INBORN ERRORS OF METABOLISM
DEFINITION
Inherited biochemical disorders.
PATHOPHYSIOLOGY
Mutations affecting proteins involved in the many metabolic pathways of the body. Typically result in deficiency of enzyme production or build-up of toxic metabolites, or both.
EPIDEMIOLOGY
Disorders involving deficiencies of enzymes are often autosomal recessive (please see noted exceptions in this chapter).
SIGNS AND SYMPTOMS
![]() Often normal at birth, but can show signs early, including metabolic acidosis, poor feeding, vomiting, lethargy, and convulsion.
Often normal at birth, but can show signs early, including metabolic acidosis, poor feeding, vomiting, lethargy, and convulsion.
![]() Mental retardation, organomegaly, unusual body odor, episodic decompensation.
Mental retardation, organomegaly, unusual body odor, episodic decompensation.
DIAGNOSIS
![]() Newborn metabolic screening:
Newborn metabolic screening:
![]() Standard in United States: Allows for early detection and treatment and can potentially prevent serious consequences.
Standard in United States: Allows for early detection and treatment and can potentially prevent serious consequences.
![]() Panel of test varies state by state but phenylketonuria (PKU), hypothyroidism, galactosemia, and hemoglobinopathies are nearly universal.
Panel of test varies state by state but phenylketonuria (PKU), hypothyroidism, galactosemia, and hemoglobinopathies are nearly universal.
![]() Tandem mass spectrometry is the usual method used for screening.
Tandem mass spectrometry is the usual method used for screening.
![]() The following conditions are screened in most states:
The following conditions are screened in most states:
![]() Galactosemia.
Galactosemia.
![]() Hypothyroidism.
Hypothyroidism.
![]() Hemoglobinopathy.
Hemoglobinopathy.
![]() Tyrosinemia.
Tyrosinemia.
![]() Biotinidase deficiency.
Biotinidase deficiency.
![]() Congenital adrenal deficiency.
Congenital adrenal deficiency.
![]() Maple syrup urine disease.
Maple syrup urine disease.
![]() Homocystinuria.
Homocystinuria.
![]() Cystic fibrosis.
Cystic fibrosis.
![]() Medium-chain acyl-CoA dehydrogenase deficiency (MCAD).
Medium-chain acyl-CoA dehydrogenase deficiency (MCAD).
![]() Urea cycle defects.
Urea cycle defects.
![]() HIV is screened in some states.
HIV is screened in some states.
![]() Classification:
Classification:
![]() Amino acid and urea cycle disorders:
Amino acid and urea cycle disorders:
![]() Homocystinuria.
Homocystinuria.
![]() Maple syrup urine disease.
Maple syrup urine disease.
![]() Phenylketonuria.
Phenylketonuria.
![]() Tyrosinemia.
Tyrosinemia.
![]() Urea cycle (arginase deficiency, argininosuccinic academia, citrullinemia, ornithine transport defect).
Urea cycle (arginase deficiency, argininosuccinic academia, citrullinemia, ornithine transport defect).
![]() Fatty acid oxidation disorders:
Fatty acid oxidation disorders:
![]() Carnitine transport defect.
Carnitine transport defect.
![]() Citrullinemia.
Citrullinemia.
![]() Glutaric acidemia type 2.
Glutaric acidemia type 2.
![]() Medium-chain acyl-CoA dehydrogenase deficiency.
Medium-chain acyl-CoA dehydrogenase deficiency.
Medium-chain acyl-CoA dehydrogenase deficiency is the most common fatty acid oxidation disorder and may be associated with intermittent severe metabolic crises or sudden death.
TABLE 8-1. Newborn Screening of Amino Acid Disorders

![]() Organic acid disorders:
Organic acid disorders:
![]() 3-hydroxy-3-methyglutaryl-CoA lyase deficiency
3-hydroxy-3-methyglutaryl-CoA lyase deficiency
![]() Glutaric acidemia type I
Glutaric acidemia type I
![]() Isovaleric acidemia
Isovaleric acidemia
![]() Methylmalonic acidemia
Methylmalonic acidemia
![]() Propionic acidemia
Propionic acidemia
![]() Many can be detected in the neonatal period or infancy, and some are included in newborn screening (see Table 8-1).
Many can be detected in the neonatal period or infancy, and some are included in newborn screening (see Table 8-1).
Most fatty acid oxidation disorders present with hypoglycemia.
TREATMENT
![]() Treatment varies but is often supportive/symptomatic.
Treatment varies but is often supportive/symptomatic.
![]() Frequently includes dietary modifications.
Frequently includes dietary modifications.
![]() Increasing availability of enzyme or gene-related treatment options.
Increasing availability of enzyme or gene-related treatment options.
DEFECTS OF AMINO ACID METABOLISM
See Table 8-2.
TABLE 8-2. Disorders of Amino Acid Metabolism

Phenylketonuria (PKU)
DEFINITION
Inherited disorder of amino acid metabolism in which phenylalanine cannot be converted to tyrosine.
ETIOLOGY
Deficiency of phenylalanine hydroxylase (or its cofactor tetrahydrobiopterin—2% of cases).

Phenylketones: phenylacetate, -lactate, and -pyruvate, in urine.
PATHOPHYSIOLOGY
![]() Accumulation of phenylalanine and its phenylketone metabolites disrupt normal metabolism and cause brain damage.
Accumulation of phenylalanine and its phenylketone metabolites disrupt normal metabolism and cause brain damage.
![]() Tyrosine becomes essential amino acid.
Tyrosine becomes essential amino acid.
EPIDEMIOLOGY
![]() Autosomal recessive.
Autosomal recessive.
![]() One in 10,000–20,000 live births.
One in 10,000–20,000 live births.
![]() Routinely screened for in the United States.
Routinely screened for in the United States.
Aspartame contains phenylalanine.
SIGNS AND SYMPTOMS
![]() Normal at birth.
Normal at birth.
![]() Severe mental retardation with IQ of 30 may develop at the end of 1 year (progressive and irreversible).
Severe mental retardation with IQ of 30 may develop at the end of 1 year (progressive and irreversible).
![]() Hypopigmentation due to low tyrosine (fair hair and skin, blue eyes).
Hypopigmentation due to low tyrosine (fair hair and skin, blue eyes).
![]() Eczema, mousy/musty body odor, hypertonia.
Eczema, mousy/musty body odor, hypertonia.
DIAGNOSIS
![]() Screened in all newborns.
Screened in all newborns.
![]() Serum tested 72 hours after initiation of first protein feed (test may be negative prior to 72 hours).
Serum tested 72 hours after initiation of first protein feed (test may be negative prior to 72 hours).
![]() If not screened neonatally, diagnosis usually made at 4–6 months of age.
If not screened neonatally, diagnosis usually made at 4–6 months of age.
![]() Prenatal and carrier testing possible.
Prenatal and carrier testing possible.
↓ pigmentation in PKU is secondary to the inhibition of tyrosinase by phenylalanine.
TREATMENT
![]() Limit dietary phenylalanine (eg, in artificial sweeteners) and ↑ tyrosine; if started within first 10 days of life, infants can have normal intelligence.
Limit dietary phenylalanine (eg, in artificial sweeteners) and ↑ tyrosine; if started within first 10 days of life, infants can have normal intelligence.
![]() Strict dietary restriction during pregnancy.
Strict dietary restriction during pregnancy.
Homocystinemia/Homocystinuria
DEFINITION
Inherited disorder of amino acid metabolism in which homocysteine is present in greater than trace amounts in the urine.
ETIOLOGY
Most commonly a deficiency of cystathionine β-synthase, but can also be a defect of methylcobalamin formation or deficiency of methyltetrahydrofolate reductase.
Lethargy, anorexia, anemia, rashes, and diarrhea are signs of tyrosine deficiency.

FIGURE 8-1. Homocysteine pathway.
PATHOPHYSIOLOGY
Homocysteine is not remethylated to methionine (see Figure 8-1).
Ectopia lentis is subluxation of the lens, signaled by iridodonesis (quivering of iris) and myopia.
EPIDEMIOLOGY
Autosomal recessive (1 in 200,000 live births).
SIGNS AND SYMPTOMS
![]() Depends on particular enzyme deficiency.
Depends on particular enzyme deficiency.
![]() Most commonly normal at birth, with failure to thrive and developmental delay subsequently occurring.
Most commonly normal at birth, with failure to thrive and developmental delay subsequently occurring.
![]() Shares several skeletal and ocular features with Marfan syndrome.
Shares several skeletal and ocular features with Marfan syndrome.
![]() Later, ectopia lentis, marfanoid body habitus, progressive mental retardation, vaso-occlusive disease, osteoporosis, or fair skin with malar flush can occur.
Later, ectopia lentis, marfanoid body habitus, progressive mental retardation, vaso-occlusive disease, osteoporosis, or fair skin with malar flush can occur.
Late complications:
![]() Astigmatism
Astigmatism
![]() Optic atrophy
Optic atrophy
![]() Glaucoma
Glaucoma
![]() Cataracts
Cataracts
![]() Retinal detachment
Retinal detachment

FIGURE 8-2. Methionine metabolism.
DIAGNOSIS
![]() Normal at birth; diagnosis usually made after 3 years of age.
Normal at birth; diagnosis usually made after 3 years of age.
![]() Elevated methionine and homocysteine in body fluids.
Elevated methionine and homocysteine in body fluids.
![]() Prenatal diagnosis possible.
Prenatal diagnosis possible.
![]() Marfan phenotype–differential diagnosis:
Marfan phenotype–differential diagnosis:
![]() Homocystinuria: Marfanoid habitus, ectopia lentis, mental retardation, osteoporosis.
Homocystinuria: Marfanoid habitus, ectopia lentis, mental retardation, osteoporosis.
![]() Ehlers-Danlos syndrome types 1 and 3: Marked joint hypermobility, mitral valve prolapse.
Ehlers-Danlos syndrome types 1 and 3: Marked joint hypermobility, mitral valve prolapse.
![]() Stickler syndrome (hereditary arthro-ophthalmopathy): Tall stature, retrognathia, mitral valve prolapse, midfacial hypoplasia, retinal detachment.
Stickler syndrome (hereditary arthro-ophthalmopathy): Tall stature, retrognathia, mitral valve prolapse, midfacial hypoplasia, retinal detachment.
![]() Klinefelter syndrome: Marfanoid habitus, small testes and genitalia, learning difficulty.
Klinefelter syndrome: Marfanoid habitus, small testes and genitalia, learning difficulty.
Branched-chain amino acids are: leucine, isoleucine, valine.
TREATMENT
![]() Pyridoxine responsive form: 50% are this form and easily missed in the neonatal period. High-dose vitamin B6.
Pyridoxine responsive form: 50% are this form and easily missed in the neonatal period. High-dose vitamin B6.
![]() Pyridoxine unresponsive form: Restriction of methionine intake and supplementation of cysteine. (May require concurrent folic acid to show response.) Betaine can also play a role in this group.
Pyridoxine unresponsive form: Restriction of methionine intake and supplementation of cysteine. (May require concurrent folic acid to show response.) Betaine can also play a role in this group.
![]() Other types may require vitamin B12 or methionine supplementation.
Other types may require vitamin B12 or methionine supplementation.
Maple Syrup Urine Disease (MSUD) or Branched-Chain Ketoaciduria
DEFINITION
Inherited disorder of branched-chain amino acid metabolism in which elevated quantities of leucine, isoleucine, valine, and corresponding oxoacids accumulate in the body fluids.
In MSUD, plasma leucine levels are usually higher than those of the other accumulating branched amino acids.
ETIOLOGY
Deficiency of branched-chain ketoacid dehydrogenase.
PATHOPHYSIOLOGY
Defect in the decarboxylation of leucine, isoleucine, and valine by a branched-chain ketoacid dehydrogenase.
Correcting the serum glucose level in MSUD does not improve the clinical state.
EPIDEMIOLOGY
One in 250,000 live births in general population.
SIGNS AND SYMPTOMS
![]() Deficiency of different subunits of enzyme account for wide clinical variability.
Deficiency of different subunits of enzyme account for wide clinical variability.
![]() Poor feeding, vomiting in first week of life, proceeding to lethargy and coma.
Poor feeding, vomiting in first week of life, proceeding to lethargy and coma.
![]() Alternating hypertonicity and flaccidity, convulsions, hypoglycemia.
Alternating hypertonicity and flaccidity, convulsions, hypoglycemia.
![]() Odor of maple syrup in urine, sweat, cerumen (burnt sugar smell).
Odor of maple syrup in urine, sweat, cerumen (burnt sugar smell).
Suspect MSUD:
![]() Intermittent symptoms (feeding difficulties and apnea) related to protein ingestion
Intermittent symptoms (feeding difficulties and apnea) related to protein ingestion
![]() Sweet-smelling cerumen
Sweet-smelling cerumen
DIAGNOSIS
![]() Elevated plasma and urine levels if leucine, isoleucine, valine, and alloisoleucine; ↓ plasma alanine.
Elevated plasma and urine levels if leucine, isoleucine, valine, and alloisoleucine; ↓ plasma alanine.
![]() Urine precipitant test.
Urine precipitant test.
![]() Neuroimaging in the acute state shows cerebral edema.
Neuroimaging in the acute state shows cerebral edema.
TREATMENT
![]() Chronically, low branched-chain amino acid diet.
Chronically, low branched-chain amino acid diet.
![]() Frequent serum level monitoring.
Frequent serum level monitoring.
![]() Acutely, intravenous administration of amino acids other than branched chain.
Acutely, intravenous administration of amino acids other than branched chain.
![]() Hemodialysis or peritoneal dialysis can save the patient’s life in acidotic crisis, but liver transplantation can definitely treat MSUD.
Hemodialysis or peritoneal dialysis can save the patient’s life in acidotic crisis, but liver transplantation can definitely treat MSUD.
Hartnup Disease
DEFINITION
Inherited defect in transport of neutral amino acids by intestinal mucosa and renal tubules.
ETIOLOGY
Deficient activity of a sodium-dependent transport system.
Urinary proline, hydroxyproline, and arginine remain normal in Hartnup disease (unlike in other causes of generalized aminoaciduria, such as Fanconi’s).
PATHOPHYSIOLOGY
Deficiency of tryptophan results in the clinical manifestations.
EPIDEMIOLOGY
Autosomal recessive.
SIGNS AND SYMPTOMS
![]() Usually asymptomatic.
Usually asymptomatic.
![]() Rarely, cutaneous photosensitivity, episodic psychiatric changes.
Rarely, cutaneous photosensitivity, episodic psychiatric changes.
![]() Marginal nutrition results in clinical manifestations in predisposed individuals.
Marginal nutrition results in clinical manifestations in predisposed individuals.
DIAGNOSIS
![]() Aminoaciduria (neutral: alanine, serine, threonine, valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, histidine).
Aminoaciduria (neutral: alanine, serine, threonine, valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, histidine).
![]() Normal plasma amino acid levels.
Normal plasma amino acid levels.
TREATMENT
Nicotinic acid/nicotinamide and a high-protein diet in symptomatic patients.
DEFECTS OF LIPID METABOLISM–LYSOSOMAL STORAGE DISEASES
Lipidoses
See Table 8-3.
Gaucher is the most common lysosomal storage disease (1 in 75,000). Splenomegaly is the most common presenting sign.
TABLE 8-3. Lysosomal Storage Diseases–Lipidoses


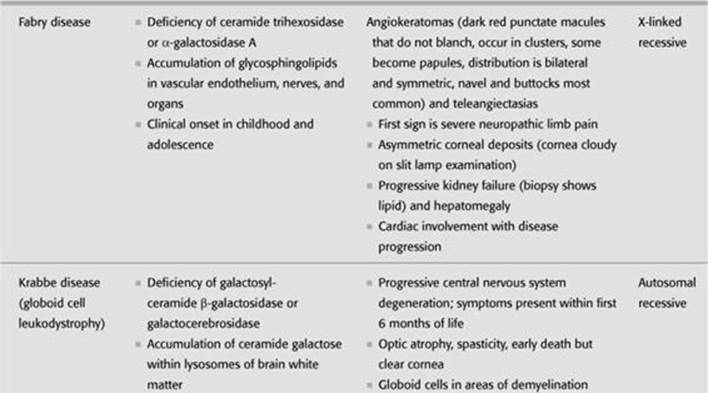

DEFINITION/ETIOLOGY/PATHOPHYSIOLOGY
Inherited deficiencies of lysosomal hydrolases cause lysosomal accumulation of sphingolipids in brain and viscera.
Fabry is X-linked recessive.
EPIDEMIOLOGY
Most are autosomal recessive.
SIGNS AND SYMPTOMS
Depends on site of abnormal accumulations:
![]() Nervous system: Neurodegeneration, ocular findings.
Nervous system: Neurodegeneration, ocular findings.
![]() Viscera: Organomegaly, skeletal abnormalities, pulmonary infiltration.
Viscera: Organomegaly, skeletal abnormalities, pulmonary infiltration.
DIAGNOSIS
Measurement of specific enzymatic activity in leukocytes or cultured fibroblasts.
TREATMENT
![]() Usually no specific treatment.
Usually no specific treatment.
![]() Supportive/symptomatic therapy.
Supportive/symptomatic therapy.
![]() Gaucher disease: Recombinant enzyme.
Gaucher disease: Recombinant enzyme.
![]() Krabbe disease: Hematopoietic stem cell transplant.
Krabbe disease: Hematopoietic stem cell transplant.
Gangliosidoses (eg, GM1 and Tay-Sachs) have cherry red spot on macula in 50% of cases, as does Niemann-Pick.
Mucopolysaccharidoses
DEFINITION/ETIOLOGY/PATHOPHYSIOLOGY
Inherited deficiencies of lysosomal enzymes needed for the degradation of glycosaminoglycans (GAGs) resulting in widespread lysosomal storage of dermatan and heparan sulfates and severe clinical abnormalities. Keratan sulfates accumulate in other mucopolysaccharidoses not mentioned. See Table 8-4.
Hepatosplenomegaly occurs in the GM1 gangliosidoses and Sandhoff disease, but not in Tay-Sachs disease.
EPIDEMIOLOGY
Most are autosomal recessive.
SIGNS AND SYMPTOMS
![]() Normal at birth, diagnosis at 1+ years.
Normal at birth, diagnosis at 1+ years.
![]() “Gargoyle” cells containing lysosomes engorged with mucopolysaccharide.
“Gargoyle” cells containing lysosomes engorged with mucopolysaccharide.
Hunter syndrome is X-linked recessive.
TABLE 8-4. Lysosomal Storage Diseases–Mucopolysaccharidoses

MPS, mucopolysaccharidosis.
![]() Excessive urinary excretion of GAGs.
Excessive urinary excretion of GAGs.
![]() Progressive mental and physical deterioration.
Progressive mental and physical deterioration.
![]() Coarse features.
Coarse features.
![]() Corneal clouding.
Corneal clouding.
![]() Stiff joints (abnormal hyalinization of collagen).
Stiff joints (abnormal hyalinization of collagen).
![]() Organomegaly.
Organomegaly.
![]() Skeletal abnormalities.
Skeletal abnormalities.
DIAGNOSIS
![]() Detection of enzyme deficiency in leukocytes or cultured fibroblasts.
Detection of enzyme deficiency in leukocytes or cultured fibroblasts.
![]() Roentgenographic changes consistent with dystosis multiplex.
Roentgenographic changes consistent with dystosis multiplex.
![]() Urinary excretion of dermatan and heparan sulfates.
Urinary excretion of dermatan and heparan sulfates.
TREATMENT
Supportive therapy. Hurler’s can be treated with bone marrow transplant.
Dysostosis Multiplex
![]() Large dolichocephalic skull
Large dolichocephalic skull
![]() Thickened calvarium
Thickened calvarium
![]() Ovoid vertebral bodies
Ovoid vertebral bodies
![]() Flared iliac bones
Flared iliac bones
![]() Shallow acetabulae
Shallow acetabulae
![]() Irregular widening of long bones
Irregular widening of long bones
DEFECTS OF CARBOHYDRATE METABOLISM–GLYCOGEN STORAGE DISEASES
See Table 8-5.
von Gierke Disease
![]()
A 3-month-old, breast-fed infant has failure to thrive, severe hepatomegaly, thin extremities, fasting hypoglycemia, lipemia, and metabolic acidosis. Think: von Gierke disease.
von Gierke is an inherited disorder that affect glycogen metabolism. It is due to the deficiency of glucose-6-phosphatase, which results in accumulation of glucose-6-phosphate, which in turns causes ↑ glycolysis and lactic acidosis. It is characterized by growth retardation, hypoglycemia, hepatomegaly, hyperlipidemia, hyperuricemia, lactic acidemia, and seizure. In neonatal period, hypoglycemia and lactic acidosis are the common presentation. Hepatomegaly becomes evident by 3–4 months of age.
TABLE 8-5. Glycogen Storage Diseases

DEFINITION
Inherited disorder of glycogen metabolism characterized by deposition of glycogen in the liver, kidney, and intestine.
ETIOLOGY
Deficiency of glucose-6-phosphatase.
PATHOPHYSIOLOGY
Glycogen-to-glucose metabolism stops at glucose-6-phosphate.
EPIDEMIOLOGY
Autosomal recessive.
SIGNS AND SYMPTOMS
![]() Fasting hypoglycemia (due to impaired gluconeogenesis, glycogenolysis, and recycling of glucose through the glucose-6-phosphate to glucose system).
Fasting hypoglycemia (due to impaired gluconeogenesis, glycogenolysis, and recycling of glucose through the glucose-6-phosphate to glucose system).
![]() Massive hepatomegaly.
Massive hepatomegaly.
![]() Elevated serum levels of lactate, uric acid, cholesterol, triglycerides.
Elevated serum levels of lactate, uric acid, cholesterol, triglycerides.
![]() Renal complications (Fanconi’s, nephrocalcinosis, focal segmental glomerulosclerosis).
Renal complications (Fanconi’s, nephrocalcinosis, focal segmental glomerulosclerosis).
![]() Slow growth, diarrhea, bleeding disorders, hypotonia, and gout.
Slow growth, diarrhea, bleeding disorders, hypotonia, and gout.
DIAGNOSIS
![]() Normal at birth; diagnosis usually at 5 months.
Normal at birth; diagnosis usually at 5 months.
![]() Administration of epinephrine, glucagons, galactose, fructose, or glycerol does not provoke normal hyperglycemic response (may precipitate acidosis).
Administration of epinephrine, glucagons, galactose, fructose, or glycerol does not provoke normal hyperglycemic response (may precipitate acidosis).
![]() DNA tests form common mutations.
DNA tests form common mutations.
![]() In cases where mutation testing is not easily done, enzyme measurements can confirm the diagnosis.
In cases where mutation testing is not easily done, enzyme measurements can confirm the diagnosis.
![]() Liver biopsy demonstrates accumulation of glycogen in cells.
Liver biopsy demonstrates accumulation of glycogen in cells.
TREATMENT
![]() Avoid fasting.
Avoid fasting.
![]() Supportive therapy aimed at maintaining normal glucose levels.
Supportive therapy aimed at maintaining normal glucose levels.
![]() Nocturnal intragastric, frequent, high-carbohydrate meals, are the main stay of treatment up to 1–2 years age.
Nocturnal intragastric, frequent, high-carbohydrate meals, are the main stay of treatment up to 1–2 years age.
![]() After 2 years of age, snacks or nocturnal intragastric feedings of uncooked cornstarch may be sufficient.
After 2 years of age, snacks or nocturnal intragastric feedings of uncooked cornstarch may be sufficient.
![]() High-protein diet is not effective.
High-protein diet is not effective.
![]() Granulocyte colony–stimulating factors to combat neutropenia and inflammation.
Granulocyte colony–stimulating factors to combat neutropenia and inflammation.
![]() Allopurinol to lower urate levels, bicarbonate or potassium citrate for lactic acidosis.
Allopurinol to lower urate levels, bicarbonate or potassium citrate for lactic acidosis.
![]() Liver transplant for refractory disease.
Liver transplant for refractory disease.
McArdle Disease
DEFINITION
Inherited disorder of glycogen metabolism characterized by deposition of glycogen in skeletal muscle.
McArdle affects the
Muscles.
ETIOLOGY
Deficiency of muscle glycogen phosphorylase (myophosphorylase).
EPIDEMIOLOGY
Autosomal recessive.
SIGNS AND SYMPTOMS
![]() Involves only skeletal muscles (accumulations of glycogen predominant in subsarcolemmal location).
Involves only skeletal muscles (accumulations of glycogen predominant in subsarcolemmal location).
![]() Temporary weakness and cramping of skeletal muscles during or after exercise.
Temporary weakness and cramping of skeletal muscles during or after exercise.
![]() No rise in blood lactate during exercise.
No rise in blood lactate during exercise.
![]() Characteristic “second wind” with initiation of fatty acid metabolism.
Characteristic “second wind” with initiation of fatty acid metabolism.
DIAGNOSIS
![]() Asymptomatic during infancy. Presents in adolescence/early childhood.
Asymptomatic during infancy. Presents in adolescence/early childhood.
![]() Muscle biopsy and assay show deficiency of enzyme.
Muscle biopsy and assay show deficiency of enzyme.
![]() Myoglobinuria, serum creatine kinase always elevated (elevated CK at rest).
Myoglobinuria, serum creatine kinase always elevated (elevated CK at rest).
TREATMENT
![]() Dietary modification (high fat and protein); sucrose prior to aerobic exercise; proper “warm-up” period.
Dietary modification (high fat and protein); sucrose prior to aerobic exercise; proper “warm-up” period.
![]() Prognosis is good with sedentary lifestyle.
Prognosis is good with sedentary lifestyle.
Pompe Disease
DEFINITION
Inherited disorder of glycogen metabolism characterized by deposition of glycogen in cardiac and skeletal muscle.
Pompe affects the “Pump.”
ETIOLOGY
Deficiency of acid α-1, 4-glucosidase (acid maltase).
PATHOPHYSIOLOGY
![]() Generalized glycogenesis because the defect is in all cells.
Generalized glycogenesis because the defect is in all cells.
![]() Results in inability to convert mannose to glucose.
Results in inability to convert mannose to glucose.
EPIDEMIOLOGY
Autosomal recessive.
SIGNS AND SYMPTOMS
![]() Rapid, progressive cardiomyopathy with massive cardiomegaly, macroglossia, hypotonia, hepatomegaly; death by 1–2 years.
Rapid, progressive cardiomyopathy with massive cardiomegaly, macroglossia, hypotonia, hepatomegaly; death by 1–2 years.
![]() Juvenile form milder, slowly progressive myopathy, little to no cardiac abnormality. Death usually secondary to respiratory failure.
Juvenile form milder, slowly progressive myopathy, little to no cardiac abnormality. Death usually secondary to respiratory failure.
Lactose = galactose + glucose.
DIAGNOSIS
![]() Electrocardiogram (ECG): May show shortened PR interval.
Electrocardiogram (ECG): May show shortened PR interval.
![]() Electromyogram (EMG).
Electromyogram (EMG).
TREATMENT
Enzyme replacement with recombinant α-glucosidase delays disease progression.
Galactosemia
![]()
A 2-week-old neonate has jaundice, hepatomegaly, and positive urinary-reducing substance. Odor of urine is normal. Think: Galactosemia.
Since galactosemia is included in the newborn screening, it is diagnosed before the symptoms develop. Jaundice, hepatomegaly, vomiting, lethargy, and feeding difficulties are the common initial presentation. Presence of a reducing substance in urine in infants with galactosemia who are ingesting lactose establishes the diagnosis.
DEFINITION
Inborn errors of carbohydrate metabolism that result in elevated galactose and metabolite levels in blood and urine.
ETIOLOGY
Three types:
![]() Classic: Absence of galactose-1-phosphate uridyltransferase (inability to process lactose/galactose).
Classic: Absence of galactose-1-phosphate uridyltransferase (inability to process lactose/galactose).
![]() Others: Galactokinase, uridine diphosphate galactose-4-epimerase.
Others: Galactokinase, uridine diphosphate galactose-4-epimerase.
When diagnosis of galactosemia is not made at birth, damage to the liver and brain become irreversible.
PATHOPHYSIOLOGY
![]() Ingestion of galactose → ↑ concentrations in the blood and urine.
Ingestion of galactose → ↑ concentrations in the blood and urine.
![]() Toxic substances, including galactitol, cause organ damage.
Toxic substances, including galactitol, cause organ damage.
EPIDEMIOLOGY
![]() Autosomal recessive.
Autosomal recessive.
![]() One in 60,000.
One in 60,000.
Neonates with galactosemia are at ↑ risk for Escherichia coli sepsis.
SIGNS AND SYMPTOMS
![]() Cataracts, hepatosplenomegaly, mental retardation, sepsis (E coli).
Cataracts, hepatosplenomegaly, mental retardation, sepsis (E coli).
![]() Triad: Liver failure (jaundice and coagulation disorder), renal tubular dysfunction (glucosuria, aminoaciduria, and acidosis), and cataract.
Triad: Liver failure (jaundice and coagulation disorder), renal tubular dysfunction (glucosuria, aminoaciduria, and acidosis), and cataract.
DIAGNOSIS
![]() Should be considered in newborn, infant, or child if jaundice, hepatomegaly, vomiting, hypoglycemia, convulsions, lethargy, irritability, feeding difficulties, poor weight gain, diarrhea, aminoaciduria, cataracts, vitreous hemorrhage, hepatic cirrhosis, ascites, splenomegaly, or mental retardation are noted.
Should be considered in newborn, infant, or child if jaundice, hepatomegaly, vomiting, hypoglycemia, convulsions, lethargy, irritability, feeding difficulties, poor weight gain, diarrhea, aminoaciduria, cataracts, vitreous hemorrhage, hepatic cirrhosis, ascites, splenomegaly, or mental retardation are noted.
![]() Presence of reducing substance in urine after ingestion of human or cow’s milk is suggestive.
Presence of reducing substance in urine after ingestion of human or cow’s milk is suggestive.
![]() Routinely screened for in the United States (Table 8-6).
Routinely screened for in the United States (Table 8-6).
Elimination of galactose from diet in galactosemia does not ensure reversal of cataract formation.
TABLE 8-6. Screening for Galactosemia and Urea Cycle Defect

TREATMENT
![]() Exclude galactose and lactose from diet (example: dairy and breast milk).
Exclude galactose and lactose from diet (example: dairy and breast milk).
![]() Soy-based formula.
Soy-based formula.
There is almost no renal threshold for fructose.
Fructosuria
DEFINITION
Inborn errors of carbohydrate metabolism that result in elevated fructose and metabolite levels in blood and urine.
ETIOLOGY
Deficiency of fructokinase.
PATHOPHYSIOLOGY
![]() Enzyme is normally found in the liver, kidney, and intestine.
Enzyme is normally found in the liver, kidney, and intestine.
![]() Ingested fructose is not metabolized.
Ingested fructose is not metabolized.
EPIDEMIOLOGY
Autosomal recessive.
SIGNS AND SYMPTOMS
![]() Asymptomatic until fructose introduced into diet
Asymptomatic until fructose introduced into diet
![]() Fructosemia and fructosuria.
Fructosemia and fructosuria.
Do not confuse fructosuria with heriditary fructose intolerance (aldolase B deficiency), which presents with failure to thrive, hypoglycemia, lactic acidosis, vomiting, and seizures. Typically discovered during infancy at time of weaning with the introduction of fructose or sucrose into diet. Also autosomal recessive.
DIAGNOSIS
Presence of urinary-reducing substrate without clinical symptoms.
TREATMENT
None indicated.
DEFECTS IN PURINE METABOLISM
Lesch-Nyhan Syndrome
DEFINITION
![]() An X-linked-recessive disorder of purine metabolism resulting in deposition of purines in tissues and subsequent clinical abnormalities.
An X-linked-recessive disorder of purine metabolism resulting in deposition of purines in tissues and subsequent clinical abnormalities.
![]() Up to 1% of patients with gout may have Lesch-Nyhan syndrome.
Up to 1% of patients with gout may have Lesch-Nyhan syndrome.
ETIOLOGY
Deficiency of hypoxanthine–guanine phosphoribosyl transferase (HGPRT).
SIGNS AND SYMPTOMS
![]() Delayed motor development.
Delayed motor development.
![]() Extrapyramidal sign resulting in choreoathetosis at approximately 1 year of age.
Extrapyramidal sign resulting in choreoathetosis at approximately 1 year of age.
![]() Spastic cerebral palsy, self-injurious behavior.
Spastic cerebral palsy, self-injurious behavior.
![]() Hyperuricemia, uricosuria, urinary tract calculi, nephropathy, tophi, gouty arthritis.
Hyperuricemia, uricosuria, urinary tract calculi, nephropathy, tophi, gouty arthritis.
Self-injurious behavior in Lesch-Nyhan syndrome can include banging head against wall and biting/mutilating one’s fingers.
DIAGNOSIS
![]() Normal at birth; diagnosis usually made at 3 months when delayed motor development becomes apparent.
Normal at birth; diagnosis usually made at 3 months when delayed motor development becomes apparent.
![]() Uric acid crystalluria may first be noted as orange crystals in the diaper during the first weeks of life.
Uric acid crystalluria may first be noted as orange crystals in the diaper during the first weeks of life.
![]() Serum uric acid levels.
Serum uric acid levels.
![]() Gout generally does not develop until puberty.
Gout generally does not develop until puberty.
TREATMENT
![]() No specific treatment; supportive therapy.
No specific treatment; supportive therapy.
![]() Allopurinol to reduce serum uric acid levels.
Allopurinol to reduce serum uric acid levels.
![]() Prevention of self-injury.
Prevention of self-injury.
![]() Death (due to infection or renal failure) in the second or third decade.
Death (due to infection or renal failure) in the second or third decade.
Think Lesch-Nyhan syndrome in the presence of self-mutilation and characteristic choreoathetosis; mental retardation.
FAMILIAL HYPERCHOLESTEROLEMIAS
See Table 8-7.
CONCEPTS IN HEREDITARY DISEASE
![]() Complex heterozygosity: Different mutations in each gene allele–each individually “silent,” but when combined produce clinical or biochemical manifestations.
Complex heterozygosity: Different mutations in each gene allele–each individually “silent,” but when combined produce clinical or biochemical manifestations.
![]() Consanguinity (children of first-degree relatives): ↑ risk for inherited disorders, as many are autosomal recessive.
Consanguinity (children of first-degree relatives): ↑ risk for inherited disorders, as many are autosomal recessive.
![]() Sudden infant death syndrome (SIDS) or apparent life-threatening event (ALTE) may be the initial presentation of an inborn error of metabolism.
Sudden infant death syndrome (SIDS) or apparent life-threatening event (ALTE) may be the initial presentation of an inborn error of metabolism.
![]() Unexplained developmental delay may indicate an underlying metabolic disease.
Unexplained developmental delay may indicate an underlying metabolic disease.
TABLE 8-7. Familial Hyperlipidemias


