INTRODUCTION
Assignment of a drug to the sedative-hypnotic class indicates that it is able to cause sedation (with concomitant relief of anxiety) or to encourage sleep. Because there is considerable chemical variation within the group, this drug classification is based on clinical uses rather than on similarities in chemical structure. Anxiety states and sleep disorders are common problems, and sedative-hypnotics are widely prescribed drugs worldwide.
I. BASIC PHARMACOLOGY OF SEDATIVE-HYPNOTICS
INTRODUCTION
An effective sedative (anxiolytic) agent should reduce anxiety and exert a calming effect. The degree of central nervous system depression caused by a sedative should be the minimum consistent with therapeutic efficacy. A hypnotic drug should produce drowsiness and encourage the onset and maintenance of a state of sleep. Hypnotic effects involve more pronounced depression of the central nervous system than sedation, and this can be achieved with many drugs in this class simply by increasing the dose. Graded dose-dependent depression of central nervous system function is a characteristic of most sedative-hypnotics. However, individual drugs differ in the relationship between the dose and the degree of central nervous system depression. Two examples of such dose-response relationships are shown in Figure 22-1. The linear slope for drug A is typical of many of the older sedative-hypnotics, including the barbiturates and alcohols. With such drugs, an increase in dose higher than that needed for hypnosis may lead to a state of general anesthesia. At still higher doses, these sedative-hypnotics may depress respiratory and vasomotor centers in the medulla, leading to coma and death. Deviations from a linear dose-response relationship, as shown for drug B, require proportionately greater dosage increments to achieve central nervous system depression more profound than hypnosis. This appears to be the case for benzodiazepines and for certain newer hypnotics that have a similar mechanism of action.
|
|
Figure 22-1. Dose-response curves for two hypothetical sedative-hypnotics. |
CHEMICAL CLASSIFICATION
Introduction
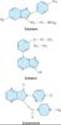
The benzodiazepines are widely used sedative-hypnotics. All of the structures shown in Figure 22-2 are 1,4-benzodiazepines, and most contain a carboxamide group in the 7-membered heterocyclic ring structure. A substituent in the 7 position, such as a halogen or a nitro group, is required for sedative-hypnotic activity. The structures of triazolam and alprazolam include the addition of a triazole ring at the 1,2-position.
The chemical structures of some older and less commonly used sedative-hypnotics, including several barbiturates, are shown in Figure 22-3. Glutethimide and meprobamate are of distinctive chemical structure but are practically equivalent to barbiturates in their pharmacologic effects. Their clinical use is rapidly declining. The sedative-hypnotic class also includes compounds of simpler chemical structure, including ethanol (see Chapter 23) and chloral hydrate.
Several drugs with novel chemical structures have been introduced more recently for use in sleep disorders. Zolpidem, an imidazopyridine, zaleplon, a pyrazolopyrimidine, and eszopiclone, a cyclopyrrolone (Figure 22-4), although structurally unrelated to benzodiazepines, share a similar mechanism of action, as described below. Eszopiclone is the (S)-enantiomer of zopiclone, a hypnotic drug that has been available outside the United States since 1989. Ramelteon, a melatonin receptor agonist, is a new hypnotic drug (see Box: Ramelteon). Buspirone is a slow-onset anxiolytic agent whose actions are quite different from those of conventional sedative-hypnotics (see Box: Buspirone).
Other classes of drugs that exert sedative effects include antipsychotics (Chapter 29), and many antidepressant drugs (Chapter 30). The latter are currently used widely in management of chronic anxiety disorders. Certain antihistaminic agents including hydroxyzine and promethazine (Chapter 16) are also sedating. These agents commonly also exert marked effects on the peripheral autonomic nervous system. Certain antihistaminics with sedative effects are available in over-the-counter sleep aids.
RAMELTEON
Melatonin receptors are thought to be involved in maintaining circadian rhythms underlying the sleep-wake cycle (Chapter 16). Ramelteon, a novel hypnotic drug prescribed specifically for patients who have difficulty in falling asleep, is an agonist at MT1 and MT2 melatonin receptors located in the suprachiasmatic nuclei of the brain. The drug has no direct effects on GABAergic neurotransmission in the central nervous system. In polysomnography studies of patients with chronic insomnia, ramelteon reportedly reduced the latency to persistent sleep with no rebound insomnia or withdrawal symptoms. The drug is rapidly absorbed after oral administration and undergoes extensive first-pass metabolism, forming an active metabolite with longer half-life (2-5 hours) than the parent drug. The CYP1A2 isoform of cytochrome P450 is mainly responsible for the metabolism of ramelteon; the drug should not be used with fluvoxamine (an inhibitor of CYP1A2) and should be used with caution in patients with liver dysfunction. Adverse effects of ramelteon include dizziness, somnolence, fatigue, and endocrine changes as well as decreases in testosterone and increases in prolactin. Ramelteon is not a controlled substance.
BUSPIRONE
Buspirone has selective anxiolytic effects, and its pharmacologic characteristics are different from those of other drugs described in this chapter. Buspirone relieves anxiety without causing marked sedative, hypnotic, or euphoric effects. Unlike benzodiazepines, the drug has no anticonvulsant or muscle relaxant properties. Buspirone does not interact directly with GABAergic systems. It may exert its anxiolytic effects by acting as a partial agonist at brain 5-HT1A receptors, but it also has affinity for brain dopamine D2 receptors. Buspirone-treated patients show no rebound anxiety or withdrawal signs on abrupt discontinuance. The drug is not effective in blocking the acute withdrawal syndrome resulting from abrupt cessation of use of benzodiazepines or other sedative-hypnotics. Buspirone has minimal abuse liability. In marked contrast to the benzodiazepines, the anxiolytic effects of buspirone may take more than a week to become established, making the drug unsuitable for management of acute anxiety states. The drug is used in generalized anxiety states but is less effective in panic disorders.
Buspirone is rapidly absorbed orally but undergoes extensive first-pass metabolism via hydroxylation and dealkylation reactions to form several active metabolites. The major metabolite is 1-(2-pyrimidyl)-piperazine (1-PP), which has a2-adrenoceptor-blocking actions and which enters the central nervous system to reach higher levels than the parent drug. It is not known what role (if any) 1-PP plays in the central actions of buspirone. The elimination half-life of buspirone is 2-4 hours, and liver dysfunction may slow its clearance. Rifampin, an inducer of cytochrome P450, decreases the half-life of buspirone; inhibitors of CYP3A4 (eg, erythromycin, ketoconazole) increase its plasma levels.
Buspirone causes less psychomotor impairment than benzodiazepines and does not affect driving skills. The drug does not potentiate effects of conventional sedative-hypnotic drugs, ethanol, or tricyclic antidepressants, and elderly patients do not appear to be more sensitive to its actions. Tachycardia, palpitations, nervousness, gastrointestinal distress, and paresthesias and a dose-dependent pupillary constriction may occur. Blood pressure may be elevated in patients receiving MAO inhibitors.
|
|
Figure 22-2. Chemical structures of benzodiazepines. |
||
|
|
Figure 22-3. Chemical structures of barbiturates and other sedative-hypnotics. |
||
|
|
Figure 22-4. Chemical structures of newer hypnotics. |
Pharmacokinetics
A. ABSORPTION AND DISTRIBUTION
The rates of oral absorption of sedative-hypnotics differ depending on a number of factors, including lipophilicity. For example, the absorption of triazolam is extremely rapid, and that of diazepam and the active metabolite of clorazepate is more rapid than other commonly used benzodiazepines. Clorazepate, a prodrug, is converted to its active form, desmethyldiazepam (nordiazepam), by acid hydrolysis in the stomach. Most of the barbiturates and other older sedative-hypnotics, as well as the newer hypnotics (eszopiclone, zaleplon, zolpidem), are absorbed rapidly into the blood following oral administration.
Lipid solubility plays a major role in determining the rate at which a particular sedative-hypnotic enters the central nervous system. This property is responsible for the rapid onset of central nervous system effects of triazolam, thiopental (Chapter 25), and the newer hypnotics.
All sedative-hypnotics cross the placental barrier during pregnancy. If sedative-hypnotics are given during the predelivery period, they may contribute to the depression of neonatal vital functions. Sedative-hypnotics are also detectable in breast milk and may exert depressant effects in the nursing infant.
B. BIOTRANSFORMATION
Metabolic transformation to more water-soluble metabolites is necessary for clearance of sedative-hypnotics from the body. The microsomal drug-metabolizing enzyme systems of the liver are most important in this regard. Few sedative-hypnotics are excreted from the body in unchanged form, so elimination half-life depends mainly on the rate of metabolic transformation.
1. Benzodiazepines¾ Hepatic metabolism accounts for the clearance of all benzodiazepines. The patterns and rates of metabolism depend on the individual drugs. Most benzodiazepines undergo microsomal oxidation (phase I reactions), including N-dealkylation and aliphatic hydroxylation catalyzed by cytochrome P450 isozymes, especially CYP3A4. The metabolites are subsequently conjugated (phase II reactions) to form glucuronides that are excreted in the urine. However, many phase I metabolites of benzodiazepines are pharmacologically active, some with long half-lives (Figure 22-5). For example, desmethyldiazepam, which has an elimination half-life of more than 40 hours, is an active metabolite of chlordiazepoxide, diazepam, prazepam, and clorazepate. Alprazolam and triazolam undergo a-hydroxylation, and the resulting metabolites appear to exert short-lived pharmacologic effects because they are rapidly conjugated to form inactive glucuronides. The short elimination half-life of triazolam (2-3 hours) favors its use as a hypnotic rather than as a sedative drug.
The formation of active metabolites has complicated studies on the pharmacokinetics of the benzodiazepines in humans because the elimination half-life of the parent drug may have little relation to the time course of pharmacologic effects. Benzodiazepines for which the parent drug or active metabolites have long half-lives are more likely to cause cumulative effects with multiple doses. Cumulative and residual effects such as excessive drowsiness appear to be less of a problem with such drugs as estazolam, oxazepam, and lorazepam, which have relatively short half-lives and are metabolized directly to inactive glucuronides. Some pharmacokinetic properties of selected benzodiazepines are listed in Table 22-1. The metabolism of several commonly used benzodiazepines including diazepam, midazolam, and triazolam is affected by inhibitors and inducers of hepatic P450 isozymes (see Chapter 4).
2. Barbiturates¾ With the exception of phenobarbital, only insignificant quantities of the barbiturates are excreted unchanged. The major metabolic pathways involve oxidation by hepatic enzymes to form alcohols, acids, and ketones, which appear in the urine as glucuronide conjugates. The overall rate of hepatic metabolism in humans depends on the individual drug but (with the exception of the thiobarbiturates) is usually slow. The elimination half-lives of secobarbital and pentobarbital range from 18 to 48 hours in different individuals. The elimination half-life of phenobarbital in humans is 4-5 days. Multiple dosing with these agents can lead to cumulative effects.
3. Newer hypnotics¾ After oral administration of the standard formulation, zolpidem reaches peak plasma levels in 1.6 hours. A biphasic release formulation extends plasma levels by approximately 2 hours. Zolpidem is rapidly metabolized to inactive metabolites via oxidation and hydroxylation by hepatic cytochromes P450 including the CYP3A4 isozyme. The elimination half-life of the drug is 1.5-3.5 hours, with clearance decreased in elderly patients. Zaleplon is metabolized to inactive metabolites mainly by hepatic aldehyde oxidase and partly by the cytochrome P450 isoform CYP3A4. The half-life of the drug is about 1 hour. Dosage should be reduced in patients with hepatic impairment and in the elderly. Cimetidine, which inhibits both aldehyde dehydrogenase and CYP3A4, markedly increases the peak plasma level of zaleplon. Eszopiclone is metabolized by hepatic cytochromes P450 (especially CYP3A4) to form the inactive N-oxide derivative and weakly active desmethyleszopiclone. The elimination half-life of eszopiclone is approximately 6 hours and is prolonged in the elderly and in the presence of inhibitors of CYP3A4 (eg, ketoconazole). Inducers of CYP3A4 (eg, rifampin) increase the hepatic metabolism of eszopiclone.
C. EXCRETION
The water-soluble metabolites of sedative-hypnotics, mostly formed via the conjugation of phase I metabolites, are excreted mainly via the kidney. In most cases, changes in renal function do not have a marked effect on the elimination of parent drugs. Phenobarbital is excreted unchanged in the urine to a certain extent (20-30% in humans), and its elimination rate can be increased significantly by alkalinization of the urine. This is partly due to increased ionization at alkaline pH, since phenobarbital is a weak acid with a pKa of 7.4.
D. FACTORS AFFECTING BIODISPOSITION
The biodisposition of sedative-hypnotics can be influenced by several factors, particularly alterations in hepatic function resulting from disease or drug-induced increases or decreases in microsomal enzyme activities (see Chapter 4).
In very old patients and in patients with severe liver disease, the elimination half-lives of these drugs are often increased significantly. In such cases, multiple normal doses of these sedative-hypnotics can result in excessive central nervous system effects.
The activity of hepatic microsomal drug-metabolizing enzymes may be increased in patients exposed to certain older sedative-hypnotics on a long-term basis (enzyme induction; see Chapter 4). Barbiturates (especially phenobarbital) and meprobamate are most likely to cause this effect, which may result in an increase in their hepatic metabolism as well as that of other drugs. Increased biotransformation of other pharmacologic agents as a result of enzyme induction by barbiturates is a potential mechanism underlying drug interactions (Appendix II). In contrast, benzodiazepines and the newer hypnotics do not change hepatic drug-metabolizing enzyme activity with continuous use.
|
|
Figure 22-5. Biotransformation of benzodiazepines. (Boldface, drugs available for clinical use in various countries; *, active metabolite.) |
Pharmacodynamics of Benzodiazepines, Barbiturates, & Newer Hypnotics
A. MOLECULAR PHARMACOLOGY OF THE GABAA RECEPTOR
The benzodiazepines, the barbiturates, zolpidem, zaleplon, eszopiclone, and many other drugs bind to molecular components of the GABAA receptor in neuronal membranes in the central nervous system. This receptor, which functions as a chloride ion channel, is activated by the inhibitory neurotransmitter GABA (see Chapter 21).
The GABAA receptor has a pentameric structure assembled from five subunits (each with four transmembrane-spanning domains) selected from multiple polypeptide classes (a, b, g, d, e, p, r, etc). Multiple subunits of several of these classes have been characterized, eg, six different a, four b, and three g. A model of the hypothetical GABAA receptor-chloride ion channel macromolecular complex is shown in Figure 22-6.
A major isoform of the GABAA receptor that is found in many regions of the brain consists of two a1 and two b2 subunits and one g2 subunit. In this isoform, the binding sites for GABA are located between adjacent a1 and b2 subunits, and the binding pocket for benzodiazepines (the BZ site of the GABAA receptor) is between an a1 and the g2 subunit. However, GABAAreceptors in different areas of the central nervous system consist of various combinations of the essential subunits, and the benzodiazepines bind to many of these, including receptor isoforms containing a2, a3, and a5 subunits. Barbiturates also bind to multiple isoforms of the GABAA receptor but at different sites from those with which benzodiazepines interact. In contrast to benzodiazepines, zolpidem, zaleplon, and eszopiclone bind more selectively because these drugs interact only with GABAA-receptor isoforms that contain a1 subunits. The heterogeneity of GABAA receptors may constitute the molecular basis for the varied pharmacologic actions of benzodiazepines and related drugs (see Box: GABA Receptor Heterogeneity & Pharmacologic Selectivity).
In contrast to GABA itself, benzodiazepines and other sedative-hypnotics have a low affinity for GABAB receptors, which are activated by the spasmolytic drug baclofen (see Chapters 21and 27).
GABA RECEPTOR HETEROGENEITY & PHARMACOLOGIC SELECTIVITY
Studies involving genetically engineered mice have demonstrated that the specific pharmacologic actions elicited by benzodiazepines and other drugs that modulate GABA actions are influenced by the composition of the subunits assembled to form the GABAA receptor. Benzodiazepines interact only with brain GABAA receptors in which the a subunits (1,2,3, and 5) have a conserved histidine residue in the N-terminal domain. Strains of mice, in which a point mutation has been inserted converting histidine to arginine in the a1 subunit, show resistance to both the sedative and amnestic effects of benzodiazepines, but anxiolytic and muscle-relaxing effects are largely unchanged. These animals are also unresponsive to the hypnotic actions of zolpidem and zaleplon, drugs that bind selectively to GABAA receptors containing a1 subunits. In contrast, mice with selective histidine-arginine mutations in the a2subunit of GABAA receptors show selective resistance to the antianxiety effects of benzodiazepines. Based on studies of this type, it has been suggested that a1 subunits in GABAAreceptors mediate sedation, amnesia, and possibly antiseizure effects of benzodiazepines, whereas a2 subunits are involved in their anxiolytic and muscle-relaxing actions. Other mutation studies have led to suggestions that an a5 subtype is involved in at least some of the memory impairment caused by benzodiazepines. It should be noted that these studies involving genetic manipulations of the GABAA receptor utilize rodent models of the anxiolytic and amnestic actions of drugs.
B. NEUROPHARMACOLOGY
GABA (gamma-aminobutyric acid) is the major inhibitory neurotransmitter in the central nervous system (Chapter 21). Electrophysiologic studies have shown that benzodiazepines potentiate GABAergic inhibition at all levels of the neuraxis, including the spinal cord, hypothalamus, hippocampus, substantia nigra, cerebellar cortex, and cerebral cortex. Benzodiazepines appear to increase the efficiency of GABAergic synaptic inhibition. The benzodiazepines do not substitute for GABA but appear to enhance GABA's effects allosterically without directly activating GABAA receptors or opening the associated chloride channels. The enhancement in chloride ion conductance induced by the interaction of benzodiazepines with GABA takes the form of an increase in the frequency of channel-opening events.
Barbiturates also facilitate the actions of GABA at multiple sites in the central nervous system, but¾in contrast to benzodiazepines¾they appear to increase the duration of the GABA-gated chloride channel openings. At high concentrations, the barbiturates may also be GABA-mimetic, directly activating chloride channels. These effects involve a binding site or sites distinct from the benzodiazepine binding sites. Barbiturates are less selective in their actions than benzodiazepines, because they also depress the actions of excitatory neurotransmitters (eg, glutamic acid) and exert nonsynaptic membrane effects in parallel with their effects on GABA neurotransmission. This multiplicity of sites of action of barbiturates may be the basis for their ability to induce full surgical anesthesia (see Chapter 25) and for their more pronounced central depressant effects (which result in their low margin of safety) compared with benzodiazepines and the newer hypnotics.
C. BENZODIAZEPINE BINDING SITE LIGANDS
The components of the GABAA receptor-chloride ion channel macromolecule that function as benzodiazepine binding sites exhibit heterogeneity (see Box: The Versatility of the Chloride Channel GABA Receptor Complex). Three types of ligand-benzodia-zepine receptor interactions have been reported: (1) Agonists facilitate GABA actions, and this occurs at multiple BZ binding sites in the case of the benzodiazepines. As noted above, the nonbenzodiazepines zolpidem, zaleplon, and eszopiclone are selective agonists at the BZ sites that contain an a1subunit. Endogenous agonist ligands for the BZ binding sites have been proposed, because benzodiazepine-like chemicals have been isolated from brain tissue of animals never exposed to these drugs. Nonbenzodiazepine molecules that have affinity for BZ sites on the GABAA receptor have also been detected in human brain. (2) Antagonists are typified by the synthetic benzodiazepine derivative flumazenil, which blocks the actions of benzodiazepines, eszopiclone, zaleplon, and zolpidem but does not antagonize the actions of barbiturates, meprobamate, or ethanol. Certain endogenous neuropeptides are also capable of blocking the interaction of benzodiazepines with BZ binding sites. (3) Inverse agonists act as negative allosteric modulators of GABA-receptor function. Their interaction with BZ sites on the GABAA receptor can produce anxiety and seizures, an action that has been demonstrated for several compounds, especially the b-carbolines, eg, n-butyl-b-carboline-3-carboxylate (b-CCB). In addition to their direct actions, these molecules can block the effects of benzodiazepines.
The physiologic significance of endogenous modulators of the functions of GABA in the central nervous system remains unclear. To date, it has not been established that the putative endogenous ligands of BZ binding sites play a role in the control of states of anxiety, sleep patterns, or any other characteristic behavioral expression of central nervous system function.
THE VERSATILITY OF THE CHLORIDE CHANNEL GABA RECEPTOR COMPLEX
The GABAA-chloride channel macromolecular complex is one of the most versatile drug-responsive machines in the body. In addition to the benzodiazepines, barbiturates, and the newer hypnotics (eg, zolpidem), many other drugs with central nervous system effects can modify the function of this important ionotropic receptor. These include alcohol; alphaxolone, etomidate, and propofol (intravenous anesthetics); volatile anesthetics (eg, halothane); several anticonvulsants (eg, gabapentin, vigabatrin); and ivermectin (an anthelmintic agent). Most of these agents facilitate or mimic the action of GABA. (It has not been shown that these drugs act exclusively or even primarily by this mechanism.) Central nervous system excitatory agents that act on the chloride channel include picrotoxin and bicuculline. These convulsant drugs block the channel directly (picrotoxin) or interfere with GABA binding (bicuculline).
D. ORGAN LEVEL EFFECTS
1. Sedation¾ Benzodiazepines, barbiturates, and most older sedative-hypnotic drugs exert calming effects with concomitant reduction of anxiety at relatively low doses. In most cases, however, the anxiolytic actions of sedative-hypnotics are accompanied by some depressant effects on psychomotor and cognitive functions. In experimental animal models, benzodiazepines and older sedative-hypnotic drugs are able to disinhibit punishment-suppressed behavior. This disinhibition has been equated with antianxiety effects of sedative-hypnotics, and it is not a characteristic of all drugs that have sedative effects, eg, the tricyclic antidepressants and antihistamines. However, the disinhibition of previously suppressed behavior may be more related to behavioral disinhibitory effects of sedative-hypnotics, including euphoria, impaired judgment, and loss of self-control, which can occur at dosages in the range of those used for management of anxiety. The benzodiazepines also exert dose-dependent anterograde amnesic effects (inability to remember events occurring during the drug's duration of action).
2. Hypnosis¾ By definition, all of the sedative-hypnotics induce sleep if high enough doses are given. The effects of sedative-hypnotics on the stages of sleep depend on several factors, including the specific drug, the dose, and the frequency of its administration. The general effects of benzodiazepines and older sedative-hypnotics on patterns of normal sleep are as follows: (1) the latency of sleep onset is decreased (time to fall asleep); (2) the duration of stage 2 NREM sleep is increased; (3) the duration of REM sleep is decreased; and (4) the duration of stage 4 NREM slow-wave sleep is decreased. The newer hypnotics all decrease the latency to persistent sleep. Zolpidem decreases REM sleep but has minimal effect on slow-wave sleep. Zaleplon decreases the latency of sleep onset with little effect on total sleep time, NREM, or REM sleep. Eszopiclone increases total sleep time, mainly via increases in stage 2 NREM sleep, and at low doses has little effect on sleep patterns. At the highest recommended dose, eszopiclone decreases REM sleep.
More rapid onset of sleep and prolongation of stage 2 are presumably clinically useful effects. However, the significance of sedative-hypnotic drug effects on REM and slow-wave sleep is not clear. Deliberate interruption of REM sleep causes anxiety and irritability followed by a rebound increase in REM sleep at the end of the experiment. A similar pattern of "REM rebound" can be detected following abrupt cessation of drug treatment with older sedative-hypnotics, especially when drugs with short durations of action (eg, triazolam) are used at high doses. With respect to zolpidem and the other newer hypnotics, there is little evidence of REM rebound when these drugs are discontinued after use of recommended doses. However, rebound insomnia occurs with both zolpidem and zaleplon if used at higher doses. Despite possible reductions in slow-wave sleep, there are no reports of disturbances in the secretion of pituitary or adrenal hormones when either barbiturates or benzodiazepines are used as hypnotics. The use of sedative-hypnotics for more than 1-2 weeks leads to some tolerance to their effects on sleep patterns.
3. Anesthesia¾ As shown in Figure 22-1, high doses of certain sedative-hypnotics depress the central nervous system to the point known as stage III of general anesthesia (see Chapter 25). However, the suitability of a particular agent as an adjunct in anesthesia depends mainly on the physicochemical properties that determine its rapidity of onset and duration of effect. Among the barbiturates, thiopental and methohexital are very lipid-soluble, penetrating brain tissue rapidly following intravenous administration, a characteristic favoring their use for the induction of anesthesia. Rapid tissue redistribution (not rapid elimination) accounts for the short duration of action of these drugs, a feature useful in recovery from anesthesia.
Benzodiazepines¾including diazepam, lorazepam, and midazolam¾are used intravenously in anesthesia (see Chapter 25), often in combination with other agents. Not surprisingly, benzodiazepines given in large doses as adjuncts to general anesthetics may contribute to a persistent postanesthetic respiratory depression. This is probably related to their relatively long half-lives and the formation of active metabolites. However, such depressant actions of the benzodiazepines are usually reversible with flumazenil.
4. Anticonvulsant effects¾ Many sedative-hypnotics are capable of inhibiting the development and spread of epileptiform electrical activity in the central nervous system. Some selectivity exists in that some members of the group can exert anticonvulsant effects without marked central nervous system depression (although psychomotor function may be impaired). Several benzodiazepines¾including clonazepam, nitrazepam, lorazepam, and diazepam¾are sufficiently selective to be clinically useful in the management of seizures (see Chapter 24). Of the barbiturates, phenobarbital and metharbital (converted to phenobarbital in the body) are effective in the treatment of generalized tonic-clonic seizures. Zolpidem, zaleplon, and eszopiclone lack anticonvulsant activity, presumably because of their more selective binding than that of benzodiazepines to GABAA receptor isoforms.
5. Muscle relaxation¾ Some sedative-hypnotics, particularly members of the carbamate (eg, meprobamate) and benzodiazepine groups, exert inhibitory effects on polysynaptic reflexes and internuncial transmission and at high doses may also depress transmission at the skeletal neuromuscular junction. Somewhat selective actions of this type that lead to muscle relaxation can be readily demonstrated in animals and have led to claims of usefulness for relaxing contracted voluntary muscle in muscle spasm (see Clinical Pharmacology). Muscle relaxation is not a characteristic action of zolpidem, zaleplon, and eszopiclone.
6. Effects on respiration and cardiovascular function¾ At hypnotic doses in healthy patients, the effects of sedative-hypnotics on respiration are comparable to changes during natural sleep. However, even at therapeutic doses, sedative-hypnotics can produce significant respiratory depression in patients with pulmonary disease. Effects on respiration are dose-related, and depression of the medullary respiratory center is the usual cause of death due to overdose of sedative-hypnotics.
At doses up to those causing hypnosis, no significant effects on the cardiovascular system are observed in healthy patients. However, in hypovolemic states, heart failure, and other diseases that impair cardiovascular function, normal doses of sedative-hypnotics may cause cardiovascular depression, probably as a result of actions on the medullary vasomotor centers. At toxic doses, myocardial contractility and vascular tone may both be depressed by central and peripheral effects, leading to circulatory collapse. Respiratory and cardiovascular effects are more marked when sedative-hypnotics are given intravenously.
|
|
Figure 22-6. A model of the GABAA receptor-chloride ion channel macromolecular complex (others could be proposed). A heterooligomeric glycoprotein, the complex consists of five or more membrane-spanning subunits. Multiple forms of a, b, and g subunits are arranged in different pentameric combinations so that GABAA receptors exhibit molecular heterogeneity. GABA appears to interact with a or b subunits triggering chloride channel opening with resulting membrane hyperpolarization. Binding of benzodiazepines to g subunits or to an area of the a unit influenced by the g unit facilitates the process of channel opening but does not directly initiate chloride current. (Modified and reproduced, with permission, from Zorumski CF, Isenberg KE: Insights into the structure and function of GABA receptors: Ion channels and psychiatry. Am J Psychiatry 1991;148:162.) |
Tolerance; Psychologic & Physiologic Dependence
Tolerance¾decreased responsiveness to a drug following repeated exposure¾is a common feature of sedative-hypnotic use. It may result in the need for an increase in the dose required to maintain symptomatic improvement or to promote sleep. It is important to recognize that partial cross-tolerance occurs between the sedative-hypnotics described here and also with ethanol (Chapter 23)¾a feature of some clinical importance, as explained below. The mechanisms responsible for tolerance to sedative-hypnotics are not well understood. An increase in the rate of drug metabolism (metabolic tolerance) may be partly responsible in the case of chronic administration of barbiturates, but changes in responsiveness of the central nervous system (pharmacodynamic tolerance) are of greater importance for most sedative-hypnotics. In the case of benzodiazepines, the development of tolerance in animals has been associated with down-regulation of brain benzodiazepine receptors. Tolerance has been reported to occur with the extended use of zolpidem. Minimal tolerance was observed with the use of zaleplon over a 5-week period and eszopiclone over a 6-month period.
The perceived desirable properties of relief of anxiety, euphoria, disinhibition, and promotion of sleep have led to the compulsive misuse of virtually all sedative-hypnotics. (See Chapter 32 for a detailed discussion.) For this reason, most sedative-hypnotic drugs are classified as Schedule III or Schedule IV drugs for prescribing purposes. The consequences of abuse of these agents can be defined in both psychologic and physiologic terms. The psychologic component may initially parallel simple neurotic behavior patterns difficult to differentiate from those of the inveterate coffee drinker or cigarette smoker. When the pattern of sedative-hypnotic use becomes compulsive, more serious complications develop, including physiologic dependence and tolerance.
Physiologic dependence can be described as an altered physiologic state that requires continuous drug administration to prevent an abstinence or withdrawal syndrome. In the case of sedative-hypnotics, this syndrome is characterized by states of increased anxiety, insomnia, and central nervous system excitability that may progress to convulsions. Most sedative-hypnotics¾including benzodiazepines¾are capable of causing physiologic dependence when used on a long-term basis. However, the severity of withdrawal symptoms differs among individual drugs and depends also on the magnitude of the dose used immediately before cessation of use. When higher doses of sedative-hypnotics are used, abrupt withdrawal leads to more serious withdrawal signs. Differences in the severity of withdrawal symptoms resulting from individual sedative-hypnotics relate in part to half-life, since drugs with long half-lives are eliminated slowly enough to accomplish gradual withdrawal with few physical symptoms. The use of drugs with very short half-lives for hypnotic effects may lead to signs of withdrawal even between doses. For example, triazolam, a benzodiazepine with a half-life of about 4 hours, has been reported to cause daytime anxiety when used to treat sleep disorders. The abrupt cessation of zolpidem, zaleplon, or eszopiclone may also result in withdrawal symptoms, though usually of less intensity than those seen with benzodiazepines.
BENZODIAZEPINE ANTAGONISTS: FLUMAZENIL
Flumazenil is one of several 1,4-benzodiazepine derivatives with a high affinity for the benzodiazepine binding site on the GABAA receptor that act as competitive antagonists. It blocks many of the actions of benzodiazepines, zolpidem, zaleplon, and eszopiclone, but does not antagonize the central nervous system effects of other sedative-hypnotics, ethanol, opioids, or general anesthetics. Flumazenil is approved for use in reversing the central nervous system depressant effects of benzodiazepine overdose and to hasten recovery following use of these drugs in anesthetic and diagnostic procedures. Although the drug reverses the sedative effects of benzodiazepines, antagonism of benzodiazepine-induced respiratory depression is less predictable. When given intravenously, flumazenil acts rapidly but has a short half-life (0.7-1.3 hours) due to rapid hepatic clearance. Because all benzodiazepines have a longer duration of action than flumazenil, sedation commonly recurs, requiring repeated administration of the antagonist.
Adverse effects of flumazenil include agitation, confusion, dizziness, and nausea. Flumazenil may cause a severe precipitated abstinence syndrome in patients who have developed physiologic benzodiazepine dependence. In patients who have ingested benzodiazepines with tricyclic antidepressants, seizures and cardiac arrhythmias may follow flumazenil administration.
II. CLINICAL PHARMACOLOGY OF SEDATIVE-HYPNOTICS
TREATMENT OF ANXIETY STATES
The psychologic, behavioral, and physiologic responses that characterize anxiety can take many forms. Typically, the psychic awareness of anxiety is accompanied by enhanced vigilance, motor tension, and autonomic hyperactivity. Anxiety is often secondary to organic disease states¾acute myocardial infarction, angina pectoris, gastrointestinal ulcers, etc¾which themselves require specific therapy. Another class of secondary anxiety states (situational anxiety) results from circumstances that may have to be dealt with only once or a few times, including anticipation of frightening medical or dental procedures and family illness or other stressful event. Even though situational anxiety tends to be self-limiting, the short-term use of sedative-hypnotics may be appropriate for the treatment of this and certain disease-associated anxiety states. Similarly, the use of a sedative-hypnotic as premedication prior to surgery or some unpleasant medical procedure is rational and proper (Table 22-2).
Excessive or unreasonable anxiety about life circumstances (generalized anxiety disorder, GAD), panic disorders, and agoraphobia are also amenable to drug therapy, sometimes in conjunction with psychotherapy. The benzodiazepines continue to be widely used for the management of acute anxiety states and for rapid control of panic attacks. They are also used in the long-term management of GAD and panic disorders. Anxiety symptoms may be relieved by many benzodiazepines, but it is not always easy to demonstrate the superiority of one drug over another. However, alprazolam is particularly effective in the treatment of panic disorders and agoraphobia and appears to be more selective in these conditions than other benzodiazepines. The choice of benzodiazepines for anxiety is based on several sound pharmacologic principles: (1) a relatively high therapeutic index (see drug B in Figure 22-1), plus availability of flumazenil for treatment of overdose; (2) a low risk of drug interactions based on liver enzyme induction; (3) minimal effects on cardiovascular or autonomic functions.
Disadvantages of the benzodiazepines include the risk of dependence, depression of central nervous system functions, and amnestic effects. In addition, the benzodiazepines exert additive central nervous system depression when administered with other drugs, including ethanol. The patient should be warned of this possibility to avoid impairment of performance of any task requiring mental alertness and motor coordination. In the treatment of generalized anxiety disorders and certain phobias, newer antidepressants, including selective serotonin reuptake inhibitors, are now considered by many authorities to be drugs of first choice (see Chapter 30). However, these agents have minimal effectiveness in acute anxiety states.
Sedative-hypnotics should be used with appropriate caution so as to minimize adverse effects. A dose should be prescribed that does not impair mentation or motor functions during waking hours. Some patients may tolerate the drug better if most of the daily dose is given at bedtime, with smaller doses during the day. Prescriptions should be written for short periods, since there is little justification for long-term therapy (defined as use of therapeutic doses for 2 months or longer). The physician should make an effort to assess the efficacy of therapy from the patient's subjective responses. Combinations of antianxiety agents should be avoided, and people taking sedatives should be cautioned about the consumption of alcohol and the concurrent use of over-the-counter medications containing antihistaminic or anticholinergic drugs (see Chapter 64).
TREATMENT OF SLEEP PROBLEMS
Sleep disorders are common and often result from inadequate treatment of underlying medical conditions or psychiatric illness. True primary insomnia is rare. Nonpharmacologic therapies that are useful for sleep problems include proper diet and exercise, avoiding stimulants before retiring, ensuring a comfortable sleeping environment, and retiring at a regular time each night. In some cases, however, the patient will need and should be given a sedative-hypnotic for a limited period. It should be noted that the abrupt discontinuance of many drugs in this class can lead to rebound insomnia.
Benzodiazepines can cause a dose-dependent decrease in both REM and slow-wave sleep, though to a lesser extent than the barbiturates. The newer hypnotics zolpidem, zaleplon, and eszopiclone are less likely than the benzodiazepines to change sleep patterns. However, so little is known about the clinical impact of these effects that statements about the desirability of a particular drug based on its effects on sleep architecture have more theoretical than practical significance. Clinical criteria of efficacy in alleviating a particular sleeping problem are more useful. The drug selected should be one that provides sleep of fairly rapid onset (decreased sleep latency) and sufficient duration, with minimal "hangover" effects such as drowsiness, dysphoria, and mental or motor depression the following day. Older drugs such as chloral hydrate, secobarbital, and pentobarbital continue to be used, but benzodiazepines, zolpidem, zaleplon, or eszopiclone are generally preferred. Daytime sedation is more common with benzodiazepines that have slow elimination rates (eg, lorazepam) and those that are biotransformed to active metabolites (eg, flurazepam, quazepam). If benzodiazepines are used nightly, tolerance can occur, which may lead to dose increases by the patient to produce the desired effect. Anterograde amnesia occurs to some degree with all benzodiazepines used for hypnosis.
Eszopiclone, zaleplon, and zolpidem have efficacies similar to those of the hypnotic benzodiazepines in the management of sleep disorders. Favorable clinical features of zolpidem and the other newer hypnotics include rapid onset of activity and modest day-after psychomotor depression with few amnestic effects. Zolpidem, currently the most frequently prescribed hypnotic drug in the United States, is available in a biphasic release formulation that provides sustained drug levels for sleep maintenance. Zaleplon acts rapidly, and because of its short half-life, the drug appears to have value in the management of patients who awaken early in the sleep cycle. At recommended doses, zaleplon and eszopiclone (despite its relatively long half-life) appear to cause less amnesia or day-after somnolence than zolpidem or benzodiazepines. The drugs commonly used for sedation and hypnosis are listed in Table 22-3 together with recommended doses. Note: Long-term use of hypnotics is an irrational and dangerous medical practice.
OTHER THERAPEUTIC USES
Table 22-2 summarizes several other important clinical uses of drugs in the sedative-hypnotic class. Drugs used in the management of seizure disorders and as intravenous agents in anesthesia are discussed in Chapters 24 and 25.
For sedative and possible amnestic effects during medical or surgical procedures such as endoscopy and bronchoscopy¾as well as for premedication prior to anesthesia¾oral formulations of shorter-acting drugs are preferred.
Long-acting drugs such as chlordiazepoxide and diazepam and, to a lesser extent, phenobarbital are administered in progressively decreasing doses to patients during withdrawal from physiologic dependence on ethanol or other sedative-hypnotics. Parenteral lorazepam is used to suppress the symptoms of delirium tremens.
Meprobamate and, more recently, the benzodiazepines have frequently been used as central muscle relaxants, though evidence for general efficacy without accompanying sedation is lacking. A possible exception is diazepam, which has useful relaxant effects in skeletal muscle spasticity of central origin (see Chapter 27).
Psychiatric uses of benzodiazepines other than treatment of anxiety states include the initial management of mania, the control of drug-induced hyperexcitability states (eg, phencyclidine intoxication), and possibly the treatment of major depressive disorders with alprazolam. Sedative-hypnotics are also used occasionally as diagnostic aids in neurology and psychiatry.
CLINICAL TOXICOLOGY OF SEDATIVE-HYPNOTICS
Direct Toxic Actions
Many of the common adverse effects of sedative-hypnotics result from dose-related depression of the central nervous system. Relatively low doses may lead to drowsiness, impaired judgment, and diminished motor skills, sometimes with a significant impact on driving ability, job performance, and personal relationships. Benzodiazepines may cause a significant dose-related anterograde amnesia; they can significantly impair ability to learn new information, particularly that involving effortful cognitive processes, while leaving the retrieval of previously learned information intact. This effect is utilized for uncomfortable clinical procedures, eg, endoscopy, because the patient is able to cooperate during the procedure but amnesic regarding it afterward. The criminal use of benzodiazepines in cases of "date rape" is based on their dose-dependent amnestic effects. Hangover effects are not uncommon following use of hypnotic drugs with long elimination half-lives. Because elderly patients are more sensitive to the effects of sedative-hypnotics, doses approximately half of those used in younger adults are safer and usually as effective. The most common reversible cause of confusional states in the elderly is overuse of sedative-hypnotics. At higher doses, toxicity may present as lethargy or a state of exhaustion or, alternatively, as gross symptoms equivalent to those of ethanol intoxication. The physician should be aware of variability among patients in terms of doses causing adverse effects. An increased sensitivity to sedative-hypnotics is more common in patients with cardiovascular disease, respiratory disease, or hepatic impairment and in older patients. Sedative-hypnotics can exacerbate breathing problems in patients with chronic pulmonary disease and in those with symptomatic sleep apnea.
Sedative-hypnotics are the drugs most frequently involved in deliberate overdoses, in part because of their general availability as very commonly prescribed pharmacologic agents. The benzodiazepines are considered to be safer drugs in this respect, since they have flatter dose-response curves. Epidemiologic studies on the incidence of drug-related deaths support this general assumption¾eg, 0.3 deaths per million tablets of diazepam prescribed versus 11.6 deaths per million capsules of secobarbital in one study. Alprazolam is purportedly more toxic in overdose than other benzodiazepines. Of course, many factors other than the specific sedative-hypnotic could influence such data¾particularly the presence of other central nervous system depressants, including ethanol. In fact, most serious cases of drug overdosage, intentional or accidental, do involve polypharmacy; and when combinations of agents are taken, the practical safety of benzodiazepines may be less than the foregoing would imply.
The lethal dose of any sedative-hypnotic varies with the patient and the circumstances (see Chapter 59). If discovery of the ingestion is made early and a conservative treatment regimen is started, the outcome is rarely fatal, even following very high doses. On the other hand, for most sedative-hypnotics¾with the exception of benzodiazepines and possibly the newer hypnotic drugs that have a similar mechanism of action¾a dose as low as ten times the hypnotic dose may be fatal if the patient is not discovered or does not seek help in time. With severe toxicity, the respiratory depression from central actions of the drug may be complicated by aspiration of gastric contents in the unattended patient¾an even more likely occurrence if ethanol is present. Cardiovascular depression further complicates successful resuscitation. In such patients, treatment consists of ensuring a patent airway, with mechanical ventilation if needed, and maintenance of plasma volume, renal output, and cardiac function. Use of a positive inotropic drug such as dopamine, which preserves renal blood flow, is sometimes indicated. Hemodialysis or hemoperfusion may be used to hasten elimination of some of these drugs.
Flumazenil reverses the sedative actions of benzodiazepines, and probably those of eszopiclone, zaleplon, and zolpidem, although experience with its use in overdose of the newer hypnotics is limited. However, its duration of action is short, its antagonism of respiratory depression is unpredictable, and there is a risk of precipitation of withdrawal symptoms in long-term users of benzodiazpines (see below). Consequently, the use of flumazenil in benzodiazepine overdose remains controversial and must be accompanied by adequate monitoring and support of respiratory function. The extensive clinical use of triazolam has led to reports of serious central nervous system effects including behavioral disinhibition, delirium, aggression, and violence. Although behavioral disinhibition may occur with sedative-hypnotic drugs, it does not appear to be more prevalent with triazolam than with other benzodiazepines. Disinhibitory reactions during benzodiazepine treatment are more clearly associated with the use of very high doses and the pretreatment level of patient hostility.
Adverse effects of the sedative-hypnotics that are not referable to their central nervous system actions occur infrequently. Hypersensitivity reactions, including skin rashes, occur only occasionally with most drugs of this class. Reports of teratogenicity leading to fetal deformation following use of piperidinediones and certain benzodiazepines justify caution in the use of these drugs during pregnancy. Because barbiturates enhance porphyrin synthesis, they are absolutely contraindicated in patients with a history of acute intermittent porphyria, variegate porphyria, hereditary coproporphyria, or symptomatic porphyria.
Alterations in Drug Response
Depending on the dosage and the duration of use, tolerance occurs in varying degrees to many of the pharmacologic effects of sedative-hypnotics. However, it should not be assumed that the degree of tolerance achieved is identical for all pharmacologic effects. There is evidence that the lethal dose range is not altered significantly by the long-term use of sedative-hypnotics. Cross-tolerance between the different sedative-hypnotics, including ethanol, can lead to an unsatisfactory therapeutic response when standard doses of a drug are used in a patient with a recent history of excessive use of these agents. However, there have been very few reports of tolerance development when eszopiclone, zolpidem, or zaleplon was used for less than 4 weeks.
With the long-term use of sedative-hypnotics, especially if doses are increased, a state of physiologic dependence can occur. This may develop to a degree unparalleled by any other drug group, including the opioids. Withdrawal from a sedative-hypnotic can have severe and life-threatening manifestations. Withdrawal symptoms range from restlessness, anxiety, weakness, and orthostatic hypotension to hyperactive reflexes and generalized seizures. Symptoms of withdrawal are usually more severe following discontinuance of sedative-hypnotics with shorter half-lives. However, eszopiclone, zolpidem, and zaleplon appear to be exceptions to this, because withdrawal symptoms are minimal following abrupt discontinuance of these newer short-acting agents. Symptoms are less pronounced with longer-acting drugs, which may partly accomplish their own "tapered" withdrawal by virtue of their slow elimination. Cross-dependence, defined as the ability of one drug to suppress abstinence symptoms from discontinuance of another drug, is quite marked among sedative-hypnotics. This provides the rationale for therapeutic regimens in the management of withdrawal states: Longer-acting drugs such as chlordiazepoxide, diazepam, and phenobarbital can be used to alleviate withdrawal symptoms of shorter-acting drugs, including ethanol.
Drug Interactions
The most common drug interactions involving sedative-hypnotics are interactions with other central nervous system depressant drugs, leading to additive effects. These interactions have some therapeutic usefulness when these drugs are used as adjuvants in anesthesia practice. However, if not anticipated, such interactions can lead to serious consequences, including enhanced depression with concomitant use of many other drugs. Additive effects can be predicted with concomitant use of alcoholic beverages, opioid analgesics, anticonvulsants, and phenothiazines. Less obvious but just as important is enhanced central nervous system depression with a variety of antihistamines, antihypertensive agents, and antidepressant drugs of the tricyclic class.
Interactions involving changes in the activity of hepatic drug-metabolizing enzyme systems have been discussed (see also Chapter 4 and Appendix II).
PREPARATIONS AVAILABLE
BENZODIAZEPINES
Alprazolam (generic, Xanax)
Oral: 0.25, 0.5, 1, 2 mg tablets, extended-release tablets, and orally disintegrating tablets; 1.0 mg/mL solution
Chlordiazepoxide (generic, Librium)
Oral: 5, 10, 25 mg capsules
Parenteral: 100 mg powder for injection
Clorazepate (generic, Tranxene)
Oral: 3.75, 7.5, 15 mg tablets and capsules
Oral sustained-release: 11.25, 22.5 mg tablets
Clonazepam (generic, Klonopin)
Oral: 0.5, 1, 2 mg tablets; 0.125, 0.25, 0.5, 1, 2 mg orally disintegrating tablets
Diazepam (generic, Valium)
Oral: 2, 5, 10 mg tablets; 1, 5 mg/mL solutions
Parenteral: 5 mg/mL for injection
Estazolam (generic, ProSom)
Oral: 1, 2 mg tablets
Flurazepam (generic, Dalmane)
Oral: 15, 30 mg capsules
Lorazepam (generic, Ativan)
Oral: 0.5, 1, 2 mg tablets; 2 mg/mL solution
Parenteral: 2, 4 mg/mL for injection
Midazolam (Versed)
Oral: 2 mg/mL syrup
Parenteral: 1, 5 mg/mL in 1, 2, 5, 10 mL vials for injection
Oxazepam (generic)
Oral: 10, 15, 30 mg capsules
Quazepam (Doral)
Oral: 7.5, 15 mg tablets
Temazepam (generic, Restoril)
Oral: 7.5, 15, 22.5, 30 mg capsules
Triazolam (generic, Halcion)
Oral: 0.125, 0.25 mg tablets
BENZODIAZEPINE ANTAGONIST
Flumazenil (Romazicon)
Parenteral: 0.1 mg/mL for IV injection
BARBITURATES
Amobarbital (Amytal)
Parenteral: powder in 250, 500 mg vials to reconstitute for injection
Mephobarbital (Mebaral)
Oral: 32, 50, 100 mg tablets
Pentobarbital (generic, Nembutal Sodium)
Oral: 50, 100 mg capsules; 4 mg/mL elixir
Rectal: 30, 60, 120, 200 mg suppositories
Parenteral: 50 mg/mL for injection
Phenobarbital (generic, Luminal Sodium)
Oral: 15, 16, 30, 60, 90, 100 mg tablets; 16 mg capsules; 15, 20 mg/5 mL elixirs
Parenteral: 30, 60, 65, 130 mg/mL for injection
Secobarbital (generic, Seconal)
Oral: 100 mg capsules
MISCELLANEOUS DRUGS
Buspirone (generic, BuSpar)
Oral: 5, 7.5, 10, 15, 30 mg tablets
Chloral hydrate (generic, Aquachloral Supprettes)
Oral: 500 mg capsules; 250, 500 mg/5 mL syrups
Rectal: 324, 648 mg suppositories
Eszopiclone (Lunesta)
Oral: 1, 2, 3 mg tablets
Hydroxyzine (generic, Atarax, Vistaril)
Oral: 10, 25, 50, 100 mg tablets; 25, 50, 100 mg capsules; 10 mg/5 mL syrup; 25 mg/5 mL suspension
Parenteral: 25, 50 mg/mL for injection
Meprobamate (generic, Equanil, Miltown)
Oral: 200, 400 mg tablets
Paraldehyde (generic)
Oral, rectal liquids: 1 g/mL
Ramelteon (Rozerem)
Oral: 8 mg tablets
Zaleplon (Sonata)
Oral: 5, 10 mg capsules
Zolpidem (Ambien, Ambien-CR)
Oral: 5, 10 mg tablets; 6.25, 12.5 mg extended-release tablets
REFERENCES
Ancoli-Israel S et al: Long-term use of sedative hypnotics in older patients with insomnia. Sleep Med 2005;6:107.
Bateson AN: The benzodiazepine site of the GABA A receptor: An old target with new potential? Sleep Med 2004;5(Suppl 1):S9.
Blednov YA et al: Deletion of the alpha1 or beta2 subunit of GABAA receptors reduces actions of alcohol and other drugs. J Pharmacol Exp Ther 2003;304:30.
Crestani F et al: Molecular targets for the myorelaxant action of diazepam. Mol Pharmacol 2001;59:442.
Drover DR: Comparative pharmacokinetics and pharmacodynamics of short-acting hypnosedatives: Zaleplon, zolpidem and zopiclone. Clin Pharmacokinet 2004;43:227.
Fricchione G: Generalized anxiety disorder. N Engl J Med 2004;351:675.
Gottesmann C: GABA mechanisms and sleep. Neuroscience 2002;111:231.
Hesse LM et al: Clinically important drug interactions with zopiclone, zolpidem and zaleplon. CNS Drugs 2003;17:513.
Israel AG, Kramer JA: Safety of zaleplon in the treatment of insomnia. Ann Pharmacother 2002;36:852.
Kato K et al: Neurochemical properties of ramelteon, a selective MT1/MT2 receptor agonist. Neuropharmacology 2005;48:301.
Kralic JE et al: GABA(A) receptor alpha-1 subunit deletion alters receptor subtype assembly, pharmacological and behavioral responses to benzodiazepines and zolpidem.Neuropharmacology 2002;43:685.
Krystal AD: The changing perspective of chronic insomnia management. J Clin Psychiatry 2004;65(suppl 8):20.
Mahmood I, Sahalwalla C: Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin Pharmacokinet 1999;36:277.
McKernan RM et al: Anxiolytic-like action of diazepam: Which GABA(A) receptor subtype is involved? Trends Pharmacol Sci 2001;22:402.
Mintzer MZ, Griffiths RR: Triazolam and zolpidem: Effects on human memory and attentional processes. Psychopharmacology (Berl) 1999;144:8.
Mohler H, Fritschy JM, Rudolph U: A new benzodiazepine pharmacology. J Pharmacol Exp Ther 2002;300:2.
Patat A, Paty I, Hindmarch I: Pharmacodynamic profile of Zaleplon, a new non-benzodiazepine hypnotic agent. Hum Psychopharmacol 2001;16:369.
Rickels K, Rynn M: Pharmacotherapy of generalized anxiety disorder. J Clin Psychiatry 2002;63(Suppl 14):9.
Rosenberg R et al: An assessment of the efficacy and safety of eszopiclone in the treatment of transient insomnia in healthy adults. Sleep Med 2005;6:15.
Silber MH: Chronic insomnia. N Engl J Med 2005;353:803.
Turek FW, Gillette MU: Melatonin, sleep and circadian rhythms: Rationale for development of specific melatonin agonists. Sleep Med 2004;5:523.
Verster JC et al: Residual effects of sleep medication on driving ability. Sleep Med Rev 2004;8:309.