INTRODUCTION
Drugs that affect skeletal muscle function as two very different therapeutic groups: those used during surgical procedures and in intensive care units to produce muscle paralysis for patients requiring ventilatory assistance (ie, neuromuscular blockers) and those used to reduce spasticity in a variety of neurologic conditions (ie, spasmolytics). Neuromuscular blocking drugs interfere with transmission at the neuromuscular end plate and lack central nervous system activity. These compounds are used primarily as adjuncts during general anesthesia to facilitate tracheal intubation and optimize surgical conditions while ensuring adequate ventilation. Drugs in the spasmolytic group have traditionally been called "centrally acting" muscle relaxants and are used primarily to treat chronic back pain and fibromyalgic conditions. Dantrolene, a spasmolytic agent that has no significant central effects and is used primarily to treat malignant hyperthermia, is also discussed in this chapter.
NEUROMUSCULAR BLOCKING DRUGS
History
During the 16th century, European explorers found that natives in the Amazon Basin of South America used curare, an arrow poison, to produce death by skeletal muscle paralysis. The active compound, d-tubocurarine, and its modern synthetic derivatives have had a significant influence on the practice of anesthesia and surgery and have proved useful in defining normal neuromuscular physiologic mechanisms.
Normal Neuromuscular Function
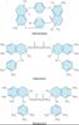
The mechanism of neuromuscular transmission at the motor end plate is similar to that described for preganglionic cholinergic nerves in Chapter 6. With the arrival of an electrical impulse at the motor nerve terminal, there is an influx of calcium and release of acetylcholine. Acetylcholine then diffuses across the synaptic cleft to the nicotinic receptor located on the motor end plate. As noted in Chapter 7, the adult NM receptor is composed of five peptides: two alpha peptides, one beta, one gamma, and one delta peptide (Figure 27-1). The binding of two acetylcholine molecules to receptors on the a-b and d-a subunits causes opening of the channel. The subsequent movement of sodium and potassium is associated with a graded depolarization of the end plate membrane (Figure 27-2). This change in voltage is termed the motor end plate potential. The magnitude of the end plate potential is directly related to the amount of acetylcholine released. If the potential is small, the permeability and the end plate potential return to normal without an impulse being propagated from the end plate region to the rest of the muscle membrane. However, if the end plate potential is large, the adjacent muscle membrane is depolarized, and an action potential will be propagated along the entire muscle fiber. Muscle contraction is then initiated by excitation-contraction coupling. The released acetylcholine is quickly removed from the end plate region by diffusion and enzymatic destruction by the local acetylcholinesterase enzyme.
At least two additional types of acetylcholine receptors are associated with the neuromuscular apparatus. One type is located on the presynaptic motor axon terminal, and activation of these receptors mobilizes additional transmitter for subsequent release by moving more acetylcholine vesicles toward the synaptic membrane. The second type of receptor is found on perijunctional cells and is not normally involved in neuromuscular transmission. However, under certain conditions (eg, prolonged immobilization, thermal burns), these receptors may proliferate sufficiently to affect subsequent neuromuscular transmission.
Skeletal muscle relaxation and paralysis can occur from interruption of function at several sites along the pathway from the central nervous system to myelinated somatic nerves, unmyelinated motor nerve terminals, nicotinic acetylcholine receptors, the motor end plate, the muscle membrane, and the intracellular contractile apparatus itself.
In practice, blockade of end plate function is accomplished by two basic mechanisms. Pharmacologic blockade of the physiologic agonist acetylcholine is characteristic of the antagonist neuromuscular blocking drugs. These drugs prevent access of the transmitter to its receptor and thereby prevent depolarization. The prototype of this nondepolarizing subgroup is d-tubocurarine. The second mechanism of blockade can be produced by an excess of a depolarizing agonist, such as acetylcholine. This paradoxical effect of acetylcholine also occurs at the ganglionic nicotinic acetylcholine receptor. The prototypical depolarizing blocking drug is succinylcholine. A similar depolarizing block can be produced by acetylcholine itself when high local concentrations are achieved in the synaptic cleft (eg, by cholinesterase inhibitor intoxication) and by nicotine and other nicotinic agonists. However, the neuromuscular block produced by these depolarizing drugs (other than succinylcholine) cannot be precisely controlled and is of no clinical value.
|
|
Figure 27-1. The adult nicotinic acetylcholine receptor (nAChR) is an intrinsic membrane protein with five distinct subunits (a2bdg) A: Cartoon of the one of five subunits of the AChR in the end plate surface of adult mammalian muscle. Each subunit contains four helical domains labeled M1 to M4. The M2 domains line the channel pore. B: Cartoon of the full AChR. The N termini of two subunits cooperate to form two distinct binding pockets for acetylcholine (ACh). These pockets occur at the a-b and the d-a subunit interfaces. |
|
|
|
Figure 27-2. Schematic representation of the neuromuscular junction. (ACh, acetylcholine; AChE, acetylcholinesterase; JF, junctional folds; M, mitochondrion; V, transmitter vesicle.) (Reproduced, with permission, from Drachman DB: Myasthenia gravis. N Engl J Med 1978;298:135.) |
|
I. BASIC PHARMACOLOGY OF NEUROMUSCULAR BLOCKING DRUGS
Chemistry
All of the available neuromuscular blocking drugs bear a structural resemblance to acetylcholine. For example, succinylcholine is two acetylcholine molecules linked end-to-end (Figure 27-3). In contrast to the single linear structure of succinylcholine and other depolarizing drugs, the nondepolarizing agents (eg, pancuronium) conceal the "double-acetylcholine" structure in one of two types of bulky, semi-rigid ring systems (Figure 27-3). The two major families of nondepolarizing blocking drugs¾the isoquinoline and steroid derivatives¾are shown in Figures 27-4 and 27-5. Another feature common to all currently used neuromuscular blockers is the presence of one or two quaternary nitrogens, which makes them poorly lipid-soluble and limits entry into the central nervous system.
|
|
Figure 27-3. Structural relationship of succinylcholine, a depolarizing agent, and pancuronium, a nondepolarizing agent, to acetylcholine, the neuromuscular transmitter. Succinylcholine, originally called diacetylcholine, is simply two molecules of acetylcholine linked through the acetate methyl groups. Pancuronium may be viewed as two acetylcholine-like fragments (outlined in color) oriented on a steroid nucleus. |
|
|
Figure 27-4. Structures of some isoquinoline neuromuscular blocking drugs. These agents are all nondepolarizing muscle relaxants. |
|
|
Figure 27-5. Structures of steroid neuromuscular blocking drugs. These agents are all nondepolarizing muscle relaxants. |
Pharmacokinetics of Neuromuscular Blocking Drugs
All of the neuromuscular blocking drugs are highly polar and inactive orally and must be administered parenterally.
A. NONDEPOLARIZING RELAXANT DRUGS
The rate of disappearance of a nondepolarizing neuromuscular blocking drug from the blood is characterized by a rapid initial distribution phase followed by a slower elimination phase. Neuromuscular blocking drugs are highly ionized and do not readily cross cell membranes. Therefore, their volume of distribution is only slightly larger than the blood volume (80-140 mL/kg).
The elimination half-life strongly correlates with the duration of action of nondepolarizing relaxants. Drugs that are excreted by the kidney typically have longer half-lives, leading to longer durations of action (> 60 minutes). Drugs eliminated by the liver tend to have shorter half-lives and durations of action (Table 27-1). All steroidal muscle relaxants are metabolized to their 3-hydroxy, 17-hydroxy, or 3,17-dihydroxy products in the liver. The 3-hydroxy metabolites are usually 40-80% as potent as the parent drug. Under normal circumstances, metabolites are not formed in sufficient quantities to produce a significant degree of neuromuscular blockade during or after anesthesia. However, if the parent compound is administered for several days in the intensive care setting, the 3-hydroxy metabolite may accumulate and cause prolonged paralysis because it has a longer half-life than the parent compound. The remaining metabolites possess minimal neuromuscular blocking properties.
The intermediate-acting steroid muscle relaxants (eg, vecuronium and rocuronium) tend to be more dependent on biliary excretion or hepatic metabolism for their elimination. These muscle relaxants are more commonly used clinically than the long-acting steroid-based drugs (eg, pancuronium, pipecuronium).
Atracurium (Figure 27-4) is an intermediate-acting isoquinoline nondepolarizing muscle relaxant. In addition to hepatic metabolism, atracurium is inactivated by a form of spontaneous breakdown known as Hofmann elimination. The main breakdown products are laudanosine and a related quaternary acid, neither of which possesses neuromuscular blocking properties. Laudanosine is slowly metabolized by the liver and has a long elimination half-life (ie, 150 minutes). It readily crosses the blood-brain barrier, and high blood concentrations may cause seizures and an increase in the volatile anesthetic requirement. During surgical anesthesia, blood levels of laudanosine range from 0.2 to 1 mcg/mL. However, with prolonged infusions of atracurium in the intensive care unit, laudanosine blood levels may exceed 5 mcg/mL.
Atracurium has several stereoisomers, and one of the more potent isomers, cisatracurium, has been approved for clinical use. It resembles atracurium but has even less dependence on hepatic inactivation, forms less laudanosine, and is less likely to release histamine. From the clinical perspective, cisatracurium has all the advantages of atracurium with fewer side effects. Therefore, cisatracurium has largely replaced atracurium in clinical practice.
Mivacurium, another isoquinoline compound, has the shortest duration of action of all nondepolarizing muscle relaxants (Table 27-1). However, its onset of action is significantly slower than that of succinylcholine. In addition, the use of a larger dose to speed the onset can be associated with profound histamine release leading to hypotension, flushing, and bronchospasm. Clearance of mivacurium by plasma cholinesterase is rapid and independent of the liver or kidney (Table 27-1). However, because patients with renal failure often have decreased levels of plasma cholinesterase, the short duration of action of mivacurium may be prolonged in patients with impaired renal function.
B. DEPOLARIZING RELAXANT DRUGS
The extremely short duration of action of succinylcholine (5-10 minutes) is due to its rapid hydrolysis by cholinesterases (eg, butyrylcholinesterase and pseudocholinesterase) in the liver and plasma. Plasma cholinesterase metabolizes succinylcholine more rapidly than mivacurium, and consequently the duration of action of succinylcholine is shorter than that of mivacurium (Table 27-1). The primary metabolite, succinylmonocholine, is rapidly broken down to succinic acid and choline. Because plasma cholinesterase has an enormous capacity to hydrolyze succinylcholine, only a small percentage of the original intravenous dose ever reaches the neuromuscular junction. There is little if any plasma cholinesterase at the motor end plate, and a succinylcholine-induced blockade is terminated by its diffusion away from the end plate into extracellular fluid. Therefore, circulating levels of plasma cholinesterase influence the duration of action of succinylcholine by determining the amount of the drug that reaches the motor end plate.
Neuromuscular blockade produced by succinylcholine and mivacurium can be prolonged in patients with a genetically abnormal variant of plasma cholinesterase. The dibucaine number is a measure of the ability of a patient to metabolize succinylcholine and can be used to identify at-risk patients. Under standardized test conditions, dibucaine inhibits the normal enzyme by 80% and the abnormal enzyme by only 20%. Many genetic variants of plasma cholinesterase have been identified, though the dibucaine-related variants are the most important. Given the rarity of these genetic variants, plasma cholinesterase testing is not a routine clinical procedure.
Mechanism of Action
The interactions of drugs with the acetylcholine receptor-end plate channel have been described at the molecular level. Several modes of action of drugs on the receptor are illustrated in Figure 27-6.
A. NONDEPOLARIZING RELAXANT DRUGS
All the neuromuscular blocking drugs in use in the USA except succinylcholine are classified as nondepolarizing agents. Tubocurarine is considered the prototype neuromuscular blocker. When small doses of nondepolarizing muscle relaxants are administered, they act predominantly at the nicotinic receptor site by competing with acetylcholine. The least potent nondepolarizing relaxants (eg, rocuronium) have the fastest onset and the shortest duration of action. In larger doses, nondepolarizing drugs can enter the pore of the ion channel (Figure 27-1) to cause a more intense motor blockade. This action further weakens neuromuscular transmission and diminishes the ability of acetylcholinesterase inhibitors (eg, neostigmine, edrophonium, pyridostigmine) to antagonize the effect of nondepolarizing muscle relaxants.
Nondepolarizing relaxants can also block prejunctional sodium channels. As a result of this action, muscle relaxants interfere with the mobilization of acetylcholine at the nerve ending. One consequence of the surmountable nature of the postsynaptic blockade produced by nondepolarizing muscle relaxants is the fact that tetanic stimulation, by releasing a large quantity of acetylcholine, is followed by transient posttetanic facilitation (ie, relief of blockade) of the twitch strength. An important clinical consequence of the same principle is the reversal of residual blockade by cholinesterase inhibitors. The characteristics of a nondepolarizing neuromuscular blockade are summarized in Table 27-2 and Figure 27-7).
B. DEPOLARIZING RELAXANT DRUGS
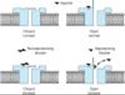
1. Phase I block (depolarizing)¾ Succinylcholine is the only clinically useful depolarizing blocking drug. Its neuromuscular effects are like those of acetylcholine except that succinylcholine produces a longer effect at the myoneural junction. Succinylcholine reacts with the nicotinic receptor to open the channel and cause depolarization of the motor end plate, and this in turn spreads to the adjacent membranes, causing contractions of muscle motor units. Data from single-channel recordings indicate that depolarizing blockers can enter the channel to produce a prolonged "flickering" of the ion conductance (Figure 27-8). Because succinylcholine is not metabolized effectively at the synapse, the depolarized membranes remain depolarized and unresponsive to subsequent impulses (ie, in a state of depolarizing block). Furthermore, because excitation-contraction coupling requires end plate repolarization ("repriming") and repetitive firing to maintain muscle tension, a flaccid paralysis results. This so-called phase I (depolarizing) block is thus augmented, not reversed, by cholinesterase inhibitors.
The characteristics of a depolarizing neuromuscular blockade are summarized in Table 27-2 and Figure 27-7.
2. Phase II block (desensitizing)¾ With continued exposure to succinylcholine, the initial end plate depolarization decreases and the membrane becomes repolarized. Despite this repolarization, the membrane cannot easily be depolarized again because it is desensitized. The mechanism for this desensitizing phase is unclear, but some evidence indicates that channel block may become more important than agonist action at the receptor in phase II of succinylcholine's neuromuscular blocking action. Regardless of the mechanism, the channels behave as if they are in a prolonged closed state (Figure 27-7). Later in phase II, the characteristics of the blockade are nearly identical to those of a nondepolarizing block (ie, a nonsustained twitch response to a tetanic stimulus) (Figure 27-7), with reversal by acetylcholinesterase inhibitors.
|
|
Figure 27-6. Schematic diagram of the interactions of drugs with the acetylcholine receptor on the end plate channel (structures are purely symbolic). Top: The action of the normal agonist, acetylcholine, in opening the channel. Bottom, left: A nondepolarizing blocker, eg, rocuronium, is shown as preventing the opening of the channel when it binds to the receptor. Right: A depolarizing blocker, eg, succinylcholine, both occupying the receptor and blocking the channel. Normal closure of the channel gate is prevented and the blocker may move rapidly in and out of the pore. Depolarizing blockers may desensitize the end plate by occupying the receptor and causing persistent depolarization. An additional effect of drugs on the end plate channel may occur through changes in the lipid environment surrounding the channel (not shown). General anesthetics and alcohols may impair neuromuscular transmission by this mechanism. |
|
|
Figure 27-7. Muscle responses to different patterns of nerve stimulation used in monitoring skeletal muscle relaxation. The alterations produced by a nondepolarizing blocker and depolarizing and desensitizing blockade by succinylcholine are shown. In the train of four (TOF) pattern, four stimuli are applied at 2 Hz. The TOF ratio (TOF-R) is calculated from the strength of the fourth contraction divided by that of the first. In the double burst pattern, three stimuli are applied at 50 Hz, followed by a 700 ms rest period and then repeated. In the posttetanic potentiation pattern, several seconds of 50 Hz stimulation are applied, followed by several seconds of rest and then by single stimuli at a slow rate (eg, 0.5 Hz). The number of detectable posttetanic twitches is the posttetanic count (PTC). (*, first posttetanic contraction.) |
|
|
Figure 27-8. Action of succinylcholine on single-channel end plate receptor currents in frog muscle. Currents through a single AchR channel were recorded using the patch clamp technique. The upper trace was recorded in the presence of a low concentration of succinylcholine; the downward deflections represent two openings of the channel and passage of inward (depolarizing) current. The lower trace was recorded in the presence of a much higher concentration of succinylcholine and shows prolonged "flickering" of the channel as it repetitively opens and closes or is "plugged" by the drug. (Reproduced, with permission, from Marshall CG, Ogden DC, Colquhoun D: The actions of suxamethonium (succinyldicholine) as an agonist and channel blocker at the nicotinic receptor of frog muscle. J Physiol [Lond] 1990;428:155.) |
II. CLINICAL PHARMACOLOGY OF NEUROMUSCULAR BLOCKING DRUGS
Skeletal Muscle Paralysis
Before the introduction of neuromuscular blocking drugs, profound skeletal muscle relaxation for intracavitary operations could be achieved only by producing deep levels of anesthesia that are accompanied by profound depressant effects on the cardiovascular and respiratory systems. The adjunctive use of neuromuscular blocking drugs makes it possible to achieve adequate muscle relaxation for all types of surgical procedures without the cardiorespiratory depressant effects produced by deep anesthesia.
Assessment of Neuromuscular Transmission
Monitoring the effect of muscle relaxants during surgery (and recovery following the administration of cholinesterase inhibitors) typically involves the use of a device that produces transdermal electrical stimulation of one of the peripheral nerves to the hand (or face) and recording of the evoked contractions (ie, twitches). The motor responses to different patterns of peripheral nerve stimulation are measured (Figure 27-7). The three most commonly used patterns include (1) single-twitch stimulation, (2) train-of-four (TOF) stimulation, and (3) tetanic stimulation. Two newer modalities are also available to monitor neuromuscular transmission: double-burst stimulation and posttetanic count.
With single-twitch stimulation, a single supramaximal electrical stimulus is applied to a peripheral nerve at frequencies from 0.1 Hz to 1.0 Hz. The higher frequency is often used during induction and reversal to more accurately determine the peak (maximal) drug effect. TOF stimulation involves four successive supramaximal stimuli given at intervals of 0.5 second (2 Hz). Each stimulus in the TOF causes the muscle to contract, and the relative magnitude of the response of the fourth twitch compared with the first twitch is the TOF ratio. With a depolarizing block, all four twitches are reduced in a dose-related fashion. With a nondepolarizing block, the TOF ratio decreases ("fades") and is inversely proportional to the degree of blockade. During recovery from nondepolarizing block, the amount of fade decreases and the TOF ratio approaches 1.0. Fade in the TOF response after administration of succinylcholine signifies the development of a phase II block.
Tetanic stimulation consists of very rapid (30-100 Hz) delivery of electrical stimuli for several seconds. During a nondepolarizing block (and a phase II block after succinylcholine), the response is not sustained and fade is observed. Fade in response to tetanic stimulation is normally considered a presynaptic event. However, the degree of fade depends primarily on the degree of neuromuscular blockade. During a partial nondepolarizing blockade, tetanic nerve stimulation is followed by an increase in the posttetanic twitch response, so-called posttetanic facilitation of neuromuscular transmission. During intense neuromuscular blockade, there is no response to either tetanic or posttetanic stimulation. As the intensity of the block diminishes, the response to posttetanic twitch stimulation reappears. The time to reappearance of the first response to TOF stimulation is related to the posttetanic count.
The double-burst stimulation pattern is a newer mode of electrical nerve stimulation developed with the goal of allowing for manual detection of residual neuromuscular blockade when it is not possible to record the responses to single-twitch, TOF, or tetanic stimulation. In this pattern, three nerve stimuli are delivered at 50 Hz followed by a 700 ms rest period and then, by two or three additional stimuli at 50 Hz. It is easier to detect fade in the responses to double-burst stimulation than to TOF stimulation. The absence of fade in response to double-burst stimulation implies that clinically significant residual neuromuscular blockade does not exist.
A. NONDEPOLARIZING RELAXANT DRUGS
During anesthesia, administration of tubocurarine, 0.1-0.4 mg/kg IV, initially causes motor weakness, followed by the skeletal muscles becoming flaccid and inexcitable to electrical stimulation (Figure 27-9). In general, larger muscles (eg, abdominal, trunk, paraspinous, diaphragm) are more resistant to blockade and recover more rapidly than smaller muscles (eg, facial, foot, hand). The diaphragm is usually the last muscle to be paralyzed. Assuming that ventilation is adequately maintained, no adverse effects occur. When administration of muscle relaxants is discontinued, recovery of muscles usually occurs in reverse order, with the diaphragm regaining function first. The pharmacologic effect of tubocurarine, 0.3 mg/kg IV, usually lasts 45-60 minutes. However, subtle evidence of residual muscle paralysis detected using a neuromuscular monitor may last for another hour.
Potency and duration of action of the other nondepolarizing drugs are shown in Table 27-1. In addition to the duration of action, the most important property distinguishing the nondepolarizing relaxants is the time to onset of effect, which determines how rapidly the patient's trachea can be intubated. Of the currently available nondepolarizing drugs, rocuronium (60-120 seconds) is the most rapid.
B. DEPOLARIZING RELAXANT DRUGS
Following the administration of succinylcholine, 0.75-1.5 mg/kg IV, transient muscle fasciculations occur over the chest and abdomen within 30 seconds, although general anesthesia tends to attenuate them. As paralysis develops rapidly (< 90 seconds), the arm, neck, and leg muscles are initially relaxed followed by the respiratory muscles. As a result of succinylcholine's rapid hydrolysis by cholinesterase in the plasma and liver, the duration of neuromuscular block typically lasts less than 10 minutes (Table 27-1).
|
|
Figure 27-9. Neuromuscular blockade from tubocurarine during equivalent levels of isoflurane and halothane anesthesia in patients. Note that isoflurane augments the block far more than does halothane. |
Cardiovascular Effects
Vecuronium, pipecuronium, doxacurium, cisatracurium, and rocuronium all have minimal, if any, cardiovascular effects. The other nondepolarizing muscle relaxants (ie, pancuronium, atracurium, mivacurium) produce cardiovascular effects that are mediated by either autonomic or histamine receptors (Table 27-3). Tubocurarine and, to a lesser extent, metocurine, mivacurium, and atracurium can produce hypotension as a result of systemic histamine release, and with larger doses ganglionic blockade may occur with tubocurarine and metocurine. Premedication with an antihistaminic compound attenuates tubocurarine- and mivacurium-induced hypotension. Pancuronium causes a moderate increase in heart rate and a smaller increase in cardiac output, with little or no change in systemic vascular resistance. Although pancuronium induced tachycardia is primarily due to a vagolytic action, release of norepinephrine from adrenergic nerve endings and blockade of neuronal uptake of norepinephrine may be secondary mechanisms. Although bronchospasm may be produced by neuromuscular blockers that release histamine (eg, mivacurium), insertion of a tracheal tube is the most common reason for bronchospasm after induction of general anesthesia.
Succinylcholine can cause cardiac arrhythmias when administered during halothane anesthesia. The drug stimulates autonomic cholinoceptors, including the nicotinic receptors at both sympathetic and parasympathetic ganglia and muscarinic receptors in the heart (eg, sinus node). The negative inotropic and chronotropic responses to succinylcholine can be attenuated by administration of an anticholinergic drug (eg, glycopyrrolate, atropine). With large doses of succinylcholine, positive inotropic and chronotropic effects may be observed. On the other hand, bradycardia has been repeatedly observed when a second dose of succinylcholine is given less than 5 minutes after the initial dose. This transient bradycardia can be prevented by thiopental, atropine, ganglionic-blocking drugs, and even nondepolarizing muscle relaxants (eg, pancuronium). Direct myocardial effects, increased muscarinic stimulation, and ganglionic stimulation contribute to this bradycardic response.
Other Adverse Effects of Depolarizing Blockade
A. HYPERKALEMIA
Patients with burns, nerve damage or neuromuscular disease, closed head injury, and other trauma can respond to succinylcholine by releasing potassium into the blood, which, on rare occasions, results in cardiac arrest.
B. INCREASED INTRAOCULAR PRESSURE
Administration of succinylcholine may be associated with the rapid onset of an increase in intraocular pressure (< 60 seconds), peaking at 2-4 minutes, and declining after 5 minutes. The mechanism may involve tonic contraction of myofibrils or transient dilation of ocular choroidal blood vessels. Despite the increase in intraocular pressure, the use of succinylcholine for ophthalmologic operations is not contraindicated unless the anterior chamber is open ("open globe") due to trauma.
C. INCREASED INTRAGASTRIC PRESSURE
In heavily muscled patients, the fasciculations associated with succinylcholine may cause an increase in intragastric pressure ranging from 5 to 40 cm H2O, increasing the risk for regurgitation and aspiration of gastric contents. This complication is more likely to occur in patients with delayed gastric emptying (eg, those with diabetes), traumatic injury, esophageal dysfunction, and morbid obesity.
D. MUSCLE PAIN
Myalgias are a common postoperative complaint of heavily muscled patients and those who receive large doses of succinylcholine. The true incidence of myalgias related to muscle fasciculations is difficult to establish because of confounding factors, including the type of surgery and positioning during the operation. However, the incidence of myalgias has been reported to vary from less than 1% to 20%. It occurs more frequently in ambulatory than in bedridden patients. The pain is thought to be secondary to the unsynchronized contractions of adjacent muscle fibers just before the onset of paralysis. However, there is controversy over whether the incidence of muscle pain following succinylcholine is actually higher than with nondepolarizing muscle relaxants when other potentially confounding factors are taken into consideration.
Interactions with Other Drugs
A. ANESTHETICS
Inhaled (volatile) anesthetics potentiate the neuromuscular blockade produced by nondepolarizing muscle relaxants in a dose-dependent fashion. Of the general anesthetics that have been studied, volatile anesthetics augment the effects of muscle relaxants in the following order: isoflurane (most); sevoflurane, desflurane, enflurane, and halothane; and nitrous oxide (least) (Figure 27-9). The most important factors involved in this interaction are the following: (1) nervous system depression at sites proximal to the neuromuscular junction (ie, central nervous system); (2) increased muscle blood flow (ie, due to peripheral vasodilation produced by volatile anesthetics), which allows a larger fraction of the injected muscle relaxant to reach the neuromuscular junction; and (3) decreased sensitivity of the postjunctional membrane to depolarization.
A rare interatction of succinylcholine with volatile anesthetics results in malignant hyperthermia, a condition caused by abnormal release of calcium from stores in skeletal muscle. This condition is treated with dantrolene and is discussed below under Spasmolytic Drugs and in Chapter 16.
B. ANTIBIOTICS
Numerous reports have described enhancement of neuromuscular blockade by antibiotics (eg, aminoglycosides). Many of the antibiotics have been shown to cause a depression of evoked release of acetylcholine similar to that caused by magnesium. The mechanism of this prejunctional effect appears to be blockade of specific P-type calcium channels.
C. LOCAL ANESTHETICS AND ANTIARRHYTHMIC DRUGS
In small doses, local anesthetics depress posttetanic potentiation via a prejunctional neural effect. In large doses, local anesthetics can block neuromuscular transmission. With higher doses, local anesthetics block acetylcholine-induced muscle contractions as a result of blockade of the nicotinic receptor ion channels. Experimentally, similar effects can be demonstrated with sodium channel-blocking antiarrhythmic drugs such as quinidine. However, at the doses used for cardiac arrhythmias, this interaction is of little or no clinical significance. Higher concentrations of bupivacaine (0.75%) have been associated with cardiac arrhythmias independent of the muscle relaxant used.
D. OTHER NEUROMUSCULAR BLOCKING DRUGS
The end plate-depolarizing effect of succinylcholine can be antagonized by administering a small dose of a nondepolarizing blocker. To prevent the fasciculations associated with succinylcholine administration, a small nonparalyzing dose of a nondepolarizing drug can be given before succinylcholine (eg, d-tubocurarine 2 mg IV or pancuronium 0.5 mg IV). Although this dose usually reduces fasciculations and postoperative myalgias, it can increase the amount of succinylcholine required for relaxation by 50-90% and can produce a feeling of weakness in awake patients. Therefore, "pre-curarization" before succinylcholine is no longer widely practiced.
Effects of Diseases & Aging on the Neuromuscular Response
Several diseases can diminish or augment the neuromuscular blockade produced by nondepolarizing muscle relaxants. Myasthenia gravis enhances the neuromuscular blockade produced by these drugs. Advanced age is associated with a prolonged duration of action from nondepolarizing relaxants as a result of decreased clearance of the drugs by the liver and kidneys. As a result, the dosage of neuromuscular blocking drugs should be reduced in elderly patients (> 70 years).
Conversely, patients with severe burns and those with upper motor neuron disease are resistant to nondepolarizing muscle relaxants. This desensitization is probably caused by proliferation of extrajunctional receptors, which results in an increased dose requirement for the nondepolarizing relaxant to block a sufficient number of receptors.
Reversal of Nondepolarizing Neuromuscular Blockade
The cholinesterase inhibitors effectively antagonize the neuromuscular blockade caused by nondepolarizing drugs. Their general pharmacology is discussed in Chapter 7. Neostigmine and pyridostigmine antagonize nondepolarizing neuromuscular blockade by increasing the availability of acetylcholine at the motor end plate, mainly by inhibition of acetylcholinesterase. To a lesser extent, these cholinesterase inhibitors also increase release of transmitter from the motor nerve terminal. In contrast, edrophonium antagonizes neuromuscular blockade purely by inhibiting acetylcholinesterase. Edrophonium may be less effective than neostigmine in reversing the effects of nondepolarizing blockers in the presence of a profound degree of neuromuscular blockade. These differences are important in determining recovery from residual block, the neuromuscular blockade remaining after completion of surgery and movement of the patient to the recovery room. Unsuspected residual block may result in hypoventilation, leading to hypoxia and even apnea, especially if patients receive central depressant medications during the early recovery period.
Since mivacurium is metabolized by plasma cholinesterase, the interaction with the reversal drugs is less predictable. On the one hand, the neuromuscular blockade is antagonized because of increased acetylcholine concentrations in the synapse. On the other hand, mivacurium concentration may be higher because of decreased plasma cholinesterase breakdown of the muscle relaxant.
A novel cyclodextrin reversal drug, sugammadex, has been recently introduced. It can rapidly inactivate steroidal neuromuscular blocking drugs by forming an inactive complex, which is excreted in the urine. This chelation process allows the practitioner to rapidly reverse even profound degrees of neuromuscular blockade at the end of surgery.
Uses of Neuromuscular Blocking Drugs
A. SURGICAL RELAXATION
By far the most important application of the neuromuscular blockers is in facilitating intracavitary surgery. This is especially important in intra-abdominal and intrathoracic procedures.
B. TRACHEAL INTUBATION
By relaxing the pharyngeal and laryngeal muscles, neuromuscular blocking drugs facilitate laryngoscopy and placement of the tracheal tube. Placement of a tracheal tube ensures an adequate airway and minimizes the risk of pulmonary aspiration during general anesthesia.
C. CONTROL OF VENTILATION
In critically ill patients who have ventilatory failure from various causes (eg, severe bronchospasm, pneumonia, chronic obstructive airway disease), it may be necessary to control ventilation to provide adequate gas exchange and to prevent atelectasis. Muscle paralysis is produced by neuromuscular blocking drugs to reduce chest wall resistance (ie, improve thoracic compliance) and ineffective spontaneous ventilation.
D. TREATMENT OF CONVULSIONS
Neuromuscular blocking drugs are occasionally used to attenuate the peripheral manifestations of convulsions associated with status epilepticus or local anesthetic toxicity. Although this approach is effective in eliminating the muscular manifestations of the seizures, it has no effect on the central processes because neuromuscular blocking drugs do not cross the blood-brain barrier.
SPASMOLYTIC DRUGS
INTRODUCTION
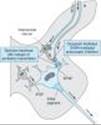
Spasticity is characterized by an increase in tonic stretch reflexes and flexor muscle spasms (ie, increased basal muscle tone) together with muscle weakness. It is often associated with cerebral palsy, multiple sclerosis, and stroke. These conditions often involve abnormal function of the bowel and bladder as well as skeletal muscle. The mechanisms underlying clinical spasticity appear to involve not only the stretch reflex arc itself but also higher centers in the central nervous system (ie, upper motor neuron lesion), with damage to descending pathways in the spinal cord resulting in hyperexcitability of the alpha motoneurons in the cord. Pharmacologic therapy may ameliorate some of the symptoms of spasticity by modifying the stretch reflex arc or by interfering directly with skeletal muscle (ie, excitation-contraction coupling). The important components involved in these processes are shown in Figure 27-10.
Drugs that modify this reflex arc may modulate excitatory or inhibitory synapses (see Chapter 21). Thus, to reduce the hyperactive stretch reflex, it is desirable to reduce the activity of the Ia fibers that excite the primary motoneuron or to enhance the activity of the inhibitory internuncial neurons. These structures are shown in greater detail in Figure 27-11.
A variety of pharmacologic agents described as depressants of the spinal "polysynaptic" reflex arc (eg, barbiturates [phenobarbital] and glycerol ethers [mephenesin]) have been used to treat these conditions of excess skeletal muscle tone. However, as illustrated in Figure 27-11, nonspecific depression of synapses involved in the stretch reflex could reduce the desired inhibitory activity, as well as the excitatory transmission. Currently available drugs can provide significant relief from painful muscle spasms, but they are less effective in improving meaningful function (eg, mobility and return to work).
|
|
Figure 27-10. Diagram of the structures involved in the stretch reflex arc. I is an inhibitory interneuron; E indicates an excitatory presynaptic terminal; Ia is a primary intrafusal afferent fiber; Ca2+ denotes activator calcium stored in the sarcoplasmic reticulum of skeletal muscle. (Reproduced, with permission, from Young RR, Delwaide PJ: Drug therapy: Spasticity. N Engl J Med 1981;304:28.) |
|
|
|
Figure 27-11. Postulated sites of spasmolytic action of diazepam and baclofen in the spinal cord. (Reproduced, with permission, from Young RR, Delwaide PJ: Drug therapy: Spasticity. N Engl J Med 1981;304:28.) |
|
DIAZEPAM
As described in Chapter 22, benzodiazepines facilitate the action of g-aminobutyric acid (GABA) in the central nervous system. Diazepam acts at GABAA synapses, and its action in reducing spasticity is at least partly mediated in the spinal cord because it is somewhat effective in patients with cord transection. Although diazepam can be used in patients with muscle spasm of almost any origin (including local muscle trauma), it produces sedation at the doses required to reduce muscle tone. The initial dosage is 4 mg/d, and it is gradually increased to a maximum of 60 mg/d. Other benzodiazepines have been used as spasmolytics (eg, midazolam), but clinical experience with them is limited.
BACLOFEN
Baclofen (p-chlorophenyl-GABA) was designed to be an orally active GABA-mimetic agent.
Baclofen exerts its spasmolytic activity at GABAB receptors. Activation of these receptors by baclofen results in hyperpolarization, probably by increased K+ conductance. It has been suggested that hyperpolarization causes presynaptic inhibition by reducing calcium influx (Figure 27-11) and reduces the release of excitatory transmitters in both the brain and the spinal cord. Baclofen may also reduce pain in patients with spasticity, perhaps by inhibiting the release of substance P (neurokinin-1) in the spinal cord.
Baclofen is at least as effective as diazepam in reducing spasticity while producing less sedation. In addition, baclofen does not reduce overall muscle strength as much as dantrolene. It is rapidly and completely absorbed after oral administration and has a plasma half-life of 3-4 hours. Dosage is started at 15 mg twice daily, increasing as tolerated to 100 mg daily. Adverse effects of this drug include drowsiness; however, patients become tolerant to the sedative effect with chronic administration. Increased seizure activity has been reported in epileptic patients. Therefore, withdrawal from baclofen must be done very slowly.
Studies have confirmed that intrathecal administration of baclofen can control severe spasticity and muscle pain that is not responsive to medication by other routes of administration. Owing to the poor egress of baclofen from the spinal cord, peripheral symptoms are rare. Therefore, higher central concentrations of the drug may be tolerated. Partial tolerance to the effect of the drug may occur after several months of therapy, but can be overcome by upward dosage adjustments to maintain the beneficial effect. Excessive somnolence, respiratory depression, and even coma have been described. Although a major disadvantage of this therapeutic approach is the difficulty of maintaining the drug delivery catheter in the subarachnoid space, long-term intrathecal baclofen therapy can improve the quality of life for patients with severe spastic disorders.
Oral baclofen has been studied in several other medical conditions. Preliminary studies suggest that it may be effective in reducing "craving" in recovering alcoholics (see Chapter 32). It has also been found to be effective in preventing migraine headaches in some patients.
TIZANIDINE
As noted in Chapter 11, a agonists such as clonidine and other imidazoline compounds have a variety of effects on the central nervous system that are not fully understood. Among these effects is the ability to reduce muscle spasm. Tizanidine is a congener of clonidine that has been studied for its spasmolytic actions. Tizanidine has significant a2-adrenoceptor agonist effects, but it reduces spasticity in experimental models at doses that cause fewer cardiovascular effects than clonidine. Neurophysiologic studies in animals and humans suggest that tizanidine reinforces both presynaptic and postsynaptic inhibition in the cord. It also inhibits nociceptive transmission in the spinal dorsal horn.
Clinical trials with oral tizanidine report comparable efficacy in relieving muscle spasm to diazepam, baclofen, and dantrolene. However, tizanidine produces a different spectrum of adverse effects, including drowsiness, hypotension, dry mouth, and asthenia. The dosage requirements vary markedly among patients, suggesting that individual dosage titration is necessary to achieve an optimal clinical effect.
OTHER CENTRALLY ACTING SPASMOLYTIC DRUGS
Gabapentin is an antiepileptic drug (see Chapter 24) that has shown considerable promise as a spasmolytic agent in several studies involving patients with multiple sclerosis. Pregabalin is a new analog of gabapentin that may also prove useful. Progabide and glycine have also been found in preliminary studies to reduce spasticity. Progabide is a GABAA and GABABagonist and has active metabolites, including GABA itself. Glycine is another inhibitory amino acid neurotransmitter (see Chapter 21). It appears to possess pharmacologic activity when given orally and readily passes the blood-brain barrier. Idrocilamide and riluzole are newer drugs for the treatment of amyotrophic lateral sclerosis that appear to have spasm-reducing effects, possibly through inhibition of glutamatergic transmission in the central nervous system.
DANTROLENE
Dantrolene is a hydantoin derivative related to phenytoin that has a unique mechanism of spasmolytic activity. In contrast to the centrally acting drugs, dantrolene reduces skeletal muscle strength by interfering with excitation-contraction coupling in the muscle fibers. The normal contractile response involves release of calcium from its stores in the sarcoplasmic reticulum (see Figures 13-2 and 27-10). This activator calcium brings about the tension-generating interaction of actin with myosin. Calcium is released from the sarcoplasmic reticulum via a calcium channel, called the ryanodine receptor (RyR) channel because the plant alkaloid ryanodine combines with a receptor on the channel protein. In the case of the skeletal muscle RyR channel, ryanodine facilitates the open configuration.
Dantrolene interferes with the release of activator calcium through this sarcoplasmic reticulum calcium channel by binding to the RyR and blocking the opening of the channel. Motor units that contract rapidly are more sensitive to the drug's effects than are slower-responding units. Cardiac muscle and smooth muscle are minimally depressed because the release of calcium from their sarcoplasmic reticulum involves a different RyR channel.
Treatment with dantrolene is usually initiated with 25 mg daily as a single dose, increasing to a maximum of 100 mg four times daily as tolerated. Only about one third of an oral dose of dantrolene is absorbed, and the elimination half-life of the drug is approximately 8 hours. Major adverse effects are generalized muscle weakness, sedation, and occasionally hepatitis.
A special application of dantrolene is in the treatment of malignant hyperthermia, a rare heritable disorder that can be triggered by a variety of stimuli, including general anesthetics (eg, volatile anesthetics) and neuromuscular blocking drugs (eg, succinylcholine; see also Chapter 16). Patients at risk for this condition have a hereditary impairment in the ability of the sarcoplasmic reticulum to sequester calcium (Figure 27-10). After administration of one of the triggering agents, there is a sudden and prolonged release of calcium, with massive muscle contraction, lactic acid production, and increased body temperature. Prompt treatment is essential to control acidosis and body temperature and to reduce calcium release. The latter is accomplished with intravenous dantrolene, starting with a dose of 1 mg/kg IV, and repeating as necessary to a maximum dose of 10 mg/kg.
|
|
Figure 13-2. Some compensatory responses that occur during congestive heart failure. In addition to the effects shown, angiotensin II increases sympathetic effects by facilitating norepinephrine release. |
BOTULINUM TOXIN
The therapeutic use of botulinum toxin for ophthalmic purposes and for local muscle spasm was mentioned in Chapter 6. Local facial injections of botulinum toxin are widely used for the short-term treatment (1-3 months per treatment) of wrinkles around the eyes and mouth. Local injection of botulinum toxin has also become a useful treatment for generalized spastic disorders (eg, cerebral palsy). Most clinical studies to date have involved administration in one or two limbs, and the benefits appear to persist for weeks to several months after a single treatment. Most studies have used type A botulinum toxin, but type B is also available.
DRUGS USED TO TREAT ACUTE LOCAL MUSCLE SPASM
A large number of drugs (eg, carisoprodol, chlorphenesin, chlorzoxazone, cyclobenzaprine, metaxalone, methocarbamol, and orphenadrine) are promoted for the relief of acute muscle spasm caused by local tissue trauma or muscle strains. It has been suggested that these drugs act primarily at the level of the brainstem. Cyclobenzaprine may be regarded as the prototype of the group. Cyclobenzaprine is structurally related to the tricyclic antidepressants and possesses antimuscarinic effects. It is ineffective in treating muscle spasm due to cerebral palsy or spinal cord injury. The drug has strong antimuscarinic actions and may cause significant sedation, as well as confusion and transient visual hallucinations. The dosage of cyclobenzaprine for acute injury-related muscle spasm is 20-40 mg/d in divided doses.
PREPARATIONS AVAILABLE
NEUROMUSCULAR BLOCKING DRUGS
Atracurium (Tracrium)
Parenteral: 10 mg/mL for injection
Cisatracurium (Nimbex)
Parenteral: 2, 10 mg/mL for IV injection
Doxacurium (Nuromax)
Parenteral: 1 mg/mL for IV injection
Metocurine (generic, Metubine Iodide)
Parenteral: 2 mg/mL for injection
Mivacurium (Mivacron)
Parenteral: 0.5, 2 mg/mL for injection
Pancuronium (generic)
Parenteral: 1, 2 mg/mL for injection
Pipecuronium (Arduan)
Parenteral: powder for 1 mg/mL for IV injection
Rocuronium (Zemuron)
Parenteral: 10 mg/mL for IV injection
Succinylcholine (generic, Anectine)
Parenteral: 20, 50, 100 mg/mL for injection; 500, 1000 mg per vial powders to reconstitute for injection
Tubocurarine (generic)
Parenteral: 3 mg (20 units)/mL for injection
Vecuronium (generic, Norcuron)
Parenteral: 10, 20 mg powder to reconstitute for injection
MUSCLE RELAXANTS (SPASMOLYTICS)
Baclofen (generic, Lioresal)
Oral: 10, 20 mg tablets
Intrathecal: 0.05, 0.5, 2 mg/mL
Botulinum toxin type A (Botox)
Parenteral: Powder for solution, 100 units/vial
Botulinum toxin type B (Myobloc)
Parenteral: 5000 units/mL for injection
Carisoprodol (generic, Soma)
Oral: 350 mg tablets
Chlorphenesin (Maolate)
Oral: 400 mg tablets
Chlorzoxazone (generic, Paraflex)
Oral: 250, 500 mg tablets, caplets
Cyclobenzaprine (generic, Flexeril)
Oral: 10 mg tablets
Dantrolene (Dantrium)
Oral: 25, 50, 100 mg capsules
Parenteral: 20 mg per vial powder to reconstitute for injection
Diazepam (generic, Valium)
Oral: 2, 5, 10 mg tablets; 5 mg/5 mL, 5 mg/mL solutions
Parenteral: 5 mg/mL for injection
Gabapentin (Neurontin)
Oral: 100, 300, 400 mg capsules; 600, 800 mg tablets; 50 mg/mL oral solution
Note: This drug is labeled for use only in epilepsy and postherpetic neuralgia
Metaxalone (Skelaxin)
Oral: 400 mg tablets
Methocarbamol (generic, Robaxin)
Oral: 500, 750 mg tablets
Parenteral: 100 mg/mL for IM, IV injection
Orphenadrine (generic, Norflex)
Oral: 100 mg tablets; 100 mg sustained-release tablets
Parenteral: 30 mg/mL for IM, IV injection
Riluzole (Rilutek)
Oral: 50 mg tablets
Note: This drug is labeled only for use in amyotrophic lateral sclerosis.
Tizanidine (Zanaflex)
Oral: 2, 4 mg tablets, capsules; 6 mg capsules
REFERENCES
Neuromuscular Blockers
Adt M, Baumert JH, Reimann HJ: The role of histamine in the cardiovascular effects of atracurium. Br J Anaesth 1992;68:155.
Atherton DP, Hunter JM: Clinical pharmacokinetics of the newer neuromuscular blocking drugs. Clin Pharmacokinet 1999;36:169.
Gibb AJ, Marshall IG: Pre- and postjunctional effects of tubocurarine and other nicotinic antagonists during repetitive stimulation in the rat. J Physiol 1984;351:275.
Jooste E et al: A mechanism for rapacuronium-induced bronchospasm. M2 muscarinic receptor antagonism. Anesthesiology 2003;98:906.
Kampe S et al: Muscle relaxants. Best Prac Res Clin Anesthesiol 2003;17:137.
Kaplan RF et al: The potency (ED50) and cardiovascular effects of rapacuronium (ORG 9487) during narcotic-nitrous oxide-propofol anesthesia in neonates, infants, and children. Anesth Analg 1999;89:1172.
Kisor DF, Schmith VD: Clinical pharmacokinetics of cisatracurium besilate. Clin Pharmacokinet 1999;36:27.
Krause T et al: Dantrolene¾a review of its pharmacology, therapeutic use and new developments. Anaesthesia 2004;59:364.
Lee C: Structure, conformation, and action of neuromuscular blocking drugs. Br J Anaesth 2001;87:755.
Marshall CG, Ogden DC, Colquhoun D: The actions of suxamethonium (succinyldicholine) as an agonist and channel blocker at the nicotinic receptor of frog muscle. J Physiol (Lond) 1990;428:155.
Meakin GH: Recent advances in myorelaxant therapy. Paed Anaesthesia 2001;11:523.
Moore EW, Hunter JM: The new neuromuscular blocking agents: Do they offer any advantages? Br J Anaesth 2001;87:912.
Naguib M et al: Advances in neurobiology of the neuromuscular junction: Implications for the anesthesiologist. Anesthesiology 2002;96:202.
Savarese JJ et al: Pharmacology of muscle relaxants and their antagonists. In: Miller RD (editor): Anesthesia, 5th ed. Churchill Livingstone, 2000.
Shields M et al: Org 25969 (sugammadex), a selective relaxant binding agent for antagonism of prolonged rocuronium-induced neuromuscular block. Br J Anaesth 2006;96:36.
Viby-Mogensen J: Neuromuscular monitoring. In: Miller RD (editor): Anesthesia, 5th ed. Churchill Livingstone, 2000.
White PF: Rapacuronium: Why did it fail as a replacement for succinylcholine? Br J Anaesth 2002;88:163.
Spasmolytics
Addolorato G et al: Ability of baclofen in reducing alcohol craving and intake: II. Preliminary clinical evidence. Alcohol Clin Exp Res 2000;24:67.
Albright AL, Cervi A, Singletary J: Intrathecal baclofen for spasticity in cerebral palsy. JAMA 1991;265:1418.
Cutter NC et al: Gabapentin effect on spasticity in multiple sclerosis: A placebo-controlled, randomized trial. Arch Phys Med Rehabil 2000;81:164.
Davidoff RA: Antispasticity drugs: Mechanisms of action. Ann Neurol 1985;17:107.
Groves L, Shellenberger MK, Davis CS: Tizanidine treatment of spasticity: A meta-analysis of controlled, double-blind, comparative studies with baclofen and diazepam. Adv Ther 1998;15:241.
Lopez JR et al: Effects of dantrolene on myoplasmic free [Ca2+] measured in vivo in patients susceptible to malignant hyperthermia. Anesthesiology 1992;76:711.
Nolan KW, Cole LL, Liptak GS: Use of botulinum toxin type A in children with cerebral palsy. Phys Ther 2006;86:573.
Verrotti A et al: Pharmacotherapy of spasticity in children with cerebral palsy. Pediatr Neurol 2006;34:1.