on the plantar aspect of the foot of a 36-year-old man.
Figure 5–77. Clinical appearance of epithelioid sarcoma on the plantar aspect of the foot of a 36-year-old man.
Epithelioid sarcoma spreads as a lumpy nodularity along tendon sheaths or fascial planes and frequently involves local lymph nodes. Local surgical resection is followed by a high local recurrence rate, and a late pulmonary metastasis is common. For this reason, early treatment should consist of an aggressive wide surgical resection.
E. Clear Cell Sarcoma: ICD-9-CM 171.x
The clear cell sarcoma is thought to be a deep, noncutaneous variant of the well-known cutaneous melanoma. It is extremely rare, affects women more often than men, and commonly occurs between 20 and 40 years of age. It arises in tendon sheaths and fascial planes, most frequently in the foot and ankle but also in the knee and arm. Clear cell sarcoma starts slowly as a painless lump and has a high potential to spread to local lymph nodes. The lesion in many cases demonstrates evidence of melanin and melanosomes and may be of neural crest origin. The microscopic clear cell appearance can cause this sarcoma to be confused with epithelioid sarcoma and synovial sarcoma.
The prognosis is poor because of a high rate of pulmonary metastasis. This tumor may spread via lymphatics as well. Treatment consists of early aggressive wide resection and may include chemotherapy and local radiation therapy.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Although most mesenchymal malignancies do not usually metastasize to lymph nodes, synovial sarcoma, epithelioid sarcoma, and rhabdomyosarcoma are exceptions.
• Soft-tissue sarcoma in the adult is generally more sensitive to radiation therapy than chemotherapy.
Ahmad SA, Patel SR, Ballo MT, et al: Extraosseous osteosarcoma: response to treatment and long-term outcome. J Clin Oncol 2002;20:521. [PMID: 11786582]
Anderson J, Gordon T, McManus A, et al: Detection of the PAX3-FKHR fusion gene in paediatric rhabdomyosarcoma: a reproducible predictor of outcome? Br J Cancer 2001;85:831. [PMID: 11556833]
Canter RJ, Qin LX, Ferrone CR, et al: Why do patients with low-grade soft tissue sarcoma die? Ann Surg Oncol 2008;15:3550. [PMID: 18830667]
Canter RJ, Qin LX, Maki RG, et al: A synovial sarcoma-specific preoperative nomogram supports a survival benefit to ifos-famide-based chemotherapy and improves risk stratification for patients. Clin Cancer Res 2008;14:8191. [PMID: 19088035]
Casper C, Wald A: The use of antiviral drugs in the prevention and treatment of Kaposi sarcoma, multicentric Castleman disease and primary effusion lymphoma. Curr Top Microbiol Immunol 2007;312:289. [PMID:17089802]
Coindre JM, Hostein I, Maire G, et al: Inflammatory malignant fibrous histiocytomas and dedifferentiated liposarcomas: histological review, genomic profile, and MDM2 and CDK4 status favour a single entity. J Pathol2004;203:822. [PMID: 15221942]
Davicioni E, Anderson MJ, Finckenstein FG, et al: Molecular classification of rhabdomyosarcoma: genotypic and phenotypic determinants of diagnosis. Am J Pathol 2009;174:550. [PMID: 19147825]
Eilber FC, Brennan MF, Eilber FR, et al: Chemotherapy is associated with improved survival in adult patients with primary extremity synovial sarcoma. Ann Surg 2007;246:105. [PMID: 17592298]
Eilber FC, Eilber FR, Eckardt J, et al: The impact of chemotherapy on the survival of patients with high-grade primary extremity liposarcoma. Ann Surg 2004;240:686. [PMID: 15383796]
Guadagnolo BA, Zagars GK, Ballo MT, et al: Long-term outcomes for synovial sarcoma treated with conservation surgery and radiotherapy. Int J Radiat Oncol Biol Phys 2007;69:1173. [PMID: 17689031].
Huang HY, Lal P, Qin J, et al: Low-grade myxofibrosarcoma: a clinicopathologic analysis of 49 cases treated at a single institution with simultaneous assessment of the efficacy of 3-tier and 4-tier grading systems. Hum Pathol2004;35:612. [PMID: 15138937]
Jones RL, Fisher C, Al-Muderis O, et al: Differential Sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer 2005;41:2853. [PMID: 16289617]
Koch M, Nielsen GP, Yoon SS: Malignant tumors of blood vessels: angiosarcomas, hemangioendotheliomas, and hemangiopericytomas. J Surg Oncol 2008;97:321. [PMID: 18286475]
Kuklo TR, Temple HT, Owens BD, et al: Preoperative versus postoperative radiation therapy for soft-tissue sarcomas. Am J Orthop 2005;34:75. [PMID: 15789525]
Ladanyi M: Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene 2001;20:5755. [PMID: 11607825]
Lazar AJ, Das P, Tuvin D, et al: Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin Cancer Res 2007;13:7314. [PMID: 18094412]
Lehnhardt M, Daigeler A, Homann HH, et al: MFH revisited: outcome after surgical treatment of undifferentiated pleomorphic or not otherwise specified (NOS) sarcomas of the extremities—an analysis of 140 patients. Langenbecks Arch Surg 2009;394:313. [PMID: 18584203]
Nakayama R, Nemoto T, Takahashi H, et al: Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous histiocytoma. Mod Pathol 2007;20:749. [PMID: 17464315]
Nascimento AF, Raut CP: Diagnosis and management of pleomorphic sarcomas (so-called “MFH”) in adults. J Surg Oncol 2008;97:330. [PMID: 18286476]
Patel KU, Szabo SS, Hernandez VS, et al: Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol 2008;39:184. [PMID: 17950782]
Pisters PW, O’Sullivan B, Maki RG: Evidence-based recommendations for local therapy for soft tissue sarcomas. J Clin Oncol 2007;25:1003. [PMID: 17350950]
Pitson G, Robinson P, Wilke D, et al: Radiation response: an additional unique signature of myxoid liposarcoma. Int J Radiat Oncol Biol Phys 2004;60:522. [PMID: 15380587]
Qualman S, Lynch J, Bridge J, et al: Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer2008;113:3242. [PMID: 18985676]
Spunt SL, Skapek SX, Coffin CM: Pediatric nonrhabdomyosarcoma soft tissue sarcomas. Oncologist 2008;13:668. [PMID: 18586922]
West RB, Harvell J, Linn SC, et al: Apo D in soft tissue tumors: a novel marker for dermatofibrosarcoma protuberans. Am J Surg Pathol 2004;28:1063. [PMID: 15252314]
Willems SM, Debiec-Rychter M, Szuhai K, et al: Local recurrence of myxofibrosarcoma is associated with increase in tumor grade and cytogenetic aberrations, suggesting a multistep tumour progression model. Mod Pathol2006;19:407. [PMID: 16415793]
![]() MANAGEMENT OF CARCINOMA METASTASIZED TO BONE
MANAGEMENT OF CARCINOMA METASTASIZED TO BONE
![]() Incidence and Natural History of Metastases: ICD-9-CM 199
Incidence and Natural History of Metastases: ICD-9-CM 199
A. Common Metastatic Carcinomas and Areas of Skeletal Involvement
Metastatic involvement of the musculoskeletal system is one of the most significant clinical issues facing orthopedic oncologists. The number of patients with metastasis to the skeletal system from a carcinoma is 15 times greater than the number of patients with primary bone tumors of all types. Approximately a third of all diagnosed adenocarcinomas include skeletal metastases, resulting in approximately 300,000 cases per year. Furthermore, 70% of patients with advanced terminal carcinoma demonstrate bone metastases at autopsy. The carcinomas that commonly metastasize to bone are prostate, breast, kidney, thyroid, and lung carcinomas. One study showed that nearly 90% of patients with these types of carcinoma had bone metastases. Among the carcinomas that less commonly metastasize to bone are cancers of the skin, oral cavity, esophagus, cervix, stomach, and colon.
The spine is the most frequent area of bone metastasis. Other common skeletal sites include the pelvis, femur, rib, proximal humerus, and skull, in that order. Metastatic lesions are rarely found distal to the elbow or knee. If lesions are found in these areas, so-called acral metastases, the lung is the most common source. Solitary bone lesions comprise only approximately 10% of cases of bone metastasis.
B. Clinical Course of Metastases
The mechanism of metastases is accounted for in a modified “seed/soil” theorem. Fewer than 1 in 10,000 neoplastic cells that escape into the circulation from the primary site are able to set up a metastatic focus, a complex multistep process by which the cell must first break free. This is a function of degradative enzymes such as collagenases, hydrolases, cathepsin D, and proteases. Once the cell invades the vascular channel, it circulates through the body. It is theorized that the cell is protected by a fibrin platelet clot. However, clinical trials with heparin do not show a significant change in metastatic outcome. Local factors such as integrins are instrumental in attracting the circulating metastatic cell to a particular remote tissue site. Once within the new tissue, the meta-static cell releases factors such as tumor angiogenesis factor, inducing neovascularization, which in turn facilitates growth of the metastatic focus.
Patients with advanced metastatic disease frequently experience dysfunction of their hematopoietic and calcium homeostasis systems. Patients may develop a normochromic, normocytic anemia with leukocytosis. In response to the anemia, the increased production of immature cells is noted on the peripheral blood smear. This is termed the leukoerythroblastic reaction. Hypercalcemia may result in up to 30% of cases with extensive metastases. This is most frequently seen in myeloma, breast cancer, and non–small cell lung cancer.
Blastic metastases are frequently painless and associated with a lower incidence of pathologic fracture because the bone is not as severely weakened (Figures 5–78 and 5–79). Not all tumors that metastasize from the prostate to the bone are blastic. The lytic variants are painful and more likely to cause pathologic fractures.

![]() Figure 5–78. Radiograph of a blastic carcinoma that metastasized from the prostate to the pelvis in an 85-year-old man.
Figure 5–78. Radiograph of a blastic carcinoma that metastasized from the prostate to the pelvis in an 85-year-old man.

![]() Figure 5–79. Skeletal specimen of a blastic carcinoma that metastasized from the prostate to the lumbar spine.
Figure 5–79. Skeletal specimen of a blastic carcinoma that metastasized from the prostate to the lumbar spine.
Most tumors that metastasize from the breast to the bone are blastic, but some demonstrate mixtures of blastic and lytic areas in the same bone. By taking serial radiographs and noting the appearance of bone metastases, it is possible to follow the progress of treatment consisting of systemic therapy with hormones or chemotherapeutic agents plus local radiation therapy. A favorable response may show a gradual conversion from a lytic to a blastic appearance as the pain decreases.
Bone destruction in lytic lesions is a response by native osteoclasts to the tumor. Neovascularity is common. Among the tumors that are characteristic for this hemorrhagic response are thyroid carcinomas (Figure 5–80), renal cell carcinoma (Figure 5–81), and multiple myeloma. Before a surgical intervention, it is beneficial to perform a prophylactic embolization of the area to reduce perioperative bleeding. If a lesion is unexpectedly found to be aneurysmal at the time of surgical exploration, it is best to debulk the friable tumor mass rapidly down to normal bone and then pack the area until it can be stabilized with bone cement.

![]() Figure 5–80. Clinical appearance (A) and radiographic appearance (B) of aneurysmal lesions in a case of carcinoma that metastasized from the thyroid to the hand.
Figure 5–80. Clinical appearance (A) and radiographic appearance (B) of aneurysmal lesions in a case of carcinoma that metastasized from the thyroid to the hand.

![]() Figure 5–81. Radiograph of a metastatic hypernephroma in the ilium.
Figure 5–81. Radiograph of a metastatic hypernephroma in the ilium.
![]() Diagnosis
Diagnosis
A. General Approach
A methodical approach is mandatory in the workup of a patient with presumed metastatic disease to bone to locate the primary tumor. A thorough history and physical examination must be completed prior to laboratory and radiographic analysis. Eight percent of patients may have their primary carcinoma detected on physical exam. Laboratory analysis should include complete blood count, ESR, renal and liver panels, alkaline phosphate, and serum protein electrophoresis.
Radiographic examination should follow with a plain chest radiograph and radiographs of known involved bones. Approximately 45% of primaries are detected in the lung on the chest radiograph. The workup should also include a staging bone scan. If this is negative, myeloma should be suspected. Furthermore, a lesion at a more convenient biopsy site may be found. Bone scan is also more sensitive than plain radiographs in detecting early lesions. CT scans of the chest, abdomen, and pelvis should be performed. Lung CT can detect up to 15% of primaries missed on the plain radiograph.
These studies in conjunction with a well-planned biopsy detect the majority of cases. Routine radiographic screening studies in search of early metastatic disease are not very helpful (Figure 5–82). Lytic changes become evident on routine radiographs only when cortical destruction approaches 30–50% (Figure 5–83).

![]() Figure 5–82. Radiograph (A) and gross appearance (B) of bone in a case of carcinoma that metastasized from the lung to the spine.
Figure 5–82. Radiograph (A) and gross appearance (B) of bone in a case of carcinoma that metastasized from the lung to the spine.

![]() Figure 5–83. Radiograph of the spine of a 45-year-old woman whose cancer had metastasized from the breast.
Figure 5–83. Radiograph of the spine of a 45-year-old woman whose cancer had metastasized from the breast.
![]() Treatment and Prognosis
Treatment and Prognosis
A. Nonsurgical Treatment
Nonsurgical management of metastatic carcinoma to bone includes observation, radiation treatment, and hormonal/cytotoxic chemotherapy. Radiation is reserved for palliative management. Each patient must be carefully evaluated as a candidate for radiation therapy. The histologic type of disease, extent of disease, prognosis, marrow reserve, and overall constitution must be assessed.
After sustaining a pathologic fracture secondary to meta-static carcinoma, the average survival time is 19 months. Each histologic type has varying lengths of survival (prostate, 29 months; breast, 23 months; renal, 12 months; lung, 4 months). Furthermore, each type of carcinoma exhibits varying radiosensitivity. Prostate and lymphoreticular types demonstrate excellent sensitivity. Breast is intermediate, and renal and gastrointestinal are poor. When used, appropriately 90% of patients gain at least minimal relief, with up to two thirds obtaining complete relief. Seventy percent of patients who are ambulatory retain this function after radiation therapy to the lower extremities. Systemic radioisotopes are also used in selected cases.
Hormonal therapy has an important role in the management of metastatic breast and prostate cancer. Fortunately, these agents are easy to administer and have few side effects.
For breast cancer, medical hormonal manipulation can be done by use of antiestrogens, progestins, luteinizing hormone–releasing hormone, or adrenal-suppressing agents. Tamoxifen is effective in 30% of all breast cancer cases but increases to 50–75% of cases when the tumor is known to be estrogen receptor, progesterone receptor positive. Surgical ablation (oophorectomy) may also have a role in certain cases.
For prostate cancer, reduction in testosterone levels via bilateral orchiectomy or administration of estrogens or antiandrogens may produce dramatic results in certain cases. Estrogens are no longer used as a first agent because of the risk of cardiovascular complication.
The use of agents aimed at decreasing bone turnover, including bisphosphonates, osteoprotegerin, and nuclear factor kappa B inhibitor, has also been introduced to limit progression of bone metastases. Although the effect on overall tumor burden is unclear, there is evidence that bisphosphonates decrease skeletal-related events. Preclinical studies also suggest that when combined with cytotoxic agents, bisphosphonates may synergistically reduce the incidence of osseous metastases and prolong survival.
Cytotoxic chemotherapy is used in adenocarcinoma treatment quite extensively. In older (>60 years) patients with advanced disease, however, the side effects of the drugs may be too severe.
![]() B. Surgical Treatment
B. Surgical Treatment
The goals for surgical intervention in the patient with meta-static carcinoma to bone are relief of pain; prevention of pathologic fractures; stabilization of realized pathologic fractures; enhancement of mobility, function, and quality of life; and perhaps improvement of survival. It is generally agreed that a patient must have a life expectancy of at least 6 weeks to warrant operative intervention. Special considerations to surgical management include noting that bone quality is attenuated and healing will be delayed if even possible. Cancer patients, irrespective of their age, may have increased difficulty protecting their fixation device/prosthesis secondary to systemic debilitation. Accordingly, rigid fixation, with polymethylmethacrylate (PMMA) augmentation as needed, is mandatory.
1. Hip—Seventy-five percent of all surgery for cancer that has metastasized to bone is performed in the hip area (Figure 5–84). Prior to 1970, surgeons attempted to stabilize these fractures with conventional hip nails or Austin Moore prostheses, but results were poor because of deficient local bone stock. After 1970, with the advent of bone cement as an adjuvant form of therapy, these same devices could be used, with improved results in most cases, along with local radiation therapy starting 2 weeks after the surgery. This technique allowed for early ambulation with less pain. However, as time passed and survival times increased, more failures were noted after 1–2 years with the hip nail and cement technique. For this reason, most surgeons currently use a cemented bipolar hemiarthroplasty for the femoral neck fractures and a longer stem calcar replacement hemiarthroplasty for the intertrochanteric fractures. Before these procedures are performed, it is wise to evaluate the entire shaft of the femur and the supraacetabular area for other lytic lesions that might require a longer stem femoral component for the shaft or a modified cemented acetabular component with a total hip replacement for acetabular lesions.

![]() Figure 5–84. Radiograph of the pathologic fractures of both hips in a 55-year-old man with lung carcinoma.
Figure 5–84. Radiograph of the pathologic fractures of both hips in a 55-year-old man with lung carcinoma.
In many cases, the diagnosis of metastasis to the proximal femur is made before a fracture occurs. In these cases, it is the responsibility of the orthopedic surgeon to decide whether the patient should receive some form of internal stabilization prior to radiation therapy. A CT scan of the involved area helps make this decision. Criteria for the performance of a prophylactic stabilization procedure include the following: (1) 50% cortical lysis, (2) a femoral lesion greater than 2.5 cm in diameter, (3) an avulsion fracture of the lesser trochanter, and (4) persistent pain in the hip area 4 weeks following the completion of radiation therapy. These criteria are not perfect, however, and large errors arise in estimation of the load-bearing capacity of the bone.
2. Supraacetabular area—In the case of a small supraace-tabular lesion with intact cortical bone, a cemented cup with a total hip system is generally most appropriate. Augmentation of the fully cemented reconstruction with threaded Steinmann pins or similar anchoring screws may be necessary in advanced cases (Figure 5–85). The principles of treatment are always the same, irrespective of the extent of disease: aggressive intralesional curettage of the area back to healthy bone, followed by the placement of large (4.76 mm) threaded Steinmann pins into the sacroiliac area. The pins are placed with an initial foundation batch of cement, leaving them exposed for a second batch of cement, on top of which the cup is placed. A routine femoral component is then cemented.


![]() Figure 5–85. Preoperative (A) and postoperative (B) radiographs of the pelvis of a 65-year-old man with metastatic transitional cell carcinoma to the right acetabular region.
Figure 5–85. Preoperative (A) and postoperative (B) radiographs of the pelvis of a 65-year-old man with metastatic transitional cell carcinoma to the right acetabular region.
3. Femoral shaft—Diaphyseal lesions that affect the femur but spare the peritrochanteric area are best handled with some form of intramedullary nail (Figure 5–86). Fixation of the entire femur, including the peritrochanteric area, with a reconstruction-type nail is preferable in the event the disease progresses within the bone. Current intramedullary fixation devices often do not need cement augmentation. However, in cases of severe bone deficiency, PMMA introduction, either directly into the defect or indirectly at the nail insertion site, is preferable.

![]() Figure 5–86. Preoperative (A) and postoperative (B) radiographs of the midshaft of the femur of a patient whose treatment involved fixation with a cemented intramedullary nail.
Figure 5–86. Preoperative (A) and postoperative (B) radiographs of the midshaft of the femur of a patient whose treatment involved fixation with a cemented intramedullary nail.
4. Humerus—The principle for the management of meta-static disease to the humerus is no different from that for the femur. In the case of diaphyseal lesions, surgeons either use a conventional intramedullary rod or they plate the lesion. PMMA may be used with either technique.
In the case of the proximal humerus involving a large amount of the humeral head and neck, it is frequently necessary to cement a long-stem prosthesis (Figure 5–87). Just as with the proximal femur, in the proximal humerus, there is no need to widely resect the tumor, and the rotator cuff is usually left intact.

![]() Figure 5–87. Preoperative (A) and postoperative (B) radiographs of the proximal humerus of a patient whose treatment involved the use of a long-stem Neer prosthesis.
Figure 5–87. Preoperative (A) and postoperative (B) radiographs of the proximal humerus of a patient whose treatment involved the use of a long-stem Neer prosthesis.
5. Spine—In most cases of metastasis to the spine, the patient’s pain can be managed adequately with local radiation therapy and medication. However, in cases of mechanical collapse associated with bony protrusion into the vertebral canal and cord compromise, surgical decompression and stabilization are frequently indicated. In the past, most of these problems were treated with posterior decompression by laminectomy alone. The results were poor because the spine was further destabilized, which resulted in increased kyphosis and anterior cord compression. With advances in the area of spinal instrumentation, the treatment shifted toward a more aggressive anterior decompression and stabilization if the patient’s general condition allows. Even in cases in which the patient’s general health does not tolerate the larger anterior approach, a less aggressive alternative might include posterior decompression supplemented by posterior spinal fixation.
The midthoracic spine is the most common area for paraplegia secondary to metastasis because of the narrow vertebral canal at this level of the spine. The ideal surgical approach to the problem in a patient with a reasonable prognosis consists of an anterior thoracotomy and anterior decompression by vertebrectomy, followed by anterior stabilization. As an alternative approach in a patient with a worse prognosis and circumferential cord compression, a posterior decompression stabilization can be considered (Figure 5–88).

![]() Figure 5–88. Preoperative T1-weighted MRI (A) and postoperative radiograph (B) of the spine of a patient whose treatment involved use of posterior rods and sublaminar wires for stabilization.
Figure 5–88. Preoperative T1-weighted MRI (A) and postoperative radiograph (B) of the spine of a patient whose treatment involved use of posterior rods and sublaminar wires for stabilization.
The second most common site for cord compression is the thoracolumbar region. The anterior reconstruction is the same in the thoracolumbar area as in the midthoracic area. A posterior stabilization may be advisable, especially in cases in which the prognosis is good.
The cervical spine is the least likely area for surgical treatment, mainly because the vertebral canal is wide at this level and cord compromise is uncommon. If surgery is needed, an ideal reconstruction is an anterior decompression and stabilization.
Radiation therapy is required postoperatively with all of these reconstructions. The use of bone graft is therefore undesirable because of inhibited osteoblastic healing.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Prostate, breast, kidney, thyroid, and lung carcinomas are the most common to metastasize to bone.
• Surgical treatment of metastatic bone disease should focus on early return to function and maximization of quality of life.
• Ionized calcium should routinely be checked in metastatic bone disease due to the morbidity and mortality of hyper-calcemia of malignancy.
• Carcinoma of the lung is the most likely to cause distal, or so-called acral, metastases.
Cappucio M, Gasbarrini A, Van Urk P, et al: Spinal metastasis: a retrospective study validating the treatment algorithm. Eur Rev Med Pharmacol Sci 2008;12:155. [PMID: 18700686]
Guise TA: Antitumor effects of bisphosphonates: promising preclinical evidence. Cancer Treat Rev 2008;34(Suppl 1):S19. [PMID: 18486348]
Hatoum HT, Lin SJ, Smith MR, et al: Zoledronic acid and skeletal complications in patients with solid tumors and bone metastases: analysis of a national medical claims database. Cancer 2008;113:1438. [PMID: 18720527]
Hipp JA, Springfield DS, Hayes WC: Predicting pathologic fracture risk in the management of metastatic bone defects. Clin Orthop Relat Res 1995;312:120. [PMID: 7634597]
Kohno N, Aogi K, Minami H, et al: Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: a randomized, placebo-controlled trial. J Clin Oncol 2005;23:3314. [PMID: 15738536]
Lu S, Zhang J, Zhou Z, et al: Synergistic inhibitory activity of zoledronate and paclitaxel on bone metastasis in nude mice. Oncol Rep 2008;20:581. [PMID: 18695909]
Manabe J, Kawaguchi N, Matsumoto S, et al: Surgical treatment of bone metastasis: indications and outcomes. Int J Clin Oncol 2005;10:103. [PMID: 15864695]
Mirels H: Metastatic disease in long bones. A proposed scoring system for diagnosing impending pathologic fractures. Clin Orthop Relat Res 1989;249:256. [PMID: 2684463]
Rougraff BT, Kneisl JS, Simon MA: Skeletal metastases of unknown origin: a prospective study of a diagnostic strategy. J Bone Joint Surg Am 1993;75:1276. [PMID: 8408149]
Wedin R, Bauer HC: Surgical treatment of skeletal metastatic lesions of the proximal femur: endoprosthesis or reconstruction nail? J Bone Joint Surg Br 2005;87:1653. [PMID: 16326880]
Wedin R, Bauer HC, Rutqvist LE: Surgical treatment for skeletal breast cancer metastases: a population-based study of 641 patients. Cancer 2001;92:257. [PMID: 11466677]
![]() DIFFERENTIAL DIAGNOSIS OF PSEUDOTUMOROUS CONDITIONS
DIFFERENTIAL DIAGNOSIS OF PSEUDOTUMOROUS CONDITIONS
In addition to benign, malignant, and metastatic neoplasms, a group of pseudotumors masquerade as bone and soft-tissue tumors. These lesions actually appear with greater frequency than either primary bone or soft-tissue tumors.
![]() Stress-Reactive Lesions
Stress-Reactive Lesions
The most common pseudotumors are those related to either bone or soft-tissue injury.
A. Stress Fracture of Bone: ICD-9-CM 733.9x
Stress fractures are common in young (<30 years) athletic individuals and can produce radiographic features that might suggest the diagnosis of a bone-forming sarcoma or Ewing sarcoma. It is important to obtain a careful history from patients regarding their physical activity both at work and at play. There will be no history of a single injury if the bone symptoms are caused by repetitive impact loading stress such as occurs with working out or cross-country running. The stress fracture usually occurs several weeks after a sudden increase in physical activity for which the patient is not properly conditioned. This is a common situation in the military, particularly during initial training.
Stress fractures are commonly located in the metaphyseal-diaphyseal areas of long weight-bearing bones. Early radiographs frequently appear normal before periosteal new bone begins to form. The most sensitive early diagnostic tool is a bone scan, which can appear hot or abnormal in the case of stress fractures, neoplasms, and infections. The MRI is sensitive to early fluid shifts in the periosteum overlying a stress fracture, but it is also sensitive to neoplastic and infectious conditions. One of the best methods to help rule out tumors and infection is to simply stop all physical stress to the injured bone for a period of 4 weeks. In patients with stress fracture, the pain should resolve spontaneously during this period, and a follow-up radiograph taken after this period reveals a typical fusiform circumferential periosteal callous formation. In patients with a tumor or infection, the pain persists, and the radiographic signs of permeative osteolysis predominate, in which case a biopsy and culture are indicated.
At times, the clinical picture of a stress fracture is confused by the preexistence of a benign stress raiser, such as a nonossifying fibroma or fibrous cortical defect (Figure 5–89).


![]() Figure 5–89. Radiograph (A), isotope bone scan (B), and CT scan (C) of the femur of a 14-year-old girl with a stress fracture. Notice the subtle increased uptake in the contralateral femur in B and the absence of a nidus, which would be seen in osteoid osteoma, but rather a linear lucency in C.
Figure 5–89. Radiograph (A), isotope bone scan (B), and CT scan (C) of the femur of a 14-year-old girl with a stress fracture. Notice the subtle increased uptake in the contralateral femur in B and the absence of a nidus, which would be seen in osteoid osteoma, but rather a linear lucency in C.
In older patients, especially in postmenopausal women, stress fractures can occur with minimal physical activity. The circumstances under which the fracture occurred might not come out in a routine history. A common location of osteoporotic stress fractures is in the sacrum (Figure 5–90).

![]() Figure 5–90. T1-weighted MRI (A), isotope bone scan (B), and CT scan (C) of the sacrum in a 71-year-old woman with stress fracture.
Figure 5–90. T1-weighted MRI (A), isotope bone scan (B), and CT scan (C) of the sacrum in a 71-year-old woman with stress fracture.
B. Myositis Ossificans: ICD-9-CM 728.12
Another common stress-reactive pseudotumor seen in the extremity is myositis ossificans, which occurs most frequently in the lower extremity in young men. The quadriceps muscle is commonly involved because of direct blows or tearing injury to this muscle. The pseudotumor mass may not arise for several months after the injury and may not be related to a specific injury. In older (>40 years), more sedentary patients, there may be no history of stress injury.
Early radiographs may not reveal soft-tissue calcification. With maturation, ossification occurs in the traumatized muscle fascial planes, which may suggest the diagnosis of a synovial sarcoma or other calcifying sarcoma. If the myositis pseudotumor is attached to the subjacent bone, it can mimic a parosteal osteosarcoma (Figure 5–91).

![]() Figure 5–91. Radiograph (A) and gross appearance of a resected specimen (B) of myositis ossificans in the adductor muscles of a 12-year-old girl.
Figure 5–91. Radiograph (A) and gross appearance of a resected specimen (B) of myositis ossificans in the adductor muscles of a 12-year-old girl.
![]() Infectious Diseases
Infectious Diseases
Bacterial, viral, tuberculous, or fungal infections of the bone or soft tissue can frequently mimic a neoplastic process. This is particularly the case with infections that are not highly virulent, do not create systemic symptoms or a febrile response, and do not cause a large alteration in acute-phase reactant laboratory work. If a tender mass is present on examination and a bone or soft-tissue tumor is suggested by imaging studies, a biopsy may be indicated and should include a tissue culture to make the correct diagnosis. Inflammatory pseudotumors can be seen in any age group but are more common in children and frequently affect the lower extremity.
A. Bacterial Infection: ICD-9-CM 730.0-2x
Bacterial infections of bone can take on the appearance of a round cell tumor such as Ewing sarcoma in children or lymphoma in adults (Figure 5–92). In contrast, tuberculous and fungal infections are less inflammatory and thus have more localized, well-marginated lesions that take on the imaging appearance of a benign tumor.

![]() Figure 5–92. Radiograph of acute osteomyelitis caused by Staphylococcus aureus in the proximal humerus of a 13-year-old boy.
Figure 5–92. Radiograph of acute osteomyelitis caused by Staphylococcus aureus in the proximal humerus of a 13-year-old boy.
B. Tuberculous or Fungal Infection: ICD-9-CM 015.x, or 730.2x
A tuberculous or fungal infection of the spine or extremity can present as a pseudotumor in children or young adults, especially in Asian or Mexican patients (Figure 5–93). The incidence of tuberculous and fungal infections, which are low-grade infections that typically have an insidious onset, is also increased in patients with AIDS.
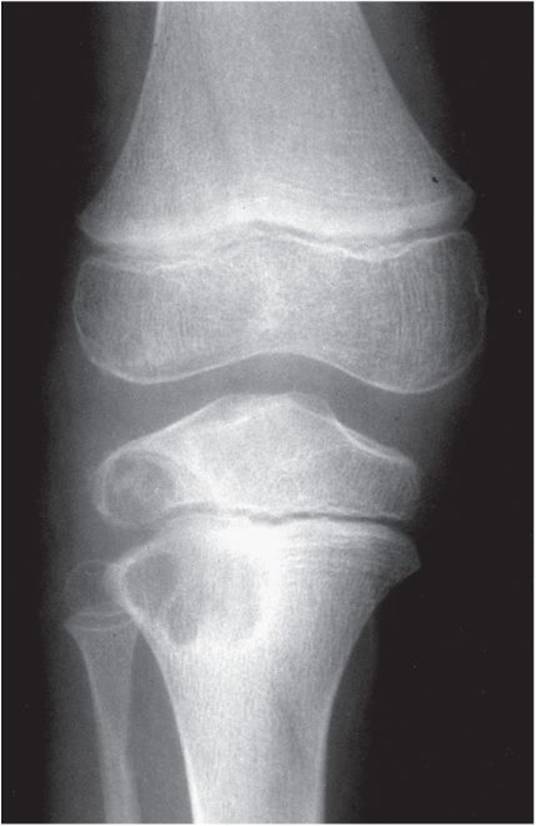
![]() Figure 5–93. Radiograph of tuberculous osteomyelitis in the proximal tibia of a 10-year-old girl.
Figure 5–93. Radiograph of tuberculous osteomyelitis in the proximal tibia of a 10-year-old girl.
C. Caffey Disease: ICD-9-CM 730.3x
Caffey disease can mimic a neoplastic process. It is an idiopathic form of periostitis that is seen in infants younger than 6 months and affects the extremities, shoulder girdle, and mandible (Figure 5–94). It may have a viral origin and is currently much rarer than it was 30 years ago. The bony changes are osteoblastic and could suggest the diagnosis of an osteosarcoma, which is rare in infants. Caffey disease is self-limiting and usually clears spontaneously without disability.

![]() Figure 5–94. Radiographs of the upper extremity (A) and the bilateral shoulders (B) of a 5-month-old infant with Caffey disease.
Figure 5–94. Radiographs of the upper extremity (A) and the bilateral shoulders (B) of a 5-month-old infant with Caffey disease.
![]() Metabolic Disorders
Metabolic Disorders
A. Brown Tumor of Primary Hyperparathyroidism: ICD-9-CM 733.90
Brown tumor is the most common metabolic disorder that mimics a neoplastic process in bone. The lytic giant cell lesions occur symmetrically in metaphyseal-epiphyseal bone as the result of increased parathyroid hormone production by a solitary parathyroid adenoma, by hyperplastic parathyroid glands, or by a solitary parathyroid carcinoma. Brown tumors occur three times more often in females than in males and are usually seen between 15 and 70 years of age. They are most common in the ends of the long bone, followed next in frequency in the pelvis, long bone diaphysis, maxillary bone, cranium, rib, and hand. Brown tumors are rarely seen in the spine. Symptoms of pain are related to the local bone destruction, but widespread pain may result from generalized osteomalacia. The hyperparathyroid condition can lead to weight loss, psychological disorders, gastrointestinal disorders, renal stones, polyuria, and polydipsia.
The radiographic features of the brown tumor in bone include a round lytic area that may be multicentric and may suggest the diagnosis of metastatic carcinoma, multiple myeloma, or histiocytic lymphoma (Figure 5–95). In the case of a solitary lesion, it may suggest the diagnosis of a nonossifying fibroma, fibrous dysplasia, giant cell tumor, or aneurysmal bone cyst. At the time of biopsy, the brown tumor has the reddish brown appearance of a giant cell tumor. Microscopically, it looks like a giant cell tumor except that the background stromal cells are more fibroblastic and the bone trabeculae demonstrate abnormally thick and poorly mineralized osteoid seams. Because of the marked similarity between the brown tumor and the giant cell tumor, clinicians should routinely order an analysis of serum calcium, phosphorus, and alkaline phosphatase levels in all patients with bone lesions that produce giant cells.

![]() Figure 5–95. Radiograph (A) and photomicrograph (B) of a brown tumor of hyperparathyroidism in the proximal humerus of a 40-year-old woman.
Figure 5–95. Radiograph (A) and photomicrograph (B) of a brown tumor of hyperparathyroidism in the proximal humerus of a 40-year-old woman.
In patients with brown tumors, the treatment consists of removing the source of the excessive parathyroid hormone. After this, the bony defects usually heal spontaneously. Bone grafting is rarely required. Although the secondary hyper-parathyroidism seen in renal failure patients does not usually develop into brown tumors, it does produce pseudotumorous calcification in soft tissue, a condition similar to tumoral calcinosis, which is discussed later in this section.
B. Paget Disease: ICD-9-CM 731.0
Paget disease is frequently included in discussions of metabolic bone disorders, although the demonstration of cytoplasmic and nuclear inclusion bodies in osteoclasts of pagetic bone similar to paramyxovirus infections may suggest a viral origin. Most clinicians are familiar with the late changes in Paget disease, which include the bowing of long bones and the finding of dense blastic changes on radiographic examination. However, many are unfamiliar with the early lytic phase of Paget disease when the radiographic findings are more suggestive of metastatic carcinoma, histiocytic lymphoma, primary sarcoma, or even primary hyperparathyroidism (Figure 5–96).

![]() Figure 5–96. Early and late radiographs of Paget disease of the tibia, taken when the patient was 45 years of age (A) and when he was 65 years of age (B).
Figure 5–96. Early and late radiographs of Paget disease of the tibia, taken when the patient was 45 years of age (A) and when he was 65 years of age (B).
C. Gaucher Disease: ICD-9-CM 272.7
Gaucher disease is a rare familial disorder in which accumulation of glucocerebroside causes enlargement of the liver, spleen, and marrow tissues. The marrow infiltration in children and young adults causes a gradual loss of bone that can mimic a neoplastic condition. The most common areas involved include the distal femur, tibia, humerus, vertebral column, skull, and mandible. Isolated focal destructive changes with endosteal scalloping and moth-eaten patterns may suggest the diagnosis of meta-static disease, myelomatosis, primary sarcoma, or fibrous dysplasia (Figure 5–97).

![]() Figure 5–97. Radiograph of a pathologic fracture secondary to Gaucher disease involving the distal femur in a 29-year-old man.
Figure 5–97. Radiograph of a pathologic fracture secondary to Gaucher disease involving the distal femur in a 29-year-old man.
![]() Hemorrhagic Conditions
Hemorrhagic Conditions
A. Pseudotumor of Hemophilia: ICD-9-CM 733.90
A hematoma in the soft tissue or bone under the periosteum may be difficult to distinguish from a tumor. Hematoma formation is frequently precipitated by some form of trauma, and the bones most commonly involved are the femur, pelvis, tibia, and small bones of the hand. It is rare to see multiple lesions. The bony lesions can be central or eccentric. The finding of lytic destruction followed by sclerotic reaction at the periphery may mimic the radiographic picture of an aneurysmal bone cyst or a giant cell tumor. In the hand bones, the osseous pseudotumors take on the appearance of a giant cell reparative granuloma or an osteoblastoma. The subperiosteal lesions bulge into the surrounding soft tissue and show reactive periosteal new bone formation and subjacent cortical erosion that may mimic Ewing sarcoma or hemorrhagic osteosarcoma (Figure 5–98).

![]() Figure 5–98. Anteroposterior (A) and lateral (B) radiographs of a pseudotumor of hemophilia in the distal femur of a 14-year-old boy.
Figure 5–98. Anteroposterior (A) and lateral (B) radiographs of a pseudotumor of hemophilia in the distal femur of a 14-year-old boy.
B. Intramuscular Hematoma: ICD-9-CM 729.92
Another hemorrhagic disorder that can produce a pseudo-tumor of soft tissue is the intramuscular hematoma. It is similar to the soft-tissue pseudotumor of hemophilia but without a bleeding abnormality. Intramuscular hematomas are almost always related to blunt trauma, but they occasionally result from a traction injury that may subsequently produce myositis ossificans. There may be no superficial signs of bruising in the overlying skin, and sometimes the hematoma grows in size at a later date, even as long as several years after the initial injury. The radiographic examination is of little help because no calcification or bony abnormality is evident. The MRI is the best imaging study, but unfortunately, the appearance of an intramuscular hematoma on MRI can mimic that of a deep soft-tissue sarcoma such as a malignant fibrous histiocytoma (Figure 5–99).

![]() Figure 5–99. Axial view T2-weighted MRI of a hematoma in the quadriceps muscles of a 46-year-old woman.
Figure 5–99. Axial view T2-weighted MRI of a hematoma in the quadriceps muscles of a 46-year-old woman.
![]() Ectopic Calcification
Ectopic Calcification
Ectopic calcification in soft tissue has many causes, most of which are related to chronic degenerative disorders in collagenous structures such as tendons or ligaments about a joint. However, in cases in which the dystrophic calcification is associated with a soft-tissue mass, the clinician must rule out the diagnosis of a soft-tissue sarcoma such as synovial sarcoma.
A. Tumoral Calcinosis: ICD-9-CM 275.49
Tumoral calcinosis, seen about the hip, shoulder, and elbow, is characterized by extensive calcium phosphate deposition in a benign fibrous mass. There is a familial form associated with a loss-of-function mutation in the FGF23gene. A gain-of-function mutation in the same gene results in autosomal dominant hypophosphatemic rickets. Otherwise, it is an idiopathic condition that affects patients between 10 and 30 years of age and occurs more frequently in males than in females. Multiple lesions occur, and the lesions cause minimal pain and tenderness.
In cases of tumoral calcinosis, the extensive central fluffy calcification might suggest the diagnosis of a synovial sarcoma, soft-tissue chondrosarcoma, or tuberculosis (Figure 5–100). At biopsy, a chalky white paste exudes from a spongelike fibrous mass. Microscopic findings include extensive amorphous calcium phosphate deposits in a fibrous stroma speckled with macrophages and inflammatory cells. If the pseudotumor is not completely removed, a recurrence is very likely.

![]() Figure 5–100. Radiograph (A) and T1-weighted MRI (B) of tumoral calcinosis in the hip of a 54-year-old woman.
Figure 5–100. Radiograph (A) and T1-weighted MRI (B) of tumoral calcinosis in the hip of a 54-year-old woman.
A similar condition is seen in patients with renal osteo-dystrophy with secondary hyperparathyroidism, and the mechanism for the deposition in this case is a high level of calcium phosphorus production.
![]() B. Compartment Syndrome: ICD-9-CM 728.12
B. Compartment Syndrome: ICD-9-CM 728.12
The ischemic calcification and even ossification that occur in traumatic compartment syndromes in the lower extremity can often mimic a tumor. The initial injury is usually a crushing type that causes increased compartment pressure from muscle swelling. This pressure eventually leads to ischemic necrosis of the compartment muscle, which several years later becomes calcific or even ossified. Because the muscle appears firm and calcified on radiographic examination, the clinician may not relate the finding to an old injury and may suspect a calcifying sarcoma such as synovial sarcoma. The most common place for this pseudotumor is in one of the muscle compartments of the leg, and it causes stiffness and muscle weakness at the ankle and foot area (Figure 5–101). This process can mimic soft-tissue calcifications secondary to a neoplastic process (Figure 5–102).

![]() Figure 5–101. Radiograph (A) and CT scan (B) of an old compartment syndrome of the anterior compartment of the leg of an 81-year-old woman who had a history of fracture treated with internal fixation 60 years prior.
Figure 5–101. Radiograph (A) and CT scan (B) of an old compartment syndrome of the anterior compartment of the leg of an 81-year-old woman who had a history of fracture treated with internal fixation 60 years prior.

![]() Figure 5–102. Radiograph of calcification in synovial sarcoma of the leg.
Figure 5–102. Radiograph of calcification in synovial sarcoma of the leg.
![]() Dysplastic Disorders
Dysplastic Disorders
Many developmental or dysplastic conditions can create bony abnormalities, which, on radiographic examination, can mimic a bone tumor. These are usually focal defects in enchondral bone formation that result from a failure to remodel primary woven bone forming at the metaphyseal end of the physis.
A. Osteoma: ICD-9-CM 213.x
Osteoma commonly occurs in the skull or maxilla and is composed of dense unorganized woven bone seen just beneath the cortex. There is no lytic component in or around the dense bone, and no symptoms are associated with the presence of osteomas. Because the lesions are commonly seen in the metaphyseal areas about the knee, the clinician may become concerned about the diagnosis of an early osteosarcoma. However, the lack of periosteal response and minimal uptake on an isotope bone scan help rule out sarcoma (Figure 5–103). In such cases, there should be no concern about future problems from the lesion, and usually no intervention is necessary.

![]() Figure 5–103. Radiograph (A) and T2-weighted MRI (B) of a dysplastic process in the distal femur of a 64-year-old woman.
Figure 5–103. Radiograph (A) and T2-weighted MRI (B) of a dysplastic process in the distal femur of a 64-year-old woman.
B. Bone Island: ICD-9-CM 213.x
The bone island, or enostosis, is an even more sharply marginated dysplastic process than the osteoma. It is most commonly located in the pelvis. It can mimic a blastic metastatic lesion in patients with prostate cancer. However, with a bone island, as with an osteoma, the bone scan shows minimal and very focal activity, and the CT scan and MRI show no reaction in the surrounding marrow. Figure 5–104shows the findings of a bone island through the pelvis of a 35-year-old man.

![]() Figure 5–104. CT scan (A) and T2-weighted MRI (B) of a bone island in the ilium of a 35-year-old man.
Figure 5–104. CT scan (A) and T2-weighted MRI (B) of a bone island in the ilium of a 35-year-old man.
![]() Bone Infarcts
Bone Infarcts
The two types of bone infarcts that can mimic bone tumors are the metaphyseal type and the epiphyseal type. They can be idiopathic in origin or secondary to increased alcohol consumption or corticosteroid use.
A. Metaphyseal Bone Infarct: ICD-9-CM 733.4x
The most common bone infarct is in the metaphyseal region, which is typically seen about the knee, hip, and shoulder in adults. Radiographically, the infarct can mimic a low-grade cartilaginous tumor such as an enchondroma. An infarct presents with a sclerotic honeycombed pattern (Figure 5–105), whereas a cartilaginous lesion presents with central flocculated calcification (Figure 5–106).

![]() Figure 5–105. Radiograph (A) and T1-weighted MRI (B) of a metaphyseal infarct in the distal femur of a 52-year-old woman.
Figure 5–105. Radiograph (A) and T1-weighted MRI (B) of a metaphyseal infarct in the distal femur of a 52-year-old woman.

![]() Figure 5–106. Radiograph of a large enchondroma in the distal femur.
Figure 5–106. Radiograph of a large enchondroma in the distal femur.
B. Epiphyseal Bone Infarct: ICD-9-CM 733.4x
Although epiphyseal bone infarcts have the same etiology as those in the metaphysis, these are most commonly found in the femoral condyles and the proximal femoral and humeral epiphyses. In these locations, the lytic change seen in the epiphyseal bone can mimic a chondroblastoma. The differential diagnosis can be difficult before the appearance of a crescent sign or other radiographic signs of subchondral collapse that usually rule out the chondroblastoma (Figure 5–107).

![]() Figure 5–107. Radiograph of an epiphyseal infarct in the femoral condyle of a 45-year-old woman.
Figure 5–107. Radiograph of an epiphyseal infarct in the femoral condyle of a 45-year-old woman.
![]() Histiocytic Disorders
Histiocytic Disorders
A. Langerhans Cell Histiocytosis: ICD-9-CM 277.89
Sometimes inappropriately called histiocytosis X, Langerhans cell histiocytosis can present in a variety of ways. Previously considered distinct diseases, including eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease, they are now considered part of the same spectrum of histiocytosis presentation. Of these, the localized granulomatous form, which is called eosinophilic granuloma or Langerhans cell granulomatosis, is the one that mimics a tumor radiographically. Eosinophilic granuloma is seen twice as often in boys as in girls and commonly occurs between 5 and 15 years of age. It is usually monostotic but in 10% of cases involves two or three separate areas. It is a histiocytic process of unknown cause but may have a viral origin. It causes local inflammatory pain and may result in low-grade fever associated with an elevated sedimentation rate. Although the most common location of eosinophilic granuloma is the skull, it is also seen in the rib, pelvis, maxilla, vertebral body (vertebra plana) (Figure 5–108), clavicle, and scapula, listed in the order of frequency. Besides affecting flat bones, it can arise in the diaphysis of long bones, followed next by the metaphysis, and it is least common in the epiphysis.

![]() Figure 5–108. Lateral radiograph of spine demonstrating characteristic vertebra plana of Langerhans cell histiocytoisis.
Figure 5–108. Lateral radiograph of spine demonstrating characteristic vertebra plana of Langerhans cell histiocytoisis.
Eosinophilic granuloma can be extremely permeative and destructive, especially in long bones (Figure 5–109) and vertebrae (Figure 5–110), thereby mimicking a more aggressive process, such as Ewing sarcoma, metastatic neuroblastoma, or osteomyelitis. It can also produce a so-called onionskin periostitis of the type seen in Ewing sarcoma. The lesion has a more aggressive pattern in younger children and later becomes more focal and granulomatous. Microscopic findings include large pale-staining histiocytes speckled with small bright-staining eosinophils and an occasional giant cell.

![]() Figure 5–109. Radiograph of a lesion of Langerhans cell histiocytosis of the humerus in a 12-year-old boy.
Figure 5–109. Radiograph of a lesion of Langerhans cell histiocytosis of the humerus in a 12-year-old boy.

![]() Figure 5–110. Radiograph of a Langerhans cell histiocytosis in the body of the C3 vertebra in a 5-year-old girl.
Figure 5–110. Radiograph of a Langerhans cell histiocytosis in the body of the C3 vertebra in a 5-year-old girl.
Eosinophilic granulomas tend to involute spontaneously without treatment, and therefore, treatment should be conservative. Simple curettement and corticosteroid injections are beneficial. In difficult areas such as the spine or pelvis, low-dose radiation treatment (10 Gy) can be considered. In more disseminated cases that do not respond to simple treatment, low-dose chemotherapy is appropriate.
B. Pigmented Villonodular Synovitis: ICD-9-CM 716.9
Although this form of synovitis can mimic a histiocytic tumor, it is thought to be a nonneoplastic condition involving histiocytic proliferation. It occurs in the subsynovial tissue about major joints of the lower extremity in patients between 20 and 40 years of age. The knee joint is the most common site of involvement, followed next by the hip, ankle, and foot. Involvement of the upper extremity is rare.
The histopathology of pigmented villonodular synovitis is similar to that of a giant cell tumor of the tendon sheath, which presents with soft-tissue tumors about the ankle and on the fingers of the hand. The usual situation involves spontaneous swelling of one knee secondary to synovial hypertrophy. The swelling can grow gradually to a massive amount and be associated with intermittent hemarthroses. The inflamed synovium can cause juxtaarticular erosion into bone at the point of attachment of the joint capsule, as is seen in any chronic proliferative synovitis, including hemophilia and coccidioidomycosis.
In fewer than 10% of cases, pigmented villonodular synovitis is more localized and presents as a focal soft-tissue mass high in the suprapatellar pouch or in the popliteal space, and no generalized swelling of the knee occurs. In these cases, the mass can mimic a soft-tissue sarcoma such as a synovial sarcoma (Figure 5–111). Cortical erosion with secondary bony changes can also be appreciated frequently (Figure 5–112).

![]() Figure 5–111. T1-weighted MRI of pigmented villonodular synovitis in the popliteal space of a 50-year-old man.
Figure 5–111. T1-weighted MRI of pigmented villonodular synovitis in the popliteal space of a 50-year-old man.

![]() Figure 5–112. Tomogram of pigmented villonodular synovitis in the proximal tibia of a young man.
Figure 5–112. Tomogram of pigmented villonodular synovitis in the proximal tibia of a young man.
![]() Essentials of Diagnosis
Essentials of Diagnosis
• Myositis ossificans exhibits peripheral or centripetal calcification as opposed to calcifying soft-tissue malignancies.
• Infection of bone can take on many appearances, but clues to the diagnosis include violation of anatomic boundaries usually respected by tumors, such as the growth plate and the intervertebral disk.
Garringer HJ, Malekpour M, Esteghamat F, et al: Molecular genetic and biochemical analyses of FGF23 mutations in familial tumoral calcinosis. Am J Physiol Endocrinol Metab 2008;295:E929. [PMID: 18682534]
Gasent Blesa JM, Alberola Candel V, Solano Vercet C, et al: Langerhans cell histiocytosis. Clin Trans Oncol 2008;10:688. [PMID: 19015065]
Mankin HJ, Rosenthal DI, Xavier R: Gaucher disease. New approaches to an ancient disease. J Bone Joint Surg Am 2001; 83-A:748. [PMID: 11379747]
Roodman GD: Studies in Paget’s disease and their relevance to oncology. Semin Oncol 2001;28(4 Suppl 11):15. [PMID: 11544571]
Seton M: Paget’s disease: epidemiology and pathophysiology. Curr Osteoporos Rep 2008;6:125. [PMID: 19032921]