INTRODUCTION
Hematopoiesis, the production from undifferentiated stem cells of circulating erythrocytes, platelets, and leukocytes, is a remarkable process that produces over 200 billion new blood cells per day in the normal person and even greater numbers of cells in people with conditions that cause loss or destruction of blood cells. The hematopoietic machinery resides primarily in the bone marrow in adults and requires a constant supply of three essential nutrients¾iron, vitamin B12, and folic acid¾as well as the presence of hematopoietic growth factors, proteins that regulate the proliferation and differentiation of hematopoietic cells. Inadequate supplies of either the essential nutrients or the growth factors result in deficiency of functional blood cells. Anemia, a deficiency in oxygen-carrying erythrocytes, is the most common and easily treated of these conditions, but thrombocytopenia and neutropenia are not rare and some forms are amenable to drug therapy. In this chapter, we first consider treatment of anemia due to deficiency of iron, vitamin B12, or folic acid and then turn to the medical use of hematopoietic growth factors to combat anemia, thrombocytopenia, and neutropenia, and to support stem cell transplantation.
AGENTS USED IN ANEMIAS
IRON
Basic Pharmacology
Iron deficiency is the most common cause of chronic anemia. Like other forms of chronic anemia, iron deficiency anemia leads to pallor, fatigue, dizziness, exertional dyspnea, and other generalized symptoms of tissue hypoxia. The cardiovascular adaptations to chronic anemia¾tachycardia, increased cardiac output, vasodilation¾can worsen the condition of patients with underlying cardiovascular disease.
Iron forms the nucleus of the iron-porphyrin heme ring, which together with globin chains forms hemoglobin. Hemoglobin reversibly binds oxygen and provides the critical mechanism for oxygen delivery from the lungs to other tissues. In the absence of adequate iron, small erythrocytes with insufficient hemoglobin are formed, giving rise to microcytic hypochromic anemia.
Pharmacokinetics
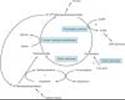
Free inorganic iron is extremely toxic, but iron is required for essential proteins such as hemoglobin; therefore, evolution has provided an elaborate system for regulating iron absorption, transport, and storage (Figure 33-1). The system uses specialized transport and storage proteins whose concentrations are controlled by the body's demand for hemoglobin synthesis and adequate iron stores (Table 33-1). Nearly all of the iron used to support hematopoiesis is reclaimed from catalysis of the hemoglobin in senescent or damaged erythrocytes. Normally, only a small amount of iron is lost from the body each day, so dietary requirements are small and easily fulfilled by the iron available in a wide variety of foods. However, in special populations with either increased iron requirements (eg, growing children, pregnant women) or increased losses of iron (eg, menstruating women), iron requirements can exceed normal dietary supplies and iron deficiency can develop.
A. ABSORPTION
The average diet in the USA contains 10-15 mg of elemental iron daily. A normal individual absorbs 5-10% of this iron, or about 0.5-1 mg daily. Iron is normally absorbed in the duodenum and proximal jejunum, although the more distal small intestine can absorb iron if necessary. Iron absorption increases in response to low iron stores or increased iron requirements. Total iron absorption increases to 1-2 mg/d in normal menstruating women and may be as high as 3-4 mg/d in pregnant women.
Iron is available in a wide variety of foods but is especially abundant in meat. The iron in meat protein can be efficiently absorbed, because heme iron in meat hemoglobin and myoglobin can be absorbed intact without first having to be dissociated into elemental iron (Figure 33-1). Iron in other foods, especially vegetables and grains, is often tightly bound to organic compounds and is much less available for absorption. Nonheme iron in foods and iron in inorganic iron salts and complexes must be reduced to ferrous iron (Fe2+) before it can be absorbed by intestinal mucosal cells.
Iron crosses the luminal membrane of the intestinal mucosal cell by two mechanisms: active transport of ferrous iron and absorption of iron complexed with heme (Figure 33-1). The divalent metal transporter, DMT1, efficiently transports ferrous iron across the luminal membrane of the intestinal enterocyte. The rate of iron uptake is regulated by mucosal cell iron stores such that more iron is transported when stores are low. Together with iron split from absorbed heme, the newly absorbed iron can be actively transported into the blood across the basolateral membrane, probably by the transporter IREG1, also known as ferroportin1. Other proteins are involved in this process, and some of them are regulated to control iron absorption and storage. Excess iron can be stored in the mucosal cell as ferritin, a water-soluble complex consisting of a core of ferric hydroxide covered by a shell of a specialized storage protein called apoferritin. In general, when total body iron stores are high and iron requirements by the body are low, newly absorbed iron is diverted into ferritin in the intestinal mucosal cells. When iron stores are low or iron requirements are high, newly absorbed iron is immediately transported from the mucosal cells to the bone marrow to support hemoglobin production.
B. TRANSPORT
Iron is transported in the plasma bound to transferrin, a b-globulin that specifically binds two molecules of ferrous iron (Figure 33-1). The transferrin-iron complex enters maturing erythroid cells by a specific receptor mechanism. Transferrin receptors¾integral membrane glycoproteins present in large numbers on proliferating erythroid cells¾bind and internalize the transferrin-iron complex through the process of receptor-mediated endocytosis. In endosomes, the iron is released and funneled into hemoglobin synthesis, whereas the transferrin-transferrin receptor complex is recycled to the plasma membrane, where the transferrin dissociates and returns to the plasma. This process provides an efficient mechanism for supplying the iron required by developing red blood cells.
Increased erythropoiesis is associated with an increase in the number of transferrin receptors on developing erythroid cells. Iron store depletion and iron deficiency anemia are associated with an increased concentration of serum transferrin.
C. STORAGE
In addition to the storage of iron in intestinal mucosal cells, iron is also stored, primarily as ferritin, in macrophages in the liver, spleen, and bone, and in parenchymal liver cells (Figure 33-1). Apoferritin synthesis is regulated by the levels of free iron. When these levels are low, apoferritin synthesis is inhibited and the balance of iron binding shifts toward transferrin. When free iron levels are high, more apoferritin is produced to sequester more iron and protect organs from the toxic effects of excess free iron.
Ferritin is detectable in serum. Since the ferritin present in serum is in equilibrium with storage ferritin in reticuloendothelial tissues, the serum ferritin level can be used to estimate total body iron stores.
D. ELIMINATION
There is no mechanism for excretion of iron. Small amounts are lost in the feces by exfoliation of intestinal mucosal cells, and trace amounts are excreted in bile, urine, and sweat. These losses account for no more than 1 mg of iron per day. Because the body's ability to excrete iron is so limited, regulation of iron balance must be achieved by changing intestinal absorption and storage of iron, in response to the body's needs. As noted below, impaired regulation of iron absorption leads to serious pathology.
|
|
Figure 33-1. Absorption, transport, and storage of iron. Intestinal epithelial cells actively absorb inorganic iron and heme iron (H). Ferrous iron that is absorbed or released from absorbed heme iron in the intestine (1) is actively transported into the blood or complexed with apoferritin (AF) and stored as ferritin. In the blood, iron is transported by transferrin (Tf) to erythroid precursors in the bone marrow for synthesis of hemoglobin (Hgb) (2) or to hepatocytes for storage as ferritin (3). The transferrin-iron complexes bind to transferrin receptors (TfR) in erythroid precursors and hepatocytes and are internalized. After release of the iron, the TfR-Tf complex is recycled to the plasma membrane and Tf is released. Macrophages that phagocytize senescent erythrocytes (RBC) reclaim the iron from the RBC hemoglobin and either export it or store it as ferritin (4). Hepatocytes use several mechanisms to take up iron and store the iron as ferritin. See text. |
Clinical Pharmacology
A. INDICATIONS FOR THE USE OF IRON
The only clinical indication for the use of iron preparations is the treatment or prevention of iron deficiency anemia. Iron deficiency is commonly seen in populations with increased iron requirements. These include infants, especially premature infants; children during rapid growth periods; pregnant and lactating women; and patients with chronic kidney disease who lose erythrocytes at a relatively high rate during hemodialysis and also form them at a high rate as a result of treatment with the erythrocyte growth factor erythropoietin (see below). Inadequate iron absorption can also cause iron deficiency. This is seen frequently after gastrectomy and in patients with severe small bowel disease that results in generalized malabsorption. Iron deficiency in these gastrointestinal conditions is due to inadequate iron absorption.
The most common cause of iron deficiency in adults is blood loss. Menstruating women lose about 30 mg of iron with each menstrual period; women with heavy menstrual bleeding may lose much more. Thus, many premenopausal women have low iron stores or even iron deficiency. In men and postmenopausal women, the most common site of blood loss is the gastrointestinal tract. Patients with unexplained iron deficiency anemia should be evaluated for occult gastrointestinal bleeding.
B. TREATMENT
Iron deficiency anemia is treated with oral or parenteral iron preparations. Oral iron corrects the anemia just as rapidly and completely as parenteral iron in most cases if iron absorption from the gastrointestinal tract is normal. An exception is the high requirement for iron of patients with advanced chronic kidney disease who are undergoing hemodialysis and treatment with erythropoietin; for these patients, parenteral iron administration is preferred.
1. Oral iron therapy¾ A wide variety of oral iron preparations are available. Because ferrous iron is most efficiently absorbed, only ferrous salts should be used. Ferrous sulfate, ferrous gluconate, and ferrous fumarate are all effective and inexpensive and are recommended for the treatment of most patients.
Different iron salts provide different amounts of elemental iron, as shown in Table 33-2. In an iron-deficient individual, about 50-100 mg of iron can be incorporated into hemoglobin daily, and about 25% of oral iron given as ferrous salt can be absorbed. Therefore, 200-400 mg of elemental iron should be given daily to correct iron deficiency most rapidly. Patients unable to tolerate such large doses of iron can be given lower daily doses of iron, which results in slower but still complete correction of iron deficiency. Treatment with oral iron should be continued for 3-6 months after correction of the cause of the iron loss. This corrects the anemia and replenishes iron stores.
Common adverse effects of oral iron therapy include nausea, epigastric discomfort, abdominal cramps, constipation, and diarrhea. These effects are usually dose-related and can often be overcome by lowering the daily dose of iron or by taking the tablets immediately after or with meals. Some patients have less severe gastrointestinal adverse effects with one iron salt than another and benefit from changing preparations. Patients taking oral iron develop black stools; this has no clinical significance in itself but may obscure the diagnosis of continued gastrointestinal blood loss.
2. Parenteral iron therapy¾ Parenteral therapy should be reserved for patients with documented iron deficiency who are unable to tolerate or absorb oral iron and for patients with extensive chronic blood loss who cannot be maintained with oral iron alone. This includes patients with various postgastrectomy conditions and previous small bowel resection, inflammatory bowel disease involving the proximal small bowel, malabsorption syndromes, and advanced chronic renal disease including hemodialysis and treatment with erythropoietin.
Iron dextran is a stable complex of ferric hydroxide and low-molecular-weight dextran containing 50 mg of elemental iron per milliliter of solution. It can be given by deep intramuscular injection or by intravenous infusion, although the intravenous route is used most commonly. Intravenous administration eliminates the local pain and tissue staining that often occur with the intramuscular route and allows delivery of the entire dose of iron necessary to correct the iron deficiency at one time. Adverse effects of intravenous iron dextran therapy include headache, light-headedness, fever, arthralgias, nausea and vomiting, back pain, flushing, urticaria, bronchospasm, and, rarely, anaphylaxis and death. Some of these effects represent a hypersensitivity reaction to the dextran component. Hypersensitivity reactions may be delayed for 48-72 hours after administration. Anaphylactic reactions to iron dextran, including fatal reactions, have been clearly documented. Owing to the risk of a hypersensitivity reaction, a small test dose of iron dextran should always be given before full intramuscular or intravenous doses are given. Patients with a strong history of allergy and patients who have previously received parenteral iron dextran are more likely to have hypersensitivity reactions after treatment with parenteral iron dextran.
Iron-sucrose complex and iron sodium gluconate complex are alternative preparations. These agents can be given only by the intravenous route. These preparations appear to be much less likely than iron dextran to cause hypersensitivity reactions.
For patients who are treated chronically with parenteral iron, it is important to periodically monitor iron storage levels to avoid the serious toxicity associated with iron overload. Unlike oral iron therapy, which is subject to the regulatory mechanism provided by the intestinal uptake system, parenteral administration, which bypasses this regulatory system, can deliver more iron than can be safely stored in intestinal cells and macrophages in the liver and tissues. Iron stores can be estimated on the basis of serum concentrations of ferritin and the transferrin saturation, which is the ratio of the total serum iron concentration to the total iron-binding capacity (TIBC).
Clinical Toxicity
A. ACUTE IRON TOXICITY
Acute iron toxicity is seen almost exclusively in young children who accidentally ingest iron tablets. Although adults are able to tolerate large doses of oral iron without serious consequences, as few as 10 tablets of any of the commonly available oral iron preparations can be lethal in young children. Adult patients taking oral iron preparations should be instructed to store tablets in child-proof containers out of the reach of children. Children who are poisoned with oral iron experience necrotizing gastroenteritis, with vomiting, abdominal pain, and bloody diarrhea followed by shock, lethargy, and dyspnea. Subsequently, improvement is often noted, but this may be followed by severe metabolic acidosis, coma, and death. Urgent treatment is necessary. Whole bowel irrigation (see Chapter 59) should be performed to flush out unabsorbed pills. Deferoxamine, a potent iron-chelating compound, can be given systemically to bind iron that has already been absorbed and to promote its excretion in urine and feces. Activated charcoal, a highly effective adsorbent for most toxins, does not bind iron and thus is ineffective. Appropriate supportive therapy for gastrointestinal bleeding, metabolic acidosis, and shock must also be provided.
B. CHRONIC IRON TOXICITY
Chronic iron toxicity (iron overload), also known as hemochromatosis, results when excess iron is deposited in the heart, liver, pancreas, and other organs. It can lead to organ failure and death. It most commonly occurs in patients with inherited hemochromatosis, a disorder characterized by excessive iron absorption, and in patients who receive many red cell transfusions over a long period of time (eg, patients with thalassemia major).
Chronic iron overload in the absence of anemia is most efficiently treated by intermittent phlebotomy. One unit of blood can be removed every week or so until all of the excess iron is removed. Iron chelation therapy using parenteral deferoxamine is much less efficient as well as more complicated, expensive, and hazardous, but it can be the only option for iron overload that cannot be managed by phlebotomy, such as the iron overload experienced by patients with thalassemia major.
Recently, an oral iron chelator deferasirox has been approved for treatment of iron overload. Deferasirox appears to be as effective as deferoxamine at reducing liver iron concentrations and is much more convenient. However, it is not clear whether deferasirox is as effective as deferoxamine at protecting the heart from iron overload.
VITAMIN B12
Introduction
Vitamin B12 serves as a cofactor for several essential biochemical reactions in humans. Deficiency of vitamin B12 leads to anemia, gastrointestinal symptoms, and neurologic abnormalities. Although deficiency of vitamin B12 due to an inadequate supply in the diet is unusual, deficiency of B12 in adults¾especially older adults¾due to inadequate absorption of dietary vitamin B12 is a relatively common and easily treated disorder.
Chemistry
Vitamin B12 consists of a porphyrin-like ring with a central cobalt atom attached to a nucleotide. Various organic groups may be covalently bound to the cobalt atom, forming different cobalamins. Deoxyadenosylcobalamin and methylcobalamin are the active forms of the vitamin in humans. Cyanocobalamin and hydroxocobalamin (both available for therapeutic use) and other cobalamins found in food sources are converted to the active forms. The ultimate source of vitamin B12 is from microbial synthesis; the vitamin is not synthesized by animals or plants. The chief dietary source of vitamin B12 is microbially derived vitamin B12 in meat (especially liver), eggs, and dairy products. Vitamin B12 is sometimes called extrinsic factor to differentiate it from intrinsic factor, a protein normally secreted by the stomach.
Pharmacokinetics
The average diet in the USA contains 5-30 mcg of vitamin B12 daily, 1-5 mcg of which is usually absorbed. The vitamin is avidly stored, primarily in the liver, with an average adult having a total vitamin B12 storage pool of 3000-5000 mcg. Only trace amounts of vitamin B12 are normally lost in urine and stool. Because the normal daily requirements of vitamin B12 are only about 2 mcg, it would take about 5 years for all of the stored vitamin B12 to be exhausted and for megaloblastic anemia to develop if B12 absorption stopped. Vitamin B12in physiologic amounts is absorbed only after it complexes with intrinsic factor, a glycoprotein secreted by the parietal cells of the gastric mucosa. Intrinsic factor combines with the vitamin B12 that is liberated from dietary sources in the stomach and duodenum, and the intrinsic factor-vitamin B12 complex is subsequently absorbed in the distal ileum by a highly specific receptor-mediated transport system. Vitamin B12 deficiency in humans most often results from malabsorption of vitamin B12 due either to lack of intrinsic factor or to loss or malfunction of the specific absorptive mechanism in the distal ileum. Nutritional deficiency is rare but may be seen in strict vegetarians after many years without meat, eggs, or dairy products.
Once absorbed, vitamin B12 is transported to the various cells of the body bound to a plasma glycoprotein, transcobalamin II. Excess vitamin B12 is transported to the liver for storage.
Pharmacodynamics
Two essential enzymatic reactions in humans require vitamin B12 (Figure 33-2). In one, methylcobalamin serves as an intermediate in the transfer of a methyl group from N5-methyltetrahydrofolate to homocysteine, forming methionine (Figure 33-2A; Figure 33-3, section 1). Without vitamin B12, conversion of the major dietary and storage folate, N5-methyltetrahydrofolate, to tetrahydrofolate, the precursor of folate cofactors, cannot occur. As a result, a deficiency of folate cofactors necessary for several biochemical reactions involving the transfer of one-carbon groups develops. In particular, the depletion of tetrahydrofolate prevents synthesis of adequate supplies of the deoxythymidylate (dTMP) and purines required for DNA synthesis in rapidly dividing cells, as shown in Figure 33-3, section 2. The accumulation of folate as N5-methyltetrahydrofolate and the associated depletion of tetrahydrofolate cofactors in vitamin B12 deficiency have been referred to as the "methylfolate trap." This is the biochemical step whereby vitamin B12 and folic acid metabolism are linked, and it explains why the megaloblastic anemia of vitamin B12 deficiency can be partially corrected by ingestion of relatively large amounts of folic acid. Folic acid can be reduced to dihydrofolate by the enzyme dihydrofolate reductase (Figure 33-3, reaction 3) and thus serve as a source of the tetrahydrofolate required for synthesis of the purines and dTMP that are needed for DNA synthesis.
The other enzymatic reaction that requires vitamin B12 is isomerization of methylmalonyl-CoA to succinyl-CoA by the enzyme methylmalonyl-CoA mutase (Figure 33-2B). In vitamin B12deficiency, this conversion cannot take place, and the substrate, methylmalonyl-CoA, accumulates. In the past, it was thought that abnormal accumulation of methylmalonyl-CoA causes the neurologic manifestations of vitamin B12 deficiency. However, newer evidence instead implicates the disruption of the methionine synthesis pathway as the cause of neurologic problems. Whatever the biochemical explanation for neurologic damage, the important point is that administration of folic acid in the setting of vitamin B12 deficiency will not prevent neurologic manifestations even though it will largely correct the anemia caused by the vitamin B12 deficiency.
|
|
Figure 33-2. Enzymatic reactions that use vitamin B12. See text for details. |
|
|
|
Figure 33-3. Enzymatic reactions that use folates. Section 1 shows the vitamin B12-dependent reaction that allows most dietary folates to enter the tetrahydrofolate cofactor pool and becomes the "folate trap" in vitamin B12 deficiency. Section 2 shows the dTMP cycle. Section 3 shows the pathway by which folic acid enters the tetrahydrofolate cofactor pool. Double arrows indicate pathways with more than one intermediate step. |
|
Clinical Pharmacology
Vitamin B12 is used to treat or prevent deficiency. There is no evidence that vitamin B12 injections have any benefit in persons who do not have vitamin B12 deficiency. The most characteristic clinical manifestation of vitamin B12 deficiency is megaloblastic anemia. The typical clinical findings in megaloblastic anemia are macrocytic anemia, often with associated mild or moderate leukopenia or thrombocytopenia (or both), and a characteristic hypercellular bone marrow with an accumulation of megaloblastic erythroid and other precursor cells. The neurologic syndrome associated with vitamin B12 deficiency usually begins with paresthesias and weakness in peripheral nerves and progresses to spasticity, ataxia, and other central nervous system dysfunctions. Correction of vitamin B12 deficiency arrests the progression of neurologic disease, but it may not fully reverse neurologic symptoms that have been present for several months. Although most patients with neurologic abnormalities caused by vitamin B12 deficiency have megaloblastic anemia when first seen, occasional patients have few if any hematologic abnormalities.
Once a diagnosis of megaloblastic anemia is made, it must be determined whether vitamin B12 or folic acid deficiency is the cause. (Other causes of megaloblastic anemia are very rare.) This can usually be accomplished by measuring serum levels of the vitamins. The Schilling test, which measures absorption and urinary excretion of radioactively labeled vitamin B12, can be used to further define the mechanism of vitamin B12 malabsorption when this is found to be the cause of the megaloblastic anemia.
The most common causes of vitamin B12 deficiency are pernicious anemia, partial or total gastrectomy, and conditions that affect the distal ileum, such as malabsorption syndromes, inflammatory bowel disease, or small bowel resection.
Pernicious anemia results from defective secretion of intrinsic factor by the gastric mucosal cells. Patients with pernicious anemia have gastric atrophy and fail to secrete intrinsic factor (as well as hydrochloric acid). The Schilling test shows diminished absorption of radioactively labeled vitamin B12, which is corrected when intrinsic factor is administered with radioactive B12, since the vitamin can then be normally absorbed.
Vitamin B12 deficiency also occurs when the region of the distal ileum that absorbs the vitamin B12-intrinsic factor complex is damaged, as when the ileum is involved with inflammatory bowel disease or when the ileum is surgically resected. In these situations, radioactively labeled vitamin B12 is not absorbed in the Schilling test, even when intrinsic factor is added. Other rare causes of vitamin B12 deficiency include bacterial overgrowth of the small bowel, chronic pancreatitis, and thyroid disease. Rare cases of vitamin B12 deficiency in children have been found to be secondary to congenital deficiency of intrinsic factor and congenital selective vitamin B12 malabsorption due to defects of the receptor sites in the distal ileum.
Almost all cases of vitamin B12 deficiency are caused by malabsorption of the vitamin; therefore, parenteral injections of vitamin B12 are required for therapy. For patients with potentially reversible diseases, the underlying disease should be treated after initial treatment with parenteral vitamin B12. Most patients, however, do not have curable deficiency syndromes and require lifelong treatment with vitamin B12.
Vitamin B12 for parenteral injection is available as cyanocobalamin or hydroxocobalamin. Hydroxocobalamin is preferred because it is more highly protein-bound and therefore remains longer in the circulation. Initial therapy should consist of 100-1000 mcg of vitamin B12 intramuscularly daily or every other day for 1-2 weeks to replenish body stores. Maintenance therapy consists of 100-1000 mcg intramuscularly once a month for life. If neurologic abnormalities are present, maintenance therapy injections should be given every 1-2 weeks for 6 months before switching to monthly injections. Oral vitamin B12-intrinsic factor mixtures and liver extracts should not be used to treat vitamin B12 deficiency; however, oral doses of 1000 mcg of vitamin B12 daily are usually sufficient to treat patients with pernicious anemia who refuse or cannot tolerate the injections. After pernicious anemia is in remission following parenteral vitamin B12 therapy, the vitamin can be administered intranasally as a spray or gel.
FOLIC ACID
Introduction
Reduced forms of folic acid are required for essential biochemical reactions that provide precursors for the synthesis of amino acids, purines, and DNA. Folate deficiency is not uncommon, even though the deficiency is easily corrected by administration of folic acid. The consequences of folate deficiency go beyond the problem of anemia because folate deficiency is implicated as a cause of congenital malformations in newborns and may play a role in vascular disease (see Box: Folic Acid Supplementation: A Public Health Dilemma).
Chemistry
Folic acid (pteroylglutamic acid) is composed of a heterocycle (pteridine), p-aminobenzoic acid, and glutamic acid (Figure 33-4). Various numbers of glutamic acid moieties may be attached to the pteroyl portion of the molecule, resulting in monoglutamates, triglutamates, or polyglutamates. Folic acid can undergo reduction, catalyzed by the enzyme dihydrofolate reductase ("folate reductase"), to give dihydrofolic acid (Figure 33-3, section 3). Tetrahydrofolate can subsequently be transformed to folate cofactors possessing one-carbon units attached to the 5-nitrogen, to the 10-nitrogen, or to both positions (Figure 33-3). The folate cofactors are interconvertible by various enzymatic reactions and serve the important biochemical function of donating one-carbon units at various levels of oxidation. In most of these, tetrahydrofolate is regenerated and becomes available for reutilization.
|
|
Figure 33-4. The structure and numbering of atoms of folic acid. (Reproduced, with permission, from Murray RK et al: Harper's Biochemistry, 24th ed. McGraw-Hill, 1996.) |
Pharmacokinetics
The average diet in the USA contains 500-700 mcg of folates daily, 50-200 mcg of which is usually absorbed, depending on metabolic requirements. Pregnant women may absorb as much as 300-400 mcg of folic acid daily. Various forms of folic acid are present in a wide variety of plant and animal tissues; the richest sources are yeast, liver, kidney, and green vegetables. Normally, 5-20 mg of folates are stored in the liver and other tissues. Folates are excreted in the urine and stool and are also destroyed by catabolism, so serum levels fall within a few days when intake is diminished. Because body stores of folates are relatively low and daily requirements high, folic acid deficiency and megaloblastic anemia can develop within 1-6 months after the intake of folic acid stops, depending on the patient's nutritional status and the rate of folate utilization.
Unaltered folic acid is readily and completely absorbed in the proximal jejunum. Dietary folates, however, consist primarily of polyglutamate forms of N5-methyltetrahydrofolate. Before absorption, all but one of the glutamyl residues of the polyglutamates must be hydrolyzed by the enzyme a-1-glutamyl transferase ("conjugase") within the brush border of the intestinal mucosa. The monoglutamate N5-methyltetrahydrofolate is subsequently transported into the bloodstream by both active and passive transport and is then widely distributed throughout the body. Inside cells, N5-methyltetrahydrofolate is converted to tetrahydrofolate by the demethylation reaction that requires vitamin B12 (Figure 33-3, section 1).
Pharmacodynamics
Tetrahydrofolate cofactors participate in one-carbon transfer reactions. As described earlier in the discussion of vitamin B12, one of these essential reactions produces the dTMP needed for DNA synthesis. In this reaction, the enzyme thymidylate synthase catalyzes the transfer of the one-carbon unit of N5, N10-methylenetetrahydrofolate to deoxyuridine monophosphate (dUMP) to form dTMP (Figure 33-3, section 2). Unlike all the other enzymatic reactions that use folate cofactors, in this reaction the cofactor is oxidized to dihydrofolate, and for each mole of dTMP produced, 1 mole of tetrahydrofolate is consumed. In rapidly proliferating tissues, considerable amounts of tetrahydrofolate are consumed in this reaction, and continued DNA synthesis requires continued regeneration of tetrahydrofolate by reduction of dihydrofolate, catalyzed by the enzyme dihydrofolate reductase. The tetrahydrofolate thus produced can then reform the cofactor N5,N10-methylenetetrahydrofolate by the action of serine transhydroxymethylase and thus allow for the continued synthesis of dTMP. The combined catalytic activities of dTMP synthase, dihydrofolate reductase, and serine transhydroxymethylase are often referred to as the dTMP synthesis cycle. Enzymes in the dTMP cycle are the targets of two anticancer drugs; methotrexate inhibits dihydrofolate reductase, and a metabolite of 5-fluorouracil inhibits thymidylate synthase (see Chapter 55).
Cofactors of tetrahydrofolate participate in several other essential reactions. N5-Methylenetetrahydrofolate is required for the vitamin B12-dependent reaction that generates methionine from homocysteine (Figure 33-2A; Figure 33-3, section 1). In addition, tetrahydrofolate cofactors donate one-carbon units during the de novo synthesis of essential purines. In these reactions, tetrahydrofolate is regenerated and can reenter the tetrahydrofolate cofactor pool.
Clinical Pharmacology
Folate deficiency results in a megaloblastic anemia that is microscopically indistinguishable from the anemia caused by vitamin B12 deficiency (see above). However, folate deficiency does not cause the characteristic neurologic syndrome seen in vitamin B12 deficiency. In patients with megaloblastic anemia, folate status is assessed with assays for serum folate or for red blood cell folate. Red blood cell folate levels are often of greater diagnostic value than serum levels, because serum folate levels tend to be labile and do not necessarily reflect tissue levels.
Folic acid deficiency, unlike vitamin B12 deficiency, is often caused by inadequate dietary intake of folates. Patients with alcohol dependence and patients with liver disease often develop folic acid deficiency because of poor diet and diminished hepatic storage of folates. Pregnant women and patients with hemolytic anemia have increased folate requirements and may become folic acid-deficient, especially if their diets are marginal. Evidence implicates maternal folic acid deficiency in the occurrence of fetal neural tube defects, eg, spina bifida. (See Box: Folic Acid Supplementation: A Public Health Dilemma.) Patients with malabsorption syndromes also frequently develop folic acid deficiency. Patients who require renal dialysis develop folic acid deficiency because folates are removed from the plasma during the dialysis procedure.
Folic acid deficiency can be caused by drugs. Methotrexate and, to a lesser extent, trimethoprim and pyrimethamine, inhibit dihydrofolate reductase and may result in a deficiency of folate cofactors and ultimately in megaloblastic anemia. Long-term therapy with phenytoin can also cause folate deficiency, but only rarely causes megaloblastic anemia.
Parenteral administration of folic acid is rarely necessary, since oral folic acid is well absorbed even in patients with malabsorption syndromes. A dose of 1 mg folic acid orally daily is sufficient to reverse megaloblastic anemia, restore normal serum folate levels, and replenish body stores of folates in almost all patients. Therapy should be continued until the underlying cause of the deficiency is removed or corrected. Therapy may be required indefinitely for patients with malabsorption or dietary inadequacy. Folic acid supplementation to prevent folic acid deficiency should be considered in high-risk patients, including pregnant women, patients with alcohol dependence, hemolytic anemia, liver disease, or certain skin diseases, and patients on renal dialysis.
FOLIC ACID SUPPLEMENTATION: A PUBLIC HEALTH DILEMMA
Starting in January 1998, all products made from enriched grains in the USA were required to be supplemented with folic acid. This FDA ruling was issued to reduce the incidence of congenital neural tube defects. Epidemiologic studies show a strong correlation between maternal folic acid deficiency and the incidence of neural tube defects such as spina bifida and anencephaly. The FDA requirement for folic acid supplementation is a public health measure aimed at the significant number of women in the USA who do not receive prenatal care and are not aware of the importance of adequate folic acid ingestion for preventing birth defects in their babies.
There may be an added benefit for adults. N5-Methyltetrahydrofolate is required for the conversion of homocysteine to methionine (Figure 33-2; Figure 33-3, reaction 1). Impaired synthesis of N5-methyltetrahydrofolate results in elevated serum concentrations of homocysteine. Data from several sources suggest a positive correlation between elevated serum homocysteine and occlusive vascular diseases such as ischemic heart disease and stroke. Clinical data suggest that the folate supplementation program has improved the folate status and reduced the prevalence of hyperhomocysteinemia in a population of middle-aged and older adults who did not use vitamin supplements. It is possible, although the evidence thus far has been negative, that the increased ingestion of folic acid will also reduce the risk of vascular disease in this population.
Although the potential benefits of supplemental folic acid during pregnancy are compelling, the decision to require folic acid in grains was¾and still is¾controversial. As described in the text, ingestion of folic acid can partially or totally correct the anemia caused by vitamin B12 deficiency. However, folic acid supplementation does not prevent the potentially irreversible neurologic damage caused by vitamin B12 deficiency. People with pernicious anemia and other forms of vitamin B12 deficiency are usually identified because of signs and symptoms of anemia, which typically occur before neurologic symptoms. The opponents of folic acid supplementation are concerned that increased folic acid intake in the general population will mask vitamin B12 deficiency and increase the prevalence of neurologic disease in our elderly population. To put this in perspective, approximately 4000 pregnancies, including 2500 live births, in the USA each year are affected by neural tube defects. In contrast, it is estimated that over 10% of the elderly population in the USA, or several million people, are at risk for the neuropsychiatric complications of vitamin B12 deficiency. In acknowledgment of this controversy, the FDA has kept its requirements for folic acid supplementation at a somewhat low level.
HEMATOPOIETIC GROWTH FACTORS
INTRODUCTION
The hematopoietic growth factors are glycoprotein hormones that regulate the proliferation and differentiation of hematopoietic progenitor cells in the bone marrow. The first growth factors to be identified were called colony-stimulating factors because they could stimulate the growth of colonies of various bone marrow progenitor cells in vitro. Many of these growth factors have been purified and cloned, and their effects on hematopoiesis have been extensively studied. Quantities of these growth factors sufficient for clinical use are produced by recombinant DNA technology.
Of the known hematopoietic growth factors, erythropoietin (epoetin alfa), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin-11 (IL-11) are currently in clinical use. Thrombopoietin and other potentially useful hematopoietic factors are still in development.
The hematopoietic growth factors have complex effects on the function of a wide variety of cell types, including nonhematologic cells. Their usefulness in other areas of medicine, particularly as potential anticancer and anti-inflammatory drugs, is being investigated.
ERYTHROPOIETIN
Chemistry & Pharmacokinetics
Erythropoietin, a 34-39 kDa glycoprotein, was the first human hematopoietic growth factor to be isolated. It was originally purified from the urine of patients with severe anemia. Recombinant human erythropoietin (rHuEPO, epoetin alfa) is produced in a mammalian cell expression system. After intravenous administration, erythropoietin has a serum half-life of 4-13 hours in patients with chronic renal failure. It is not cleared by dialysis. It is measured in international units (IU). Darbepoetin alfa is a glycosylated form of erythropoietin and differs from it functionally only in having a twofold to threefold longer half-life.
Pharmacodynamics
Erythropoietin stimulates erythroid proliferation and differentiation by interacting with specific erythropoietin receptors on red cell progenitors. The erythropoietin receptor is a member of the JAK/STAT superfamily of cytokine receptors that use protein phosphorylation and transcription factor activation to regulate cellular function (see Chapter 2). Erythropoietin also induces release of reticulocytes from the bone marrow. Endogenous erythropoietin is primarily produced in the kidney. In response to tissue hypoxia, more erythropoietin is produced through an increased rate of transcription of the erythropoietin gene. This results in correction of the anemia, provided that the bone marrow response is not impaired by red cell nutritional deficiency (especially iron deficiency), primary bone marrow disorders (see below), or bone marrow suppression from drugs or chronic diseases.
Normally, an inverse relationship exists between the hematocrit or hemoglobin level and the serum erythropoietin level. Nonanemic individuals have serum erythropoietin levels of less than 20 IU/L. As the hematocrit and hemoglobin levels fall and anemia becomes more severe, the serum erythropoietin level rises exponentially. Patients with moderately severe anemias usually have erythropoietin levels in the 100-500 IU/L range, and patients with severe anemias may have levels of thousands of IU/L. The most important exception to this inverse relationship is in the anemia of chronic renal failure. In patients with renal disease, erythropoietin levels are usually low because the kidneys cannot produce the growth factor. These are the patients most likely to respond to treatment with exogenous erythropoietin. In most primary bone marrow disorders (aplastic anemia, leukemias, myeloproliferative and myelodysplastic disorders, etc) and most nutritional and secondary anemias, endogenous erythropoietin levels are high, so there is less likelihood of a response to exogenous erythropoietin (but see below).
Clinical Pharmacology
The availability of erythropoietin has had a significant positive impact for patients with anemia of chronic renal failure (Table 33-3). Erythropoietin consistently improves the hematocrit and hemoglobin level and usually eliminates the need for transfusions in these patients. An increase in reticulocyte count is usually observed in about 10 days and an increase in hematocrit and hemoglobin levels in 2-6 weeks. Most patients can maintain a hematocrit of about 35% with erythropoietin doses of 50-150 IU/kg intravenously or subcutaneously three times a week. Failure to respond to erythropoietin is most commonly due to concurrent iron deficiency, which can be corrected by giving oral or parenteral iron. Folate supplementation may also be necessary in some patients.
In selected patients, erythropoietin may also be useful for the treatment of anemia due to primary bone marrow disorders and secondary anemias. This includes patients with aplastic anemia and other bone marrow failure states, myeloproliferative and myelodysplastic disorders, multiple myeloma and perhaps other chronic bone marrow malignancies, and the anemias associated with chronic inflammation, AIDS, and cancer. Patients with these disorders who have disproportionately low serum erythropoietin levels for their degree of anemia are most likely to respond to treatment with this growth factor. Patients with endogenous erythropoietin levels of less than 100 IU/L have the best chance of response, although patients with erythropoietin levels between 100 and 500 IU/L respond occasionally. These patients generally require higher erythropoietin doses (150-300 IU/kg three times a week) to achieve a response, and responses are often incomplete.
Erythropoietin has been used successfully to offset the anemia produced by zidovudine treatment in patients with HIV infection and in the treatment of the anemia of prematurity. It can also be used to accelerate erythropoiesis after phlebotomies for autologous transfusion for elective surgery, or for treatment of iron overload (hemochromatosis).
Erythropoietin is one of the drugs banned by the International Olympic Committee. The use of erythropoietin by athletes is based on their hope that increased red blood cell concentration will increase oxygen delivery and improve performance.
Toxicity
The most common adverse effects of erythropoietin are associated with a rapid increase in hematocrit and hemoglobin and include hypertension and thrombotic complications. These difficulties can be minimized by raising the hematocrit and hemoglobin slowly and by adequately monitoring and treating hypertension. Allergic reactions have been infrequent and mild.
MYELOID GROWTH FACTORS
Chemistry & Pharmacokinetics
G-CSF and GM-CSF, the two myeloid growth factors currently available for clinical use, were originally purified from cultured human cell lines (Table 33-3). Recombinant human G-CSF (rHuG-CSF; filgrastim) is produced in a bacterial expression system. It is a nonglycosylated peptide of 175 amino acids, with a molecular weight of 18 kDa. Recombinant human GM-CSF (rHuGM-CSF; sargramostim) is produced in a yeast expression system. It is a partially glycosylated peptide of 127 amino acids, with three molecular species with molecular weights of 15,500; 15,800; and 19,500. These preparations have serum half-lives of 2-7 hours after intravenous or subcutaneous administration. Pegfilgrastim, a covalent conjugation product of filgrastim and a form of polyethylene glycol, has a much longer serum half-life than recombinant G-CSF, and so it can be injected once per myelosuppressive chemotherapy cycle instead of daily for several days.
Pharmacodynamics
The myeloid growth factors stimulate proliferation and differentiation by interacting with specific receptors found on various myeloid progenitor cells. Like the erythropoietin receptor, these receptors are members of the JAK/STAT superfamily (see Chapter 2). G-CSF stimulates proliferation and differentiation of progenitors already committed to the neutrophil lineage. It also activates the phagocytic activity of mature neutrophils and prolongs their survival in the circulation. G-CSF also has a remarkable ability to mobilize hematopoietic stem cells, ie, to increase their concentration in peripheral blood. This biologic effect underlies a major advance in transplantation¾the use of peripheral blood stem cells (PBSCs) rather than bone marrow stem cells for autologous and allogeneic hematopoietic stem cell transplantation (see below).
GM-CSF has broader biologic actions than G-CSF. It is a multipotential hematopoietic growth factor that stimulates proliferation and differentiation of early and late granulocytic progenitor cells as well as erythroid and megakaryocyte progenitors. Like G-CSF, GM-CSF also stimulates the function of mature neutrophils. GM-CSF acts together with interleukin-2 to stimulate T-cell proliferation and appears to be a locally active factor at the site of inflammation. GM-CSF mobilizes peripheral blood stem cells, but it is significantly less efficacious than G-CSF in this regard.
Clinical Pharmacology
A. CANCER CHEMOTHERAPY-INDUCED NEUTROPENIA
Neutropenia is a common adverse effect of the cytotoxic drugs used to treat cancer and increases the risk of serious infection in patients receiving chemotherapy. Unlike the treatment of anemia and thrombocytopenia, transfusion of neutropenic patients with granulocytes collected from donors is performed rarely and with limited success. The introduction of G-CSF in 1991 represented a milestone in the treatment of chemotherapy-induced neutropenia. This growth factor dramatically accelerates the rate of neutrophil recovery after dose-intensive myelosuppressive chemotherapy (Figure 33-5). It reduces the duration of neutropenia and usually raises the nadir count, the lowest neutrophil count seen following a cycle of chemotherapy.
The ability of G-CSF to increase neutrophil counts after myelosuppressive chemotherapy is nearly universal, but its impact on clinical outcomes is more variable. Some clinical trials have shown that G-CSF reduces episodes of febrile neutropenia, requirements for broad-spectrum antibiotics, and days of hospitalization; however, other trials failed to find these favorable outcomes. To date, no clinical trial has shown improved survival in cancer patients treated with G-CSF. Clinical guidelines for the use of G-CSF after cytotoxic chemotherapy recommend reserving G-CSF for patients with a prior episode of febrile neutropenia after cytotoxic chemotherapy, patients receiving dose-intensive chemotherapy, patients at high risk for febrile neutropenia, and patients who are unlikely to survive an episode of febrile neutropenia. Pegfilgrastim is an alternative to G-CSF for prevention of chemotherapy-induced febrile neutropenia. Pegfilgrastim can be administered less frequently, and it may shorten the period of severe neutropenia slightly more than G-CSF.
Like G-CSF and pegfilgrastim, GM-CSF also reduces the duration of neutropenia after cytotoxic chemotherapy. It has been more difficult to show that GM-CSF reduces the incidence of febrile neutropenia, probably because GM-CSF itself can induce fever. In the treatment of chemotherapy-induced neutropenia, G-CSF, 5 mcg/kg/d, or GM-CSF, 250 mcg/m2/d, is usually started within 24-72 hours after completing chemotherapy and is continued until the absolute neutrophil count is > 10,000 cells/u L. Pegfilgrastim is given as a single dose instead of daily injections.
The utility and safety of the myeloid growth factors in the postchemotherapy supportive care of patients with acute myeloid leukemia (AML) have been the subject of a number of clinical trials. Because leukemic cells arise from progenitors whose proliferation and differentiation are normally regulated by hematopoietic growth factors, including GM-CSF and G-CSF, there was concern that myeloid growth factors could stimulate leukemic cell growth and increase the rate of relapse. The results of randomized clinical trials suggest that both G-CSF and GM-CSF are safe following induction and consolidation treatment of myeloid and lymphoblastic leukemia. There has been no evidence that these growth factors reduce the rate of remission or increase relapse rate. On the contrary, the growth factors accelerate neutrophil recovery and reduce infection rates and days of hospitalization. Both G-CSF and GM-CSF have FDA approval for treatment of patients with AML.
B. OTHER APPLICATIONS
G-CSF and GM-CSF have also proved to be effective in treating the neutropenia associated with congenital neutropenia, cyclic neutropenia, myelodysplasia, and aplastic anemia. Many patients with these disorders respond with a prompt and sometimes dramatic increase in neutrophil count. In some cases, this results in a decrease in the frequency of infections. Because neither G-CSF nor GM-CSF stimulates the formation of erythrocytes and platelets, they are sometimes combined with other growth factors for treatment of pancytopenia.
The myeloid growth factors play an important role in autologous stem cell transplantation for patients undergoing high-dose chemotherapy. High-dose chemotherapy with autologous stem cell support is increasingly used to treat patients with tumors that are resistant to standard doses of chemotherapeutic drugs. The high-dose regimens produce extreme myelosuppression; the myelosuppression is then counteracted by reinfusion of the patient's own hematopoietic stem cells (which are collected prior to chemotherapy). The administration of G-CSF or GM-CSF early after autologous stem cell transplantation has been shown to reduce the time to engraftment and to recovery from neutropenia in patients receiving stem cells obtained either from bone marrow or from peripheral blood. These effects are seen in patients being treated for lymphoma or for solid tumors. G-CSF and GM-CSF are also used to support patients who have received allogeneic bone marrow transplantation for treatment of hematologic malignancies or bone marrow failure states. In this setting, the growth factors speed the recovery from neutropenia without increasing the incidence of acute graft-versus-host disease.
Perhaps the most important role of the myeloid growth factors in transplantation is for mobilization of peripheral blood stem cells (PBSCs). Stem cells collected from peripheral blood have nearly replaced bone marrow as the hematopoietic preparation used for autologous transplantation, and the use of PBSCs for allogeneic transplantation is also being investigated. The cells can be collected in an outpatient setting with a procedure that avoids much of the risk and discomfort of bone marrow collection, including the need for general anesthesia. In addition, there is evidence that PBSC transplantation results in more rapid engraftment of all hematopoietic cell lineages and in reduced rates of graft failure or delayed platelet recovery.
G-CSF is the cytokine most commonly used for PBSC mobilization because of its increased efficacy and reduced toxicity compared with GM-CSF. To mobilize stem cells, patients or donors are given 5-10 mcg/kg/d subcutaneously for 4 days. On the fifth day, they undergo leukapheresis. The success of PBSC transplantation depends on transfusion of adequate numbers of stem cells. CD34, an antigen present on early progenitor cells and absent from later, committed, cells, is used as a marker for the requisite stem cells. The goal is to reinfuse at least 5 ´ 106 CD34 cells/kg; this number of CD34 cells usually results in prompt and durable engraftment of all cell lineages. It can take several separate leukaphereses to collect enough CD34 cells, especially from older patients and patients who have been exposed to radiation therapy or chemotherapy.
|
|
Figure 33-5. Effects of G-CSF (color) or placebo (black line) on absolute neutrophil count (ANC) after cytotoxic chemotherapy for lung cancer. Doses of chemotherapeutic drugs were administered on days 1 and 3. G-CSF or placebo injections were started on day 4 and continued daily through day 12 or 16. The first peak in ANC reflects the recruitment of mature cells by G-CSF. The second peak reflects a marked increase in new neutrophil production by the bone marrow under stimulation by G-CSF. (Normal ANC is 2.2-8.6 ´ 109/L.) (Modified and reproduced, with permission, from Crawford et al: Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med 1991;325:164.) |
Toxicity
Although the two growth factors have similar effects on neutrophil counts, G-CSF is used more frequently because it is better tolerated. G-CSF can cause bone pain, which clears when the drug is discontinued. GM-CSF can cause more severe side effects, particularly at higher doses. These include fever, malaise, arthralgias, myalgias, and a capillary leak syndrome characterized by peripheral edema and pleural or pericardial effusions. Allergic reactions may occur but are infrequent. Splenic rupture is a rare but serious complication of the use of G-CSF for PBSC.
MEGAKARYOCYTE GROWTH FACTORS
Chemistry & Pharmacokinetics
Interleukin-11 (IL-11) is a 65-85 kDa protein produced by fibroblasts and stromal cells in the bone marrow. Oprelvekin, the recombinant form of interleukin-11 approved for clinical use (Table 33-3), is produced by expression in E coli. The half-life of IL-11 is 7-8 hours when the drug is injected subcutaneously.
Thrombopoietin, a 65-85 kDa glycosylated protein, is constitutively expressed by a variety of organs and cell types. Hepatocytes appear to be the major source of human thrombopoietin, and patients with cirrhosis and thrombocytopenia have low serum thrombopoietin levels. Recombinant thrombopoietin is produced by expression in human cells; the recombinant product contains two intramolecular disulfide bonds and a number of carbohydrate side chains.
Pharmacodynamics
Interleukin-11 acts through a specific cell surface cytokine receptor to stimulate the growth of multiple lymphoid and myeloid cells. It acts synergistically with other growth factors to stimulate the growth of primitive megakaryocytic progenitors and, most importantly, increases the number of peripheral platelets and neutrophils.
Acting through its own cytokine receptor, thrombopoietin also independently stimulates the growth of primitive megakaryocytic progenitors. In addition, it stimulates mature megakaryocytes and even activates mature platelets to respond to aggregation-inducing stimuli. The critical in vivo role of thrombopoietin has been demonstrated in genetically engineered knockout mice who lack either thrombopoietin or its receptor. These mice have marked thrombocytopenia but do not display anemia or leukopenia.
Clinical Pharmacology
Patients with thrombocytopenia have a high risk of hemorrhage. Although platelet transfusion is commonly used to treat thrombocytopenia, this procedure can cause adverse reactions in the recipient; furthermore, a significant number of patients fail to exhibit the expected increase in platelet count.
Interleukin-11 is the first growth factor to gain FDA approval for treatment of thrombocytopenia. It is approved for the secondary prevention of thrombocytopenia in patients receiving cytotoxic chemotherapy for treatment of nonmyeloid cancers. Clinical trials show that it reduces the number of platelet transfusions required by patients who experience severe thrombocytopenia after a previous cycle of chemotherapy. Although IL-11 has broad stimulatory effects on hematopoietic cell lineages in vitro, it does not appear to have significant effects on the leukopenia caused by myelosuppressive chemotherapy. Interleukin-11 is given by subcutaneous injection at a dose of 50 mcg/kg/d. It is started 6-24 hours after completion of chemotherapy and continued for 14-21 days or until the platelet count passes the nadir and rises to > 50,000 cells/uL.
Recombinant thrombopoietin is still an investigational agent. The primary focus of current clinical trials is for the treatment of chemotherapy-induced thrombocytopenia and thrombocytopenia accompanying hematologic stem cell transplantation.
Toxicity
The most common adverse effects of interleukin-11 are fatigue, headache, dizziness, and cardiovascular effects. The cardiovascular effects include anemia (due to hemodilution), dyspnea (due to fluid accumulation in the lungs), and transient atrial arrhythmias. Hypokalemia has also been seen in some patients. All of these adverse effects appear to be reversible. In the limited clinical trial data available thus far, recombinant thrombopoietin appears to be well tolerated.
PREPARATIONS AVAILABLE
Darbepoetin alfa (Aranesp)
Parenteral: 25, 40, 60, 100, 200, 300, 500 mcg/mL for IV or SC injection
Deferasirox (Exjade)
Oral: 125, 250, 500 mg tablets
Deferoxamine (generic, Desferal)
Parenteral: 500, 2000 mg vials for IM, SC, or IV injection
Epoetin alfa (erythropoietin, EPO) (Epogen, Procrit)
Parenteral: 2000, 3000, 4000, 10000, 20000, 40000 IU/mL vials for IV or SC injection
Filgrastim (G-CSF) (Neupogen)
Parenteral: 300 mcg vials for IV or SC injection
Folic acid (folacin, pteroylglutamic acid) (generic)
Oral: 0.4, 0.8, 1 mg tablets
Parenteral: 5 mg/mL for injection
Iron (generic)
Oral: See Table 33-2.
Parenteral (Iron dextran) (InFeD, DexFerrum): 50 mg elemental iron/mL
Parenteral (Sodium ferric gluconate complex) (Ferrlecit): 12.5 mg elemental iron/mL
Parenteral (Iron sucrose)(Venofer): 20 mg elemental iron/mL
Oprelvekin (interleukin-11) (Neumega)
Parenteral: 5 mg vials for SC injection
Pegfilgrastim (Neulasta)
Parenteral: 10 mg/mL solution in single-dose syringe
Sargramostim (GM-CSF) (Leukine)
Parenteral: 250, 500 mcg vials for IV infusion
Vitamin B12 (generic cyanocobalamin or hydroxocobalamin)
Oral (cyanocobalamin): 100, 500, 1000, 5000 mcg tablets, 100, 250, 500 mcg lozenges
Nasal (Nascobal): 5000 mcg/mL (500 mcg/spray)
Parenteral (cyanocobalamin): 100, 1000 mcg/mL for IM or SC injection
Parenteral (hydroxocobalamin): 1000 mcg/mL for IM injection only
REFERENCES
Anderson GJ et al: Mechanisms of haem and non-haem iron absorption: Lessons from inherited disorders of iron metabolism. Biometals 2005;18:339.
American Society of Clinical Oncology Supportive Care and Quality of Life Practice Guidelines Website: http://www.asco.org/portal/site/ASCO/menuitem.509189bfd2c2bf5ca7ffa807320041a0/?vgnextoid=04d61f886024a010VgnVCM100000ed730ad1RCRD
Crawford J et al: Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med 1991;325:164.
Cook JD: Diagnosis and management of iron-deficiency anaemia. Best Pract Res Clin Haematol 2005;18:309.
Du X, Williams DA: Interleukin-11: Review of molecular, cell biology and clinical use. Blood 1997;89:3897.
Eschbach JW: Iron requirements in erythropoietin therapy. Best Pract Res Clin Haematol 2005;18:347.
Fetscher S, Mertelsmann R: Supportive care in hematological malignancies: Hematopoietic growth factors, infections, transfusion therapy. Curr Opin Hematol 1999;6:262.
Fisher JW: Erythropoietin: Physiology and pharmacology update. Exp Biol Med 2003;228:1.
Gazitt Y: Comparison between granulocyte colony-stimulating factor and granulocyte-macrophage colony stimulating factor in the mobilization of peripheral blood stem cells. Curr Opin Hematol 2002;9:190.
Green R, Miller JW: Folate deficiency beyond megaloblastic anemia: Hyperhomocysteinemia and other manifestations of dysfunctional folate status. Semin Hematol 1999;36:47.
Harker LA: Physiology and clinical applications of platelet growth factors. Curr Opin Hematol 1999;6:127.
Jacques PF et al: The effects of folic acid fortification on plasma folate and total homocysteine concentrations. N Engl J Med 1999;340:1449.
Kuter DJ, Begley CG: Recombinant human thrombopoietin: Basic biology and evaluation of clinical studies. Blood 2002;100:3457.
Rothenberg SP: Increasing the dietary intake of folate: Pros and cons. Semin Hematol 1999;36:65.
Shpall EJ: The utilization of cytokines in stem cell mobilization strategies. Bone Marrow Transplant 1999;23(Suppl 2):S13.