INTRODUCTION
Viruses are obligate intracellular parasites; their replication depends primarily on synthetic processes of the host cell. Therefore, to be effective, antiviral agents must either block viral entry into or exit from the cell or be active inside the host cell. As a corollary, nonselective inhibitors of virus replication may interfere with host cell function and produce toxicity.
Progress in antiviral chemotherapy began in the early 1950s, when the search for anticancer drugs generated several new compounds capable of inhibiting viral DNA synthesis. The two first-generation antivirals, 5-iododeoxyuridine and trifluorothymidine, had poor specificity (ie, they inhibited host cell DNA as well as viral DNA) that rendered them too toxic for systemic use. However, both agents are effective when used topically for the treatment of herpes keratitis.
Acronyms & Other Names
3TC Lamivudine
AZT Zidovudine (previously azidothymidine)
CMV Cytomegalovirus
CYP Cytochrome P450
d4T Stavudine
ddC Zalcitabine
ddI Didanosine
EBV Epstein-Barr virus
FTC Emtricitabine
HAART Highly active antiretroviral therapy
HBV Hepatitis B virus
HCV Hepatitis C virus
HHV-6 Human herpesvirus-6
HIV Human immunodeficiency virus
HPV Human papillomavirus
HSV Herpes simplex virus
IFN Interferon
KSHV Kaposi's sarcoma-associated herpesvirus
NNRTI Nonnucleoside reverse transcriptase inhibitor
NRTI Nucleoside reverse transcriptase inhibitor
PI Protease inhibitor
RSV Respiratory syncytial virus
SVR Sustained antiviral response
VZV Varicella-zoster virus
Recent research has focused on the identification of agents with greater selectivity, in vivo stability, and lack of toxicity. Selective antiretroviral agents that inhibit a critical HIV-1 enzyme such as reverse transcriptase or the protease required for final packaging of the virus particle have become available. However, because replication of the virus peaks at or before the manifestation of clinical symptoms in many viral infections, chemoprophylaxis or early initiation of therapy may be key. In chronic illnesses such as viral hepatitis or HIV infection, potent inhibition of viral replication is crucial in limiting the extent of systemic damage.
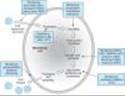
Viral replication consists of several steps (Figure 49-1): (1) attachment of the virus to the host cell; (2) entry of the virus through the host cell membrane; (3) uncoating of viral nucleic acid; (4) synthesis of early regulatory proteins, eg, nucleic acid polymerases; (5) synthesis of RNA or DNA; (6) synthesis of late, structural proteins; (7) assembly (maturation) of viral particles; and (8) release from the cell. Antiviral agents can potentially target any of these steps.
|
|
Figure 49-1. The major sites of antiviral drug action. Note: Interferon alfas are speculated to have multiple sites of action. (Modified and reproduced, with permission, from Trevor AT, Katzung BG, Masters SM: Pharmacology: Examination & Board Review, 6th ed. McGraw-Hill, 2002.) |
I. AGENTS TO TREAT HERPES SIMPLEX VIRUS (HSV) & VARICELLA-ZOSTER VIRUS (VZV) INFECTIONS
INTRODUCTION
Three oral nucleoside analogs are licensed for the treatment of HSV and VZV infections: acyclovir, valacyclovir, and famciclovir. They have similar mechanisms of action and similar indications for clinical use; all are well tolerated. Acyclovir has been the most extensively studied; it was licensed first and is the only one of the three available for intravenous use in the United States. Comparative trials have demonstrated similar efficacies of these three agents for the treatment of HSV but modest superiority of famciclovir and valacyclovir over acyclovir for the treatment of herpes zoster. Neither valacyclovir nor famciclovir have been fully evaluated in pediatric patients; thus, neither is indicated for the treatment of varicella infection.
ACYCLOVIR
Acyclovir (Figure 49-2) is an acyclic guanosine derivative with clinical activity against HSV-1, HSV-2, and VZV. In vitro activity against Epstein-Barr virus (EBV), cytomegalovirus (CMV), and human herpesvirus-6 (HHV-6) is present but comparatively weaker.
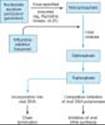
Acyclovir requires three phosphorylation steps for activation. It is converted first to the monophosphate derivative by the virus-specified thymidine kinase and then to the di- and triphosphate compounds by host cell enzymes (Figure 49-3). Because it requires the viral kinase for initial phosphorylation, acyclovir is selectively activated, and the active metabolite accumulates, only in infected cells. Acyclovir triphosphate inhibits viral DNA synthesis by two mechanisms: competition with deoxyGTP for the viral DNA polymerase, resulting in binding to the DNA template as an irreversible complex; and chain termination following incorporation into the viral DNA.
The bioavailability of oral acyclovir is 15-20% and is unaffected by food. An intravenous formulation is available. Topical formulations produce high concentrations in herpetic lesions, but systemic concentrations are undetectable by this route.
Acyclovir is cleared primarily by glomerular filtration and tubular secretion. The half-life is approximately 3 hours in patients with normal renal function and 20 hours in patients with anuria. Acyclovir is readily cleared by hemodialysis but not by peritoneal dialysis. Acyclovir diffuses readily into most tissues and body fluids. Cerebrospinal fluid concentrations are 50% of serum values.
Oral acyclovir has multiple uses. In first episodes of genital herpes, oral acyclovir shortens the duration of symptoms by approximately 2 days, the time to lesion healing by 4 days, and the duration of viral shedding by 7 days. In recurrent genital herpes, the time course is shortened by 1-2 days. Treatment of first-episode genital herpes does not alter the frequency or severity of recurrent outbreaks. Long-term suppression of genital herpes with oral acyclovir in patients with frequent recurrences decreases the frequency of symptomatic recurrences and of asymptomatic viral shedding, thus decreasing the rate of sexual transmission. However, outbreaks may resume upon discontinuation of suppressive acyclovir. Oral acyclovir is only modestly beneficial in recurrent herpes labialis. It significantly decreases the total number of lesions, duration of symptoms, and viral shedding in patients with varicella (if begun within 24 hours after the onset of rash) or cutaneous zoster (if begun within 72 hours). However, because VZV is less susceptible to acyclovir than HSV, higher doses are required (Table 49-1). When given prophylactically to patients undergoing organ transplantation, oral acyclovir (200 mg every 8 hours or 800 mg every 12 hours) or intravenous acyclovir (5 mg/kg every 8 hours) prevents reactivation of HSV infection.
Intravenous acyclovir is the treatment of choice for herpes simplex encephalitis, neonatal HSV infection, and serious HSV or VZV infections (Table 49-1). In immunocompromised patients with VZV infection, intravenous acyclovir reduces the incidence of cutaneous and visceral dissemination.
Topical acyclovir is substantially less effective than oral therapy for primary HSV infection. It is of no benefit in treating recurrent genital herpes.
Resistance to acyclovir can develop in HSV or VZV through alteration in either the viral thymidine kinase or the DNA polymerase, and clinically resistant infections have been reported in immunocompromised hosts. Most clinical isolates are resistant on the basis of deficient thymidine kinase activity and thus are cross-resistant to valacyclovir, famciclovir, and ganciclovir. Agents such as foscarnet, cidofovir, and trifluridine do not require activation by viral thymidine kinase and thus have preserved activity against the most prevalent acyclovir-resistant strains (Figure 49-3).
Acyclovir is generally well tolerated. Nausea, diarrhea, and headache have occasionally been reported. Intravenous infusion may be associated with reversible renal dysfunction (due to crystalline nephropathy) or neurologic toxicity (eg, tremors, delirium, seizures). However, these are uncommon with adequate hydration and avoidance of rapid infusion rates. High doses of acyclovir cause chromosomal damage and testicular atrophy in rats, but there has been no evidence of teratogenicity, reduction in sperm production, or cytogenetic alterations in peripheral blood lymphocytes in patients receiving chronic daily suppression of genital herpes for more than 10 years.
|
|
Figure 49-2. Chemical structures of some antiviral nucleoside and nucleotide analogs. |
|
|
|
Figure 49-3. Mechanism of action of antiherpes agents. |
|
VALACYCLOVIR
Valacyclovir is the L-valyl ester of acyclovir (Figure 49-2). It is rapidly converted to acyclovir after oral administration via intestinal and hepatic first-pass metabolism, resulting in serum levels that are three to five times greater than those achieved with oral acyclovir and approximate those achieved with intravenous acyclovir administration. Oral bioavailability is 54%, and cerebrospinal fluid levels are 50% of those in serum. Elimination half-life is 2.5-3.3 hours.
Approved uses of valacyclovir include treatment of first or recurrent genital herpes, suppression of frequently recurring genital herpes, and as a 1-day treatment for orolabial herpes (Table 49-1). Once-daily dosing of valacyclovir (500 mg) for chronic suppression in persons with recurrent genital herpes has been shown to markedly decrease the risk of sexual transmission. In comparative trials with acyclovir for the treatment of patients with zoster, rates of cutaneous healing were similar, but valacyclovir was associated with a shorter duration of zoster-associated pain. Valacyclovir has also been shown to be effective in preventing cytomegalovirus disease after organ transplantation when compared with placebo.
Valacyclovir is generally well tolerated, although nausea, vomiting, or rash occasionally occur. Agitation, dizziness, headache, liver enzyme elevation, anemia, and neutropenia are rare. At high doses, confusion, hallucinations, and seizures have been reported. AIDS patients who received high-dosage valacyclovir chronically (ie, 8 g/d) had an increased incidence of gastrointestinal intolerance as well as thrombotic microangiopathies (thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome).
FAMCICLOVIR
Famciclovir is the diacetyl ester prodrug of 6-deoxypenciclovir, an acyclic guanosine analog (Figure 49-2). After oral administration, famciclovir is rapidly converted by first-pass metabolism to penciclovir. It is active in vitro against HSV-1, HSV-2, VZV, EBV, and HBV. As with acyclovir, activation by phosphorylation is catalyzed by the virus-specified thymidine kinase in infected cells, followed by competitive inhibition of the viral DNA polymerase to block DNA synthesis. Unlike acyclovir, however, penciclovir does not cause chain termination. Penciclovir triphosphate has lower affinity for the viral DNA polymerase than acyclovir triphosphate, but it achieves higher intracellular concentrations and has a more prolonged intracellular effect in experimental systems. The most commonly encountered clinical mutants of HSV are thymidine kinase-deficient; these are cross-resistant to acyclovir and famciclovir.
The bioavailability of penciclovir from orally administered famciclovir is 70%. Penciclovir triphosphate has an intracellular half-life of 10 hours in HSV-1-infected cells, 20 hours in HSV-2-infected cells, and 7 hours in VZV-infected cells in vitro. Penciclovir is excreted primarily in the urine.
Oral famciclovir is effective for the treatment of first and recurrent genital herpes, for chronic daily suppression of genital herpes, and for the treatment of acute zoster (Table 49-1). One-day usage of famciclovir (1000 mg twice daily) significantly accelerates time to healing of recurrent genital herpes compared with placebo, by approximately 2 days. A single dose of 1500 mg or two 750 mg doses (BID) accelerates herpes labialis healing time. Comparison of famciclovir to valacyclovir for treatment of herpes zoster in immunocompetent patients showed similar rates of cutaneous healing and pain resolution, although both agents were associated with a shortened duration of zoster-associated pain compared with acyclovir.
Oral famciclovir is generally well tolerated, although headache, diarrhea, and nausea may occur. As with acyclovir, testicular toxicity has been demonstrated in animals receiving repeated doses. However, men receiving daily famciclovir (250 mg every 12 hours) for 18 weeks had no changes in sperm morphology or motility. The incidence of mammary adenocarcinoma was increased in female rats receiving famciclovir for 2 years.
PENCICLOVIR
The guanosine analog penciclovir, the active metabolite of famciclovir, is also available for topical use. One percent penciclovir cream is effective for the treatment of recurrent herpes labialis in immunocompetent adults (Table 49-1). When therapy was initiated within 1 hour after the onset of signs or symptoms and continued every 2 hours during waking hours for 4 days, treatment with topical penciclovir resulted in a shortening of the median time until healing by approximately 0.7 days compared with placebo. Side effects are uncommon.
DOCOSANOL
Docosanol is a saturated 22-carbon aliphatic alcohol that inhibits fusion between the plasma membrane and the HSV envelope, thereby preventing viral entry into cells and subsequent viral replication. Topical docosanol 10% cream is available without a prescription. When therapy is initiated within 12 hours of the onset of prodromal symptoms and applied five times daily, the healing time is decreased by approximately 18 hours compared with placebo in recurrent orolabial herpes.
TRIFLURIDINE
Trifluridine (trifluorothymidine) is a fluorinated pyrimidine nucleoside that inhibits viral DNA synthesis in HSV-1, HSV-2, vaccinia, and some adenoviruses. It is phosphorylated intracellularly to its active form by host cell enzymes, and then competes with thymidine triphosphate for incorporation by the viral DNA polymerase (Figure 49-3). Incorporation of trifluridine triphosphate into both viral and host DNA prevents its systemic use. Application of a 1% solution is effective in treating keratoconjunctivitis and recurrent epithelial keratitis due to HSV-1 and HSV-2. Topical application of trifluridine solution, alone or in combination with interferon alfa, has been used successfully in the treatment of acyclovir-resistant HSV infections.
II. AGENTS TO TREAT CYTOMEGALOVIRUS (CMV) INFECTIONS
INTRODUCTION
CMV infections occur primarily in the setting of advanced immunosuppression and are typically due to reactivation of latent infection. Dissemination of infection results in end-organ disease, including retinitis, colitis, esophagitis, central nervous system disease, and pneumonitis. Although the incidence in HIV-infected patients has markedly decreased with the advent of potent antiretroviral therapy, reactivation of CMV infection after organ transplantation is still clinically prevalent.
The availability of oral valganciclovir and the ganciclovir intraocular implant has decreased the usage of intravenous ganciclovir, intravenous foscarnet, and intravenous cidofovir for the treatment of end-organ CMV disease (Table 49-2). Oral valganciclovir has largely replaced oral ganciclovir because of its lower pill burden. The choice of therapy in patients with CMV retinitis must also take into account the location of the lesion and the extent of immediate visual impairment or threat.
GANCICLOVIR
Ganciclovir is an acyclic guanosine analog (Figure 49-2) that requires activation by triphosphorylation before inhibiting the viral DNA polymerase. Initial phosphorylation is catalyzed by the virus-specified protein kinase phosphotransferase UL97 in CMV-infected cells. The activated compound competitively inhibits viral DNA polymerase and causes termination of viral DNA elongation (Figure 49-3).
Ganciclovir has in vitro activity against CMV, HSV, VZV, EBV, HHV-6, and KSHV (Kaposi's sarcoma-associated herpesvirus). Its activity against CMV is up to 100 times greater than that of acyclovir.
Ganciclovir may be administered intravenously, orally, or via intraocular implant. Cerebrospinal fluid concentrations are approximately 50% of those in serum. The elimination half-life is 4 hours with normal renal function and the intracellular half-life is 18 hours. Clearance of the drug is linearly related to creatinine clearance. Ganciclovir is readily cleared by hemodialysis. The bioavailability of oral ganciclovir is poor. In patients with an intraocular implant, ganclovir is released into the vitreous cavity at a rate of approximately 1.4 mcg/h.
Intravenous ganciclovir has been shown to delay progression of CMV retinitis in patients with AIDS when compared with no treatment. Dual therapy with foscarnet and ganciclovir has been shown to be more effective in delaying progression of retinitis than either drug administered alone (see Foscarnet), although side effects are compounded. Intravenous ganciclovir is also used to treat CMV colitis and esophagitis. The risk of Kaposi's sarcoma is reduced in AIDS patients receiving long-term ganciclovir. Intravenous ganciclovir, followed by either oral ganciclovir or high-dose oral acyclovir, reduces the risk of CMV infection in transplant recipients. Intravenous ganciclovir for CMV pneumonitis in immunocompromised patients may be beneficial, particularly in combination with intravenous cytomegalovirus immunoglobulin. Oral ganciclovir is indicated for prevention of end-organ CMV disease in AIDS patients and as maintenance therapy of CMV retinitis after induction. Although less effective than intravenous ganciclovir, the risk of myelosuppression and of catheter-related complications is diminished.
Ganciclovir may also be administered intraocularly to treat CMV retinitis, either by direct intravitreal administration or via an intraocular implant. The implant, which achieves high and prolonged intraocular levels of ganciclovir, has been shown to delay progression of retinitis to a greater degree than systemic therapy with ganciclovir. Surgical replacement is required at intervals of 5-8 months. Concurrent therapy with a systemic anti-CMV agent is recommended.
Resistance to ganciclovir increases with duration of usage. The more common mutation is in UL97, resulting in decreased levels of the triphosphorylated (ie, active) form of ganciclovir. The less common UL54 mutation in DNA polymerase results in higher levels of resistance and potential cross-resistance with cidofovir and foscarnet. Antiviral susceptibility testing is recommended in patients in whom resistance is suspected clinically, as is the substitution of alternative therapies and concomitant reduction in immunosuppressive therapies, if possible. The addition of CMV hyperimmune globulin may also be considered.
The most common adverse effect of systemic ganciclovir treatment, particularly after intravenous administration, is myelosuppression. Myelosuppression may be additive in patients receiving concurrent zidovudine, azathioprine, or mycophenolate mofetil. Other potential adverse effects are nausea, diarrhea, fever, rash, headache, insomnia, and peripheral neuropathy, as well as retinal detachment in patients with CMV retinitis. Central nervous system toxicity (confusion, seizures, psychiatric disturbance) and hepatotoxicity have been rarely reported. Ganciclovir is mitogenic in mammalian cells and carcinogenic and embryotoxic at high doses in animals and causes aspermatogenesis; the clinical significance of these preclinical data is unclear.
Levels of ganciclovir may rise in patients concurrently taking probenecid or trimethoprim. Concurrent use of ganciclovir with didanosine may result in increased levels of didanosine.
VALGANCICLOVIR
Valganciclovir is an L-valyl ester prodrug of ganciclovir that exists as a mixture of two diastereomers (Figure 49-2). After oral administration, both diastereomers are rapidly hydrolyzed to ganciclovir by intestinal and hepatic esterases.
Valganciclovir is well absorbed and rapidly metabolized in the intestinal wall and liver to ganciclovir; no other metabolites have been detected. The absolute bioavailability of oral valganciclovir is 60%; it is recommended that the drug be taken with food. The AUC0-24h resulting from valganciclovir (900 mg once daily) is similar to that after 5 mg/kg once daily of intravenous ganciclovir, and approximately 1.65 times that of oral ganciclovir. Plasma protein binding is less than 2%. The major route of elimination is renal¾through glomerular filtration and active tubular secretion. Plasma concentrations of valganciclovir are reduced approximately 50% by hemodialysis.
Valganciclovir is indicated for the treatment of CMV retinitis in patients with AIDS and for the prevention of CMV disease in high-risk kidney, heart, and kidney-pancreas transplant patients. Adverse effects, drug interactions, and resistance patterns are the same as those associated with ganciclovir.
FOSCARNET
Foscarnet (phosphonoformic acid) is an inorganic pyrophosphate compound (Figure 49-2) that inhibits viral DNA polymerase, RNA polymerase, and HIV reverse transcriptase directly without requiring activation by phosphorylation. It has in vitro activity against HSV, VZV, CMV, EBV, HHV-6, KSHV, and HIV-1.
Foscarnet is available in an intravenous formulation only; poor oral bioavailability and gastrointestinal intolerance preclude oral use. Cerebrospinal fluid concentrations are 43-67% of steady-state serum concentrations. Although the mean plasma half-life is 3-6.8 hours, up to 30% of foscarnet may be deposited in bone, with a half-life of several months. The clinical repercussions of this are unknown. Clearance of foscarnet is primarily by the kidney and is directly proportional to creatinine clearance. Serum drug concentrations are reduced approximately 50% by hemodialysis.
An effective treatment for CMV retinitis, foscarnet has an efficacy approximately equal to that of ganciclovir. Foscarnet is also used for treatment of CMV colitis, CMV esophagitis, acyclovir-resistant HSV infection, and acyclovir-resistant VZV infection. The dosage of foscarnet must be titrated according to the patient's calculated creatinine clearance before each infusion. Use of an infusion pump to control the rate of infusion is important to avoid toxicity, and relatively large volumes of fluid are required because of the drug's poor solubility. The combination of ganciclovir and foscarnet is synergistic in vitro against CMV and has been shown to be superior to either agent alone in delaying progression of retinitis; however, toxicity is also increased when both agents are administered concurrently. As with ganciclovir, a decrease in the incidence of Kaposi's sarcoma has been observed in patients who have received long-term foscarnet.
Foscarnet has been administered intravitreally for the treatment of CMV retinitis in patients with AIDS, but data regarding efficacy and safety are lacking.
Resistance to foscarnet in HSV and CMV isolates is due to point mutations in the DNA polymerase gene and is typically associated with prolonged or repeated exposure to the drug. Mutations in the HIV-1 reverse transcriptase gene have also been described. Although foscarnet-resistant CMV isolates are typically cross-resistant to ganciclovir, foscarnet activity is usually maintained against ganciclovir- and cidofovir-resistant isolates of CMV.
Potential adverse effects of foscarnet include renal impairment, hypo- or hypercalcemia, hypo- or hyperphosphatemia, hypokalemia, and hypomagnesemia. Saline preloading helps to prevent nephrotoxicity, as does avoidance of concomitant administration of drugs with nephrotoxic potential (eg, amphotericin B, pentamidine, aminoglycosides). The risk of severe hypocalcemia is increased with concomitant use of pentamidine. Penile ulcerations associated with foscarnet therapy may be due to high levels of ionized drug in the urine. Nausea, vomiting, anemia, elevation of liver enzymes, and fatigue have been reported; the risk of anemia may be additive in patients receiving concurrent zidovudine. Central nervous system toxicities include headache, hallucinations, and seizures; seizures may be increased with concurrent use of imipenem. Foscarnet caused chromosomal damage in preclinical studies.
CIDOFOVIR
Cidofovir (Figure 49-2) is a cytosine nucleotide analog with in vitro activity against CMV, HSV-1, HSV-2, VZV, EBV, HHV-6, KSHV, adenovirus, poxviruses, polyomaviruses, and human papillomavirus. In contrast to ganciclovir, phosphorylation of cidofovir to the active diphosphate is independent of viral enzymes (Figure 49-3). After phosphorylation, cidofovir acts both as a potent inhibitor of and as an alternative substrate for viral DNA polymerase, competitively inhibiting DNA synthesis and becoming incorporated into the viral DNA chain. Isolates with resistance to cidofovir have been selected in vitro; these isolates tend to be cross-resistant with ganciclovir but retain susceptibility to foscarnet. Clinical resistance to cidofovir has not been reported to date in patients.
Although the terminal half-life of cidofovir is about 2.6 hours, the active metabolite, cidofovir diphosphate, has a prolonged intracellular half-life of 17-65 hours, thus allowing widely spaced administration. A separate metabolite, cidofovir phosphocholine, has a half-life of at least 87 hours and may serve as an intracellular reservoir of active drug. Cerebrospinal fluid penetration is poor. Elimination involves active renal tubular secretion. High-flux hemodialysis has been shown to reduce the serum levels of cidofovir by approximately 75%.
Intravenous cidofovir is effective for the treatment of CMV retinitis and is used experimentally to treat adenovirus infections. Intravenous cidofovir must be administered with probenecid (2 g at 3 hours before the infusion and 1 g at 2 and 8 hours after), which blocks active tubular secretion and decreases nephrotoxicity. Cidofovir dosage must be adjusted for alterations in the calculated creatinine clearance or the presence of urine protein before each infusion, and aggressive adjunctive hydration is required. Initiation of cidofovir therapy is contraindicated in patients with existing renal insufficiency. Direct intravitreal administration of cidofovir is not recommended because of ocular toxicity.
The primary adverse effect of intravenous cidofovir is a dose-dependent nephrotoxicity, which may be reduced with prehydration using normal saline. Concurrent administration of other potentially nephrotoxic agents (eg, amphotericin B, aminoglycosides, nonsteroidal anti-inflammatory drugs, pentamidine, foscarnet) should be avoided. Prior administration of foscarnet may increase the risk of nephrotoxicity. Other potential side effects include uveitis, ocular hypotony, neutropenia (15%), and metabolic acidosis. Gastrointestinal intolerance, fever, and rash due to probenecid may occur. The drug caused mammary adenocarcinomas in rats and is embryotoxic.
III. ANTIRETROVIRAL AGENTS
INTRODUCTION
Substantial advances have been made in antiretroviral therapy since the introduction of the first agent, zidovudine, in 1987, and many antiretroviral agents are now available (Table 49-3). In addition, greater knowledge of viral dynamics through the use of viral load and resistance testing has made clear that combination therapy with maximally efficacious and potent agents will reduce viral replication to the lowest possible level and decrease the likelihood of emergence of resistance. Thus, administration of highly active antiretroviral therapy (HAART), typically comprising a combination of 3-4 antiretroviral agents, has become the standard of care. Such regimens may be composed of nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, protease inhibitors, and a fusion inhibitor (see below). Viral susceptibility to specific agents varies among patients and may change with time, owing to development of resistance. Therefore, such combinations must be chosen with care and tailored to the individual, as must changes to a given regimen. In addition to potency and susceptibility, important factors in the selection of agents for any given patient are tolerability, convenience, and optimization of adherence (see Box: Treatment of HIV-Infected Individuals: Importance of Pharmacokinetic Knowledge).
Four classes of antiretroviral agents are available for use: nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), and fusion inhibitors. As new agents have become available, several older ones have had diminished usage, because of either suboptimal safety profile or inferior antiviral potency.
TREATMENT OF HIV-INFECTED INDIVIDUALS: IMPORTANCE OF PHARMACOKINETIC KNOWLEDGE
Concurrent use of many medications is necessary in most HIV-infected patients. These medications include combinations of antiretroviral agents, prophylaxis or treatment for opportunistic infections, antiemetics, neuropsychiatric drugs, and opioid pain medications. Such extreme polypharmacy necessitates awareness of pharmacokinetic and pharmacodynamic interactions.
Perhaps the most important of the pharmacokinetic complications results from the metabolism of the NNRTI and PI agents by the CYP450 enzyme system, primarily the 3A4 isoform. Because many are inducers or inhibitors of CYP3A4 as well as substrates, drug-drug interactions may have marked clinical ramifications. However, variable effects on different CYP450 isoforms may make interactions somewhat unpredictable. For example, in the treatment of tuberculosis, the use of rifampin, a standard antimycobacterial agent but also one of the most potent 3A4 inducers, may either decrease efficacy (eg, atazanavir, lopinavir) or increase toxicity (eg, saquinavir) of concurrent antiretroviral agents, owing to alteration of serum levels. Increased levels of rifabutin (associated with uveitis) or trazodone (causing hypotension, syncope), when co-administered with ritonavir, may markedly increase toxicity. Increased levels of clarithromycin used for treatment or prophylaxis of Mycobacterium avium infection or as an antibacterial agent, when co-administered with indinavir, ritonavir, and atazanavir, may increase the potential for QT interval prolongation. Conversely, decreased levels of clarithromycin with efavirenz may reduce antibacterial efficacy. Most recently, these types of interactions have been used to advantage in the form of dual protease inhibitor regimens (boosted regimens), based on resultant increased plasma concentrations of the substrate (eg, lopinavir, saquinavir) when co-administered with an inducer (most often ritonavir). Improved drug exposure, increased antiviral potency, more convenient dosing, and improved tolerability result, thus improving patient adherence.
NUCLEOSIDE & NUCLEOTIDE REVERSE TRANSCRIPTASE INHIBITORS
INTRODUCTION
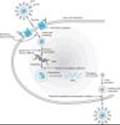
The NRTIs act by competitive inhibition of HIV-1 reverse transcriptase and can also be incorporated into the growing viral DNA chain to cause termination (Figure 49-4). Each requires intracytoplasmic activation via phosphorylation by cellular enzymes to the triphosphate form. Most have activity against HIV-2 as well as HIV-1.
Nucleoside analogs may be associated with mitochondrial toxicity, probably owing to inhibition of mitochondrial DNA polymerase gamma, and they can increase the risk of lactic acidosis with hepatic steatosis, which may be fatal, as well as disorders of lipid metabolism. NRTI treatment should be suspended in the setting of rapidly rising aminotransferase levels, progressive hepatomegaly, or metabolic acidosis of unknown cause.
|
|
Figure 49-4. Life cycle of HIV. Binding of viral glycoproteins to host cell CD4 and chemokine receptors precedes fusion and entry into the cell. After uncoating, reverse transcription copies the single-stranded HIV RNA genome into double-stranded DNA, which is integrated into the host cell genome. Gene transcription by host cell enzymes produces messenger RNA, which is translated into proteins that assemble into immature noninfectious virions that bud from the host cell membrane. Maturation into fully infectious virions is through proteolytic cleavage. |
ABACAVIR
Abacavir is a guanosine analog (Figure 49-2) that is well absorbed following oral administration (83%) and unaffected by food. The elimination half-life is 1.5 hours, and the intracellular half-life ranges from 12 to 26 hours. Cerebrospinal fluid levels are approximately one third those of plasma.
High-level resistance to abacavir appears to require at least two or three concomitant mutations (eg, M184V, L74V, D67N) and thus tends to develop slowly. The K65R mutation is associated with reduced susceptibility to lamivudine, abacavir, tenofovir, and emtricitabine.
Hypersensitivity reactions, occasionally fatal, have been reported in approximately 5% of patients receiving abacavir. Symptoms, which generally occur within the first 6 weeks of therapy, include fever, malaise, nausea, vomiting, diarrhea, and anorexia. Respiratory symptoms such as dyspnea, pharyngitis, and cough may also be present, and skin rash occurs in about 50% of patients. Laboratory abnormalities such as mildly elevated serum aminotransferase or creatine kinase levels may be present but are not specific for the hypersensitivity reaction. Although the syndrome tends to resolve quickly with discontinuation of medication, rechallenge with abacavir results in return of symptoms within hours and may be fatal. Other potential adverse events are rash, fever, nausea, vomiting, diarrhea, headache, dyspnea, fatigue, and pancreatitis (rare).
DIDANOSINE
Didanosine (ddI) is a synthetic analog of deoxyadenosine (Figure 49-2). Oral bioavailability is 30-40%; dosing on an empty stomach is required. Cerebrospinal fluid concentrations of the drug are approximately 20% of serum concentrations. The elimination half-life is 1.5 hours, but the intracellular half-life of the activated compound is as long as 20-24 hours. The drug is eliminated by glomerular filtration and tubular secretion. Dosage reduction is therefore required for low creatinine clearance and for low body weight (Table 49-3).
Buffered powder for oral solution and chewable tablets are taken twice daily; enteric-coated capsules can be taken once daily because of greater bioavailability. The buffer in the tablets and powder interferes with absorption of indinavir, delavirdine, dapsone, and itraconazole; therefore, concurrent administration is to be avoided. Because the tablets contain both phenylalanine (36.5 mg) and sodium (1380 mg), caution should be exercised in patients with phenylketonuria and those on sodium-restricted diets.
Resistance to didanosine is typically associated with the L74V mutation, although decreased susceptibility may also occur in the presence of K65R and multiple thymidine analog mutations (TAMs). These may partially restore susceptibility to zidovudine but may confer cross-resistance to abacavir, zalcitabine, and lamivudine. The M184V mutation is found in a significant proportion of isolates selected by didanosine and may confer resistance to lamivudine.
The major clinical toxicity associated with didanosine therapy is dose-dependent pancreatitis. Other risk factors for pancreatitis (eg, alcoholism, hypertriglyceridemia) are relative contraindications to administration of didanosine, and other drugs with the potential to cause pancreatitis, including zalcitabine and stavudine, should be avoided (Table 49-3). Other reported adverse effects include painful peripheral distal neuropathy, diarrhea (particularly with tablets and powder), hepatitis, esophageal ulceration, cardiomyopathy, and central nervous system toxicity (headache, irritability, insomnia). Asymptomatic hyperuricemia may precipitate attacks of gout in susceptible individuals. Reports of retinal changes and optic neuritis in patients receiving didanosine, particularly in adults receiving high doses and in children, mandate periodic retinal examinations.
Fluoroquinolones and tetracyclines should be administered at least 2 hours before or after didanosine to avoid decreased antibiotic plasma concentrations due to chelation. Serum levels of didanosine are increased when co-administered with tenofovir and ganciclovir, thus increasing the risk of toxicity; they are decreased by atazanavir, delavirdine, ritonavir, tipranavir, and methadone (Table 49-4).
EMTRICITABINE
Emtricitabine (formerly called FTC) is a fluorinated analog of lamivudine with a long intracellular half-life (> 39 hours), allowing for once-daily dosing (Figure 49-2). Oral bioavailability of the capsules is 93% and is unaffected by food, but penetration into the cerebrospinal fluid is low. Elimination is by both glomerular filtration and active tubular secretion. The mean plasma elimination half-life is 8-9 hours.
The oral solution, which contains propylene glycol, is contraindicated in young children, pregnant women, patients with renal or hepatic failure, and those using metronidazole or disulfiram. Also, because of its in vitro activity against HBV, patients co-infected with HIV and HBV should be closely monitored if treatment with emtricitabine is interrupted or discontinued, owing to the likelihood of hepatitis flares.
Like lamivudine, the M184V/I mutation is most frequently associated with emtricitabine use and may emerge rapidly in patients receiving HAART regimens that are not fully suppressive. There is cross-resistance to lamivudine but not to other NRTI agents. Isolates with the K65R mutation may have decreased susceptibility to emtricitabine. Because of their similar mechanisms of action and resistance profiles, the combination of lamivudine and emtricitabine is not recommended.
The most common adverse effects observed in patients receiving emtricitabine are headache, diarrhea, nausea, and asthenia. In addition, hyperpigmentation of the palms and/or soles may be observed (~ 3%), particularly in blacks (up to 13%). No drug-drug interactions of note have been reported to date.
LAMIVUDINE
Lamivudine (3TC) is a cytosine analog (Figure 49-2) with in vitro activity against HIV-1 that is synergistic with a variety of antiretroviral nucleoside analogs¾including zidovudine and stavudine¾against both zidovudine-sensitive and zidovudine-resistant HIV-1 strains. Activity against HBV is described below.
Oral bioavailability exceeds 80% and is not food-dependent. In children, the mean cerebrospinal fluid: plasma ratio of lamivudine was 0.2. Mean elimination half-life is 2.5 hours, whereas the intracellular half-life of the active 5'-triphosphate metabolite in HIV-1-infected cell lines is 10.5-15.5 hours. The majority of lamivudine is eliminated unchanged in the urine, and the dose should be reduced in patients with renal insufficiency or low body weight (Table 49-3).
Lamivudine therapy rapidly selects for the M184V mutation in regimens that are not fully suppressive; this mutation confers high-level resistance as well as a reduction in susceptibility to abacavir, didanosine, and zalcitabine. Conversely, the M184V mutation may restore phenotypic susceptibility to zidovudine, indicating that this two-drug combination regimen may be particularly beneficial. However, HIV-1 strains resistant to both lamivudine and zidovudine have been isolated. The K65R mutation is associated with reduced susceptibility to lamivudine, abacavir, tenofovir, and emtricitabine.
Potential adverse effects are headache, insomnia, fatigue, and gastrointestinal discomfort, although these are typically mild. Lamivudine's bioavailability increases when it is co-administered with trimethoprim-sulfamethoxazole. Lamivudine and zalcitabine may inhibit the intracellular phosphorylation of one another in vitro, thus decreasing potency; therefore, their concurrent use should be avoided if possible.
STAVUDINE
The thymidine analog stavudine (d4T) (Figure 49-2) has high oral bioavailability (86%) that is not food-dependent. The plasma half-life is 1.2 hours, the intracellular half-life is approximately 3.5 hours, and mean cerebrospinal fluid concentrations are 55% of those of plasma. Excretion is by active tubular secretion and glomerular filtration. The dosage of stavudine should be reduced in patients with renal insufficiency and low body weight (Table 49-3).
A number of mutations are associated with reduced susceptibility to stavudine; the predominant mutations are M41L, D67N, K70R, L210W, T215Y/F, and K219O.
The major dose-limiting toxicity is a dose-related peripheral sensory neuropathy. The incidence of neuropathy may be increased when stavudine is administered with other neuropathy-inducing drugs such as didanosine and zalcitabine. Symptoms typically resolve completely upon discontinuation of stavudine; in such cases, a reduced dosage may be cautiously restarted. Other potential adverse effects include pancreatitis, arthralgias, and elevation in serum aminotransferases. Lactic acidosis with hepatic steatosis, as well as fat atrophy, appears to occur more frequently in patients receiving stavudine than in those receiving other NRTI agents. Moreover, because the co-administration of stavudine and didanosine may increase the incidence of lactic acidosis and pancreatitis, concurrent use should be avoided, if possible. This combination has been implicated in several deaths in HIV-infected pregnant women. A rare side effect is a rapidly progressive ascending neuromuscular weakness. Since zidovudine may reduce the phosphorylation of stavudine, these two drugs should generally not be used together.
TENOFOVIR
Tenofovir is an acyclic nucleoside phosphonate (ie, nucleotide) analog of adenosine (Figure 49-2). Like the nucleoside analogs, tenofovir competitively inhibits HIV reverse transcriptase and causes chain termination after incorporation into DNA.
Tenofovir disopoxilfumarate is a water-soluble prodrug of active tenofovir. The oral bioavailability in fasted patients is approximately 25% and increases to 39% after a high-fat meal. Serum half-life is 17 hours and intracellular half-life is prolonged at more than 60 hours. Elimination occurs by a combination of glomerular filtration and active tubular secretion, and the dosage must be adjusted in patients with renal insufficiency.
The primary mutation associated with resistance to tenofovir is K65R, although varying degrees of decreased susceptibility to tenofovir may be conferred by zidovudine-associated mutations (eg, M41L, L210W), according to the number of specific mutations present.
Gastrointestinal complaints (eg, nausea, diarrhea, vomiting, flatulence) are the most common side effects but rarely require discontinuation of therapy. Other potential adverse effects include headache and asthenia. Preclinical studies in several animal species have demonstrated bone toxicity (eg, osteomalacia); however, to date there has been no evidence of bone toxicity in humans. Cases of renal impairment, including acute renal failure and Fanconi's syndrome, have been reported in patients receiving tenofovir. Tenofovir may compete with other drugs that are actively secreted by the kidneys, such as cidofovir, acyclovir, and ganciclovir. The combination of tenofovir with didanosine is associated with both decreased virologic efficacy and increased toxicity (due to increased didanosine levels) and therefore should be avoided.
ZALCITABINE
Zalcitabine (ddC) is a cytosine analog with high oral bioavailability (> 80%) and a relatively long intracellular half-life (10 hours) despite its elimination half-life of 1-2 hours (Figure 49-2). However, plasma levels decrease by 25-39% when the drug is administered with food or antacids. Cerebrospinal fluid concentrations are approximately 20% of those in the plasma.
Although a variety of mutations associated with in vitro resistance to zalcitabine have been described (eg, T69D, K65R, M184V, L74V), phenotypic resistance appears to be rare, particularly in combination regimens.
Zalcitabine therapy is associated with a dose-dependent peripheral neuropathy that can be treatment-limiting in 10-20% of patients but appears to be slowly reversible if treatment is stopped promptly. The potential for causing peripheral neuropathy constitutes a relative contraindication to use with other drugs that may cause this toxicity, including stavudine, didanosine, and isoniazid. Decreased renal clearance caused by amphotericin B, foscarnet, and aminoglycosides may increase the risk of zalcitabine neuropathy. The other major reported toxicity is oral and esophageal ulcerations. Pancreatitis occurs less frequently than with didanosine administration, but co-administration of other drugs that cause pancreatitis may increase the frequency of this adverse effect. Headache, nausea, rash, and arthralgias may occur but tend to be mild or resolve during therapy. Cardiomyopathy has rarely been reported. Zalcitabine causes thymic lymphomas in rodents, but no clinical correlates have been observed in humans.
The AUC of zalcitabine increases when co-administered with probenecid or cimetidine, and bioavailability decreases with concurrent antacids or metoclopramide. Lamivudine inhibits the phosphorylation of zalcitabine in vitro, potentially interfering with its efficacy.
ZIDOVUDINE
Zidovudine (azidothymidine; AZT) is a deoxythymidine analog (Figure 49-2) that is well absorbed from the gut and distributed to most body tissues and fluids, including the cerebrospinal fluid, where drug levels are 60-65% of those in serum. The serum half-life averages 1 hour, and the intracellular half-life of the phosphorylated compound is 3-7 hours. Zidovudine is eliminated primarily by renal excretion following glucuronidation in the liver. Clearance of zidovudine is reduced by approximately 50% in uremic patients, and toxicity may increase in patients with advanced hepatic insufficiency.
Zidovudine was the first antiretroviral agent to be approved and has been well studied. The drug has been shown to decrease the rate of clinical disease progression and prolong survival in HIV-infected individuals. Efficacy has also been demonstrated in the treatment of HIV-associated dementia and thrombocytopenia. In pregnancy (Table 49-5), a regimen of oral zidovudine beginning between 14 and 34 weeks of gestation (100 mg five times a day), intravenous zidovudine during labor (2 mg/kg over 1 hour, then 1 mg/kg/h by continuous infusion), and zidovudine syrup to the neonate from birth through 6 weeks of age (2 mg/kg every 6 hours) has been shown to reduce the rate of vertical (mother-to-newborn) transmission of HIV by up to 23%.
As with other NRTI agents, resistance may limit clinical efficacy. High-level zidovudine resistance is generally seen in strains with three or more of the five most common mutations: M41L, D67N, K70R, T215F, and K219Q. However, the emergence of certain mutations that confer decreased susceptibility to one drug (eg, L74V for didanosine and M184V for lamivudine) seems to enhance zidovudine susceptibility in previously zidovudine-resistant strains. Withdrawal of zidovudine exposure may permit the reversion of zidovudine-resistant HIV-1 isolates to the susceptible wild-type phenotype.
The most common adverse effect of zidovudine is myelosuppression, resulting in macrocytic anemia (1-4%) or neutropenia (2-8%). Gastrointestinal intolerance, headaches, and insomnia may occur but tend to resolve during therapy. Less frequent toxicities include thrombocytopenia, hyperpigmentation of the nails, and myopathy. Very high doses can cause anxiety, confusion, and tremulousness. Zidovudine causes vaginal neoplasms in mice; however, no human cases of genital neoplasms have been reported to date.
Increased serum levels of zidovudine may occur with concomitant administration of probenecid, phenytoin, methadone, fluconazole, atovaquone, valproic acid, and lamivudine, either through inhibition of first-pass metabolism or through decreased clearance. Zidovudine may decrease phenytoin levels, and this warrants monitoring of serum phenytoin levels in epileptic patients taking both agents. Hematologic toxicity may be increased during co-administration of other myelosuppressive drugs such as ganciclovir, ribavirin, and cytotoxic agents. Combination regimens containing zidovudine and stavudine should be avoided; antagonism has been demonstrated in vitro.
NONNUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS
INTRODUCTION
The NNRTIs bind directly to HIV-1 reverse transcriptase (Figure 49-4), resulting in blockade of RNA- and DNA-dependent DNA polymerase. The binding site of NNRTIs is near to but distinct from that of NRTIs. Unlike the NRTI agents, NNRTIs neither compete with nucleoside triphosphates nor require phosphorylation to be active. NNRTI resistance occurs rapidly with monotherapy and can be due to a single mutation (eg, K103N). The K103N and Y181C mutations confer resistance across the entire class of NNRTIs, whereas L100I, Y188C, and G190A confer resistance to both nevirapine and efavirenz.
As a class, NNRTI agents tend to be associated with varying levels of gastrointestinal intolerance and skin rash, the latter of which may infrequently be serious (eg, Stevens-Johnson syndrome). A further limitation to use of NNRTI agents as a component of HAART is their metabolism by the CYP450 system, leading to innumerable potential drug-drug interactions (Tables 49-3 and 49-4). NNRTI agents are all substrates for CYP3A4 and can act as inducers (nevirapine), inhibitors (delavirdine), or mixed inducers and inhibitors (efavirenz). Given the large number of non-HIV medications that are also metabolized by this pathway (see Chapter 4); drug-drug interactions must be expected and looked for.
DELAVIRDINE
Delavirdine has an oral bioavailability of about 85%, but this is reduced by antacids or H2-blockers. It is extensively bound (~ 98%) to plasma proteins and has correspondingly low cerebrospinal fluid levels.
Skin rash occurs in approximately 18% of patients receiving delavirdine; it typically occurs during the first 1-3 weeks of therapy and does not preclude rechallenge. However, severe rash such as erythema multiforme and Stevens-Johnson syndrome have rarely been reported. Other possible adverse effects are headache, fatigue, nausea, diarrhea, and increased serum aminotransferase levels. Delavirdine has been shown to be teratogenic in rats, causing ventricular septal defects and other malformations at dosages not unlike those achieved in humans. Thus, pregnancy should be avoided when taking delavirdine.
Delavirdine is extensively metabolized to inactive metabolites by the CYP3A and CYP2D6 enzymes and also inhibits CYP3A4 and 2C9. Therefore, there are numerous potential drug-drug interactions to consider (Tables 49-3 and 49-4). Because of this effect, however, co-administration with delavirdine may allow indinavir or saquinavir to be dosed twice daily rather than the usual three times a day. The concurrent use of delavirdine with amprenavir/fosamprenavir and rifabutin is not recommended because of decreased delavirdine levels.
EFAVIRENZ
Efavirenz can be given once daily because of its long half-life (40-55 hours). It is moderately well absorbed following oral administration (45%). Since toxicity may increase owing to increased bioavailability after a high-fat meal, efavirenz should be taken on an empty stomach. Peak plasma concentrations occur 3-5 hours after administration of daily doses; steady-state plasma concentrations are reached in 6-10 days. Efavirenz is principally metabolized by CYP3A4 and CYP2B6 to inactive hydroxylated metabolites; the remainder is eliminated in the feces as unchanged drug. It is highly bound to albumin (~ 99%), and cerebrospinal fluid levels range from 0.3% to 1.2% of plasma levels.
The principal adverse effects of efavirenz involve the central nervous system (dizziness, drowsiness, insomnia, headache, confusion, amnesia, agitation, delusions, depression, nightmares, euphoria); these may occur in up to 50% of patients and may be severe. However, they tend to resolve after the first month of treatment. Skin rash has also been reported early in therapy in up to 28% of patients, is usually mild to moderate in severity, and typically resolves despite continuation. Other potential adverse reactions are nausea, vomiting, diarrhea, crystalluria, elevated liver enzymes, and an increase in total serum cholesterol by 10-20%. High rates of fetal abnormalities occurred in pregnant monkeys exposed to efavirenz in doses roughly equivalent to the human dosage; several cases of congenital anomalies have been reported in humans. Therefore, efanirenz should be avoided in pregnant women, particularly in the first trimester.
Efavirenz is both an inducer and an inhibitor of CYP3A4, thus inducing its own metabolism and interacting with the metabolism of many other drugs (Tables 49-3 and 49-4).
NEVIRAPINE
The oral bioavailability of nevirapine is excellent (~ 90%) and is not food-dependent. The drug is highly lipophilic and achieves cerebrospinal fluid levels that are 45% of those in plasma. Serum half-life is 25-30 hours. It is extensively metabolized by the CYP3A isoform to hydroxylated metabolites and then excreted, primarily in the urine.
In addition to its use as a component of a combination antiretroviral regimen, a single dose of nevirapine (200 mg) has been shown to be effective in the prevention of transmission of HIV from mother to newborn when administered to women at the onset of labor and followed by a 2-mg/kg oral dose to the neonate within 3 days after delivery. However, resistance has been documented after this single dose.
Rash occurs in approximately 17% of patients, most typically in the first 4-6 weeks of therapy, and is dose-limiting in about 7% of patients. When initiating therapy, gradual dose escalation over 14 days is recommended to decrease the incidence of rash. Women may have a greater propensity for rash. Severe and life-threatening skin rashes have been rarely reported, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Nevirapine therapy should be immediately discontinued in patients with severe rash and in those with accompanying constitutional symptoms. Hepatotoxicity occurs in about 4% of patients and appears to occur more frequently in those with higher pre-therapy CD4 cell counts (ie, > 250 cells/mm3 in women and > 400 cells/mm3 in men), in women, and in those with hepatitis B or C co-infection. Fulminant hepatitis may rarely occur, typically within the first 18 weeks of therapy and can be fatal. Other adverse effects associated with nevirapine therapy are fever, nausea, headache, and somnolence.
Nevirapine is a moderate inducer of CYP3A metabolism, resulting in decreased levels of amprenavir, indinavir, lopinavir, saquinavir, efavirenz, and methadone if administered concurrently (Table 49-4). Drugs that induce the CYP3A system, such as tipranavir, rifampin, rifabutin, and St. John's wort, can decrease levels of nevirapine, whereas those that inhibit CYP3A activity, such as fluconazole, ketoconazole, and clarithromycin, can increase nevirapine levels.
PROTEASE INHIBITORS
INTRODUCTION
During the later stages of the HIV growth cycle, the Gag and Gag-Pol gene products are translated into polyproteins, and these become immature budding particles. Protease is responsible for cleaving these precursor molecules to produce the final structural proteins of the mature virion core. By preventing cleavage of the Gag-Pol polyprotein, protease inhibitors (PIs) result in the production of immature, noninfectious viral particles (Figure 49-4). Unfortunately, specific genotypic alterations that confer phenotypic resistance are fairly common with these agents, thus contraindicating monotherapy. Some of the most common mutations conferring resistance to most if not all of the PI agents are L10I/R/V, M46I/L, I54V/M/L, V82A/F/T/S, and I84V.
A syndrome of redistribution and accumulation of body fat that results in central obesity, dorsocervical fat enlargement (buffalo hump), peripheral and facial wasting, breast enlargement, and a cushingoid appearance has been observed in patients receiving antiretroviral therapy. These abnormalities may be particularly associated with the use of PIs, although the recently licensed atazanavir appears to be an exception (see below). Concurrent increases in triglyceride and LDL levels, along with glucose intolerance and insulin resistance, have also been noted. The cause is not yet known.
Protease inhibitors have been associated with increased spontaneous bleeding in patients with hemophilia A or B.
All of the antiretroviral PIs are substrates and inhibitors of CYP3A4, with ritonavir having the most pronounced inhibitory effect and saquinavir the least. Some PI agents such as amprenavir and ritonavir are also inducers of specific CYP isoforms. As a result, there is enormous potential for drug-drug interactions with other antiretroviral agents and other commonly used medications (Tables 49-3 and 49-4). It is noteworthy that the potent CYP3A4 inhibitory properties of ritonavir have been utilized to clinical advantage by having it "boost" the levels of other PI agents when given in combination.
AMPRENAVIR
Amprenavir is rapidly absorbed from the gastrointestinal tract and can be taken with or without food. However, high-fat meals decrease absorption and thus should be avoided. The plasma half-life is relatively long (7-10.6 hours). Amprenavir is metabolized in the liver by CYP3A4 and should be used with caution in the setting of hepatic insufficiency.
The key mutation conferring resistance to amprenavir appears to be I50V. Evidence to date suggests that cross-resistance to other members of the PI class of drugs may be less prevalent with amprenavir than with previously available compounds.
The most common adverse effects of amprenavir are nausea, diarrhea, vomiting, perioral paresthesias, depression, and rash. Up to 3% of patients in clinical trials to date have had rashes (including Stevens-Johnson syndrome) severe enough to warrant drug discontinuation.
Amprenavir is both an inducer and an inhibitor of CYP3A4 and is contraindicated with numerous other drugs (Tables 49-3 and 49-4). The oral solution, which contains propylene glycol, is contraindicated in young children, pregnant women, patients with renal or hepatic failure, and those using metronidazole or disulfiram. Also, the oral solutions of amprenavir and ritonavir should not be co-administered because the propylene glycol in one and the ethanol in the other may compete for the same metabolic pathway, leading to accumulation of either. Because the oral solution also contains vitamin E at several times the recommended daily dosage, supplemental vitamin E should be avoided. Amprenavir is contraindicated in patients with a history of sulfa allergy because it is itself a sulfonamide. Lopinavir/ritonavir should not be co-administered with amprenavir owing to decreased amprenavir and increased lopinavir exposures. An increased dosage of amprenavir is recommended when co-administered with efavirenz (with or without the addition of ritonavir to boost levels).
ATAZANAVIR
Atazanavir is a newer azapeptide PI with a pharmacokinetic profile that allows once-daily dosing. Its oral bioavailability is approximately 60-68%; the drug should be taken with food. Atazanavir requires an acidic medium for absorption and exhibits pH-dependent aqueous solubility; therefore, separation of ingestion from acid-reducing agents by at least 12 hours is recommended. Atazanavir is able to penetrate both the cerebrospinal and seminal fluids. The plasma half-life is 6-7 hours, which increases to approximately 11 hours when co-administered with ritonavir. The primary route of elimination is biliary; atazanavir should not be given to patients with severe hepatic insufficiency.
Resistance to atazanavir has been associated with various known PI mutations including the novel I50L substitution, which has been associated with increased susceptibility to other PIs.
The most common adverse effects in patients receiving atazanavir in clinical trials were nausea, vomiting, diarrhea, abdominal pain, headache, peripheral neuropathy, and skin rash. As with indinavir, indirect hyperbilirubinemia with overt jaundice may occur, in all likelihood owing to inhibition of the UGT1A1 enzyme. Although bilirubinemia is not regularly associated with hepatic injury, elevation of hepatic enzymes has also been observed, usually in patients with underlying hepatitis B or C infection. In contrast to the other PIs, atazanavir does not appear to be associated with dyslipidemias, fat redistribution, or the metabolic syndrome. Atazanavir may be associated with electrocardiographic PR interval prolongation, which is usually inconsequential but may be exacerbated by other causative agents such as calcium channel blockers. Also, a possible concentration-dependent increase in the QTc interval may occur in patients receiving atazanavir in dosages greater than 400 mg/d or in conjunction with the CYP3A4 inhibitor clarithromycin.
As an inhibitor of CYP3A4 and CYP2C9, the potential for drug-drug interactions with atazanavir is great (see Tables 49-3 and 49-4). Atazanavir AUC is reduced by 76% on average when combined with omeprazole; thus, the combination is to be avoided. In addition, co-administration of atazanavir with other drugs that inhibit the glucuronidation enzyme UGT1A1, such as indinavir and irinotecan, is contraindicated because of enhanced toxicity. Tenofovir and efavirenz should not be co-administered with atazanavir unless ritonavir is added to boost levels.
FOSAMPRENAVIR
Fosamprenavir is a prodrug of amprenavir that is rapidly hydrolyzed by enzymes in the intestinal epithelium. Tablets may be taken with or without food. Because of its significantly lower daily pill burden, fosamprenavir tablets have replaced amprenavir capsules for adults. All pharmacokinetic and pharmacodynamic attributes are those of amprenavir (see above).
INDINAVIR
Indinavir must be consumed on an empty stomach for maximal absorption; however, if co-administered with ritonavir, it may be taken without regard to food. Oral bioavailability is about 65%, and the drug has a high level of cerebrospinal fluid penetration (up to 76% of serum levels). Serum half-life is 1.5-2 hours. Excretion is primarily fecal. An increase in AUC by 60% and in half-life to 2.8 hours in the setting of hepatic insufficiency necessitates dose reduction.
Resistance may be associated with multiple mutations, particularly at positions 46 and 82, and the number of codon alterations (typically substitutions) tends to predict the level of phenotypic resistance. Resistance to indinavir is associated with a loss of susceptibility to ritonavir.
The most common adverse effects are indirect hyperbilirubinemia and nephrolithiasis due to crystallization of the drug. Nephrolithiasis can occur within days after initiating therapy, with an estimated incidence of 10-20%, and it may be associated with renal failure. Consumption of at least 48 ounces of water daily is important to maintain adequate hydration and prevent nephrolithiasis. Thrombocytopenia, elevations of serum aminotransferase levels, nausea, diarrhea, and irritability have also been reported. Insulin resistance may be more common with indinavir than with the other PI agents, occurring in 3-5% of patients. There have also been rare cases of acute hemolytic anemia. In rats, high doses of indinavir are associated with development of thyroid adenomas.
Since indinavir is an inhibitor of CYP3A4, numerous and complex drug interactions can occur (Tables 49-3 and 49-4). Combination with ritonavir (boosting) allows for twice-daily rather than thrice-daily dosing and eliminates the food restriction associated with use of indinavir. However, there is potential for an increase in nephrolithiasis with this combination compared with indinavir alone; thus, a high fluid intake (1.5-2 L/d) is advised.
LOPINAVIR/RITONAVIR
Lopinavir 100/ritonavir 400 is a licensed combination in which subtherapeutic doses of ritonavir inhibit the CYP3A-mediated metabolism of lopinavir, thereby resulting in increased exposure to lopinavir. Trough levels of lopinavir are greater than the median HIV-1 wild-type 50% inhibitory concentration, thus maintaining potent viral suppression as well as providing a pharmacologic barrier to the emergence of resistance. In this combination, therefore, ritonavir is acting as a pharmacokinetic enhancer rather than an antiretroviral agent. In addition to improved patient compliance due to reduced pill burden, lopinavir/ritonavir is generally well tolerated.
Absorption of lopinavir is enhanced with food. The oral solution contains alcohol. Lopinavir is extensively metabolized by the CYP3A isozyme of the hepatic cytochrome P450 system, which is inhibited by ritonavir. Serum levels of lopinavir may be increased in patients with hepatic impairment.
The most common adverse effects of lopinavir are diarrhea, abdominal pain, nausea, vomiting, and asthenia. Potential drug-drug interactions are extensive (see ritonavir and Tables 49-3 and 49-4). Increased dosage of lopinavir/ritonavir is recommended when co-administered with efavirenz or nevirapine, which induce lopinavir metabolism. Concurrent use of fosamprenavir should be avoided owing to increased exposure to lopinavir with decreased levels of amprenavir.
NELFINAVIR
Nelfinavir has higher absorption in the fed state (increased AUC by two- to threefold), undergoes metabolism by CYP3A, and is excreted primarily in the feces. The plasma half-life in humans is 3.5-5 hours. The D30N mutation appears to be particularly closely linked with phenotypic resistance in isolates obtained from clinical trials.
The most common adverse effects associated with nelfinavir are diarrhea and flatulence. Diarrhea often responds to antidiarrheal medications but can be dose-limiting. Like other PIs, nelfinavir is an inhibitor of the CYP3A system, and multiple drug interactions may occur (Tables 49-3 and 49-4). An increased dosage of nelfinavir is recommended when co-administered with rifabutin (with a decreased dose of rifabutin), whereas a decrease in saquinavir dose is suggested with concurrent nelfinavir. Nelfinavir has a favorable safety and pharmacokinetic profile for pregnant women compared with that of other PIs (Table 49-5).
RITONAVIR
Ritonavir is an inhibitor of HIV-1 and HIV-2 proteases with high bioavailability (about 75%) that increases when the drug is given with food. Metabolism to an active metabolite occurs via the CYP3A and CYP2D6 isoforms; excretion is primarily in the feces. Caution is advised when administering the drug to persons with impaired hepatic function.
Resistance is associated with mutations at positions 84, 82, 71, 63, and 46, of which the I84V mutation appears to be the most critical.
The most common adverse effects of ritonavir are gastrointestinal disturbances, paresthesias (circumoral and peripheral), elevated serum aminotransferase levels, altered taste, and hypertriglyceridemia. Nausea, vomiting, and abdominal pain typically occur during the first few weeks of therapy. Slow dose escalation over 4-5 days is recommended to decrease the dose-limiting side effects. Liver adenomas and carcinomas have been induced in male mice receiving ritonavir; no similar effects have been observed to date in humans.
Ritonavir is a potent inhibitor of CYP3A4; as such, co-administration with agents heavily metabolized by CYP3A must be approached with caution (Tables 49-3 and 49-4). In addition, therapeutic levels of digoxin and theophylline should be monitored when co-administered with ritonavir owing to likely increase in their concentrations. However, the CYP3A4 inhibitory properties of ritonavir have been exploited to raise the trough concentration and prolong the half-life of more potent and less toxic PI agents. Thus, lower than therapeutic doses of ritonavir are commonly given in combination with agents such as lopinavir, indinavir, or amprenavir to reduce the risk of resistance by increasing the time of drug exposure. Moreover, the prolonged half-life allows for less frequent dosing of the other PI agent, thus enhancing adherence.
SAQUINAVIR
In its original formulation as a hard gel capsule (saquinavir-H; Invirase), oral saquinavir is poorly bioavailable (only about 4% after food). It was therefore largely replaced in clinical use by a soft gel capsule formulation (saquinavir-S; Fortovase) in which absorption was increased approximately threefold. However, reformulation of saquinavir-H for once-daily dosing in combination with low-dose ritonavir (see below) has both improved antiviral efficacy and decreased the gastrointestinal side effects typically associated with saquinavir-S. Moreover, co-administration of saquinavir-H with ritonavir results in blood levels of saquinavir similar to those associated with saquinavir-S, thus capitalizing on the pharmacokinetic interaction of the two agents. The manufacturer announced plans to discontinue the manufacture of saquinavir-S in early 2006.
The most common critical resistance mutations are L90M and G48V, conferring an approximately tenfold decrease in susceptibility to saquinavir.
Both formulations of saquinavir should be taken within 2 hours after a fatty meal for enhanced absorption. Saquinavir has a large volume of distribution, but penetration into the cerebrospinal fluid is negligible. The elimination half-life is 12 hours. Excretion is primarily in the feces. Reported adverse effects include gastrointestinal discomfort (nausea, diarrhea, abdominal discomfort, dyspepsia; these are more common with saquinavir-S) and rhinitis.
Saquinavir is subject to extensive first-pass metabolism by CYP3A4, and functions as a CYP3A4 inhibitor as well as a substrate; thus, it should be used with the same precautions regarding drug-drug interactions as are the other PIs (Table 49-4). Co-administration with the CYP3A4 inhibitor ritonavir has been adopted by clinicians because of the higher¾and thus more efficacious¾levels of saquinavir while enabling reduction in daily dose and frequency of saquinavir. A decreased dose of saquinavir is recommended when co-administered with nelfinavir. Liver function tests should be monitored if saquinavir is co-administered with delavirdine or rifampin.
TIPRANAVIR
Tipranavir is another newer PI. Bioavailability is poor but is increased when taken with a high-fat meal. The drug is metabolized by the liver microsomal system. Tipranavir must be taken in combination with ritonavir to achieve effective serum levels. It is contraindicated in patients with hepatic insufficiency. Tipranavir contains a sulfonamide moiety and should not be administered to patients with known sulfa allergy.
The most common adverse effects are diarrhea, nausea, vomiting, abdominal pain, and rash; the latter is more common in women. Liver toxicity, including life-threatening hepatic decompensation, has been observed and is more common in patients with chronic hepatitis B or C. In 2006 a black box warning was added noting a possible increase in intracranial hemorrhage in patients taking tipranavir. Other potential adverse effects include depression; elevations in total cholesterol, triglycerides, and amylase; and decreased white blood cell count.
Tipranavir both inhibits and induces the CYP3A4 system. When used in combination with ritonavir, its net effect is inhibition. Tipranavir also induces P-glycoprotein transporter and thus may alter the disposition of many other drugs (see Table 49-4). Concurrent administration of tipranavir with amprenavir or saquinavir should be avoided owing to decreased blood levels of the latter drugs.
FUSION INHIBITORS¾ENFUVIRTIDE
Enfuvirtide (formerly called T-20) is the first representative of a new class of antiretroviral agents: It is a fusion inhibitor that blocks entry into the cell (Figure 49-4). Enfuvirtide, a synthetic 36-amino-acid peptide, binds to the gp41 subunit of the viral envelope glycoprotein, preventing the conformational changes required for the fusion of the viral and cellular membranes. Enfuvirtide must be administered by subcutaneous injection. Metabolism appears to be by proteolytic hydrolysis without involvement of the CYP450 system. Elimination half-life is 3.8 hours.
Resistance to enfuvirtide can occur, and the frequency and mechanisms of this phenomenon are being investigated. However, enfuvirtide lacks cross-resistance to the other currently approved antiretroviral drug classes.
The most common adverse effects associated with enfuvirtide therapy are local injection site reactions. Hypersensitivity reactions may rarely occur, are of varying severity, and may recur on rechallenge. Eosinophilia has also been noted. No interactions have been identified that would require the alteration of the dosage of other antiretroviral drugs.
INVESTIGATIONAL ANTIRETROVIRAL AGENTS
New therapies are being sought that offer convenient dosing, lower incidence of adverse effects, new viral targets, and activity against resistant viruses. Agents under evaluation or reformulation for once-daily dosing include stavudine and nevirapine. New agents currently in advanced stages of clinical development include the NRTI agent elvucitabine, the NNRTI agents TMC-125 and TMC-278, the PI agents TMC-114* and GSK-640385, and chemokine co-receptor inhibitors to block virus entry such as maraviroc. In addition, new drug classes such as maturation inhibitors and integrase inhibitors are under clinical investigation.
IV. ANTIHEPATITIS AGENTS
INTRODUCTION
Several agents effective against hepatitis B virus (HBV) and hepatitis C virus (HCV) are now available (Table 49-6). Although treatment is suppressive rather than curative, the high prevalence of these infections worldwide, with their concomitant morbidity and mortality, reflect a critical need for improved therapeutics.
INTERFERON ALFA
Interferons are host cytokines that exert complex antiviral, immunomodulatory, and antiproliferative activities (see Chapter 56). Interferon (IFN)-alfa appears to function by induction of intracellular signals following binding to specific cell membrane receptors, resulting in inhibition of viral penetration, translation, transcription, protein processing, maturation, and release, as well as increased expression of major histocompatibility complex antigens, enhanced phagocytic activity of macrophages, and augmentation of the proliferation and survival of cytotoxic T cells.
Injectable preparations of interferon alfa are available for treatment of both HBV and HCV virus infections (Table 49-6). Interferon alfa-2a and interferon alfa-2b
*TMC-114 was licensed as darunavir in the USA in 2006, to be co-administered with ritonavir in treatment-experienced patients with resistance to other PIs (Table 49-3).
may be administered subcutaneously or intramuscularly, whereas interferon alfacon-1 is administered subcutaneously. Elimination half-life is 2-5 hours for interferon alfa-2a and -2b, depending on the route of administration. The half-life of interferon alfacon-1 in patients with chronic hepatitis C ranges from 6 hours to 10 hours. Alfa interferons are filtered at the glomeruli and undergo rapid proteolytic degradation during tubular reabsorption, such that detection in the systemic circulation is negligible. Liver metabolism and subsequent biliary excretion are considered minor pathways.
Pegylated interferon alfa-2a and pegylated interferon alfa-2b have recently been introduced for the treatment of patients with HBV and HCV infections. Slower clearance of these agents results in substantially longer terminal half-lives and steadier drug concentrations, allowing for less frequent dosing. Renal elimination accounts for about 30% of clearance, and clearance is approximately halved in subjects with impaired renal function; dosage must therefore be adjusted.
In patients with chronic HBV infection, a recent meta-analysis of clinical trials showed that treatment with interferon alfa is associated with a higher incidence of hepatitis e antigen (HBeAg) seroconversion and undetectable HBV DNA levels than placebo. The addition of the pegylated moiety results in further increases in the proportion of patients with HBeAg seroconversion (~ 30%) and a decline by approximately 4 log copies/mL (99.99%) in HBV DNA after 1 year. Trials of the pegylated interferon alfas in chronic HCV infection are discussed below.
Typical side effects of interferon alfa include a flu-like syndrome (ie, headache, fevers, chills, myalgias, and malaise) that occurs within 6 hours after dosing in more than 30% of patients during the first week of therapy and tends to resolve upon continued administration. Transient hepatic enzyme elevations may occur in the first 8-12 weeks of therapy and appear to be more common in responders. Potential adverse effects during chronic therapy include neurotoxicities (mood disorders, depression, somnolence, confusion, seizures), myelosuppression, profound fatigue, weight loss, rash, cough, myalgia, alopecia, tinnitus, reversible hearing loss, retinopathy, pneumonitis, and possibly cardiotoxicity. Induction of autoantibodies may occur, causing exacerbation or unmasking of autoimmune or thyroid disease. The polyethylene glycol molecule is a nontoxic polymer that is readily excreted in the urine.
Contraindications to interferon alfa therapy include hepatic decompensation, autoimmune disease, and history of cardiac arrhythmia. Caution is advised in the setting of psychiatric disease, epilepsy, thyroid disease, ischemic cardiac disease, severe renal insufficiency, and cytopenia. Alfa interferons are abortifacient in primates and should not be administered in pregnancy. Potential drug-drug interactions include increased theophylline levels and increased methadone levels. Combination therapy with NRTI agents may cause hepatic failure; in particular, co-administration with didanosine is not recommended. Co-administration with zidovudine may exacerbate cytopenias.
TREATMENT OF HEPATITIS B VIRUS INFECTION
INTRODUCTION
The most common efficacy end points in clinical trials of hepatitis B virus infection are seroconversion from HBeAg from positive to negative and suppression of HBV DNA to undetectable levels. These end points are correlated with improvement in necroinflammatory disease, a decreased risk of hepatocellular carcinoma and cirrhosis, and a decreased need for liver transplantation. However, because current therapies suppress HBV replication rather than eradicate the virus, initial responses may not be durable. The covalently closed circular (ccc) DNA exists in stable form indefinitely within the cell, serving as a reservoir for HBV throughout the life of the cell and resulting in the capacity to reactivate. Relapse is more common in patients co-infected with HBV and hepatitis D virus.
As of 2006 there were three oral nucleoside/nucleotide analogs and two injectable interferon drugs available in the United States for the treatment of chronic HBV infection. Although three of the current antiretroviral NRTIs (emtricitabine, lamivudine, and tenofovir) have potent activity against HBV, only lamivudine is approved for clinical treatment. Although not FDA-approved, tenofovir is recommended by recent consensus guidelines for the treatment of patients co-infected with HBV and HIV-1. The anti-herpes agent famciclovir also has anti-HBV activity, but it is relatively weak and requires thrice-daily dosing. Because NRTI agents may be used in patients co-infected with hepatitis B and HIV, it is important to note that acute exacerbation of hepatitis may occur upon discontinuation or interruption of these agents.
LAMIVUDINE
The pharmacokinetics of lamivudine are described earlier in this chapter (see page 804). The more prolonged intracellular half-life in HBV cell lines (17-19 hours) than in HIV-infected cell lines (10.5-15.5 hours) allows for lower doses and less frequent administration. Lamivudine can be safely administered to patients with decompensated liver disease.
Lamivudine inhibits HBV DNA polymerase and HIV reverse transcriptase by competing with deoxycytidine triphosphate for incorporation into the viral DNA, resulting in chain termination. Lamivudine achieves 3-4 log decreases in viral replication in most patients and suppression of HBV DNA to undetectable levels in about 44% of patients. Seroconversion of HBeAg from positive to negative occurs in about 17% of patients and is durable at 3 years in about 70% of responders. Continuation of treatment for 4-8 months after seroconversion may improve the durability of response. Response in HBeAg-negative patients is initially high but less durable.
Chronic therapy with lamivudine in patients with hepatitis may ultimately be limited by the emergence of lamivudine-resistant HBV isolates (eg, with the YMDD mutation). Resistance has been associated with flares of hepatitis and progressive liver disease. Cross-resistance between lamivudine and either emtricitabine or entecavir may occur; however, cross-resistance between lamivudine and adefovir has not been reported.
In the doses used for HBV infection, lamivudine has an excellent safety profile. Headache, nausea, and dizziness are rare. Co-infection with HIV may increase the risk of pancreatitis. No evidence of mitochondrial toxicity has been reported.
ADEFOVIR DIPIVOXIL
Although initially and abortively developed for treatment of HIV infection, adefovir dipivoxil gained approval, at lower and less toxic doses, for treatment of HBV infection. Adefovir dipivoxil is the diester prodrug of adefovir, an acyclic phosphonated adenine nucleotide analog (Figure 49-2). It is phosphorylated by cellular kinases to the active diphosphate metabolite and then competitively inhibits HBV DNA polymerase to result in chain termination after incorporation into the viral DNA. Naturally occuring (ie, primary) adefovir-resistant rt233 HBV mutants have recently been described.
Oral bioavailability is about 59% and is unaffected by meals. The terminal elimination half-life is approximately 7.5 hours. Adefovir is excreted by a combination of glomerular filtration and active tubular secretion and thus may be administered to patients with decompensated liver disease.
Recent placebo-controlled trials showed that adefovir resulted in a mean of 3.5 logs reduction of HBV DNA copies/mL, normalization of aspartate aminotransferase in 48%-72% of patients, and improvement in liver histology and fibrosis in 53%-64% of patients at 48 weeks. More prolonged therapy results in higher rates of response, with anti-HBeAg seroconversion in 23% by 72 weeks and improved inflammation and fibrosis on liver biopsy at 5 years. Although emergence of resistance during therapy is rare (~ 4% after 3 years of use), specific mutations have been associated with viral rebound. There is no cross-resistance between adefovir and lamivudine.
Adefovir dipivoxil is well tolerated. A dose-dependent nephrotoxicity has been observed in clinical trials, manifested by increased serum creatinine with decreased serum phosphorous and more common in patients with baseline renal insufficiency. Other potential adverse effects are headache, diarrhea, asthenia, and abdominal pain. As with other NRTI agents, lactic acidosis and hepatic steatosis are considered a risk owing to mitochondrial dysfunction. Adefovir is embryotoxic in rats.
ENTECAVIR
Entecavir is an orally administered guanosine nucleoside analog (Figure 49-2) that competitively inhibits all three functions of HBV DNA polymerase, including base priming, reverse transcription of the negative strand, and synthesis of the positive strand of HBV DNA. Oral bioavailability approaches 100% but is decreased by food; therefore, entecavir should be taken on an empty stomach. The intracellular half-life of the active phosphorylated compound is 15 hours. It is excreted by the kidney, undergoing both glomerular filtration and net tubular secretion.
Comparison with lamivudine in patients with chronic HBV infection demonstrated similar rates of HBeAg seroconversion but significantly higher rates of HBV DNA viral suppression with entecavir, normalization of serum alanine aminotransferase levels, and histologic improvement in the liver. No primary resistance was observed to emerge after entecavir use for up to 48 weeks. However, decreased susceptibility to entecavir occurs in association with lamivudine resistance. Adefovir maintains activity against entecavir-resistant strains.
Entecavir is well tolerated. The most frequently reported adverse events are headache, fatigue, dizziness, and nausea. Lung adenomas and carcinomas in mice, hepatic adenomas and carcinomas in rats and mice, vascular tumors in mice, and brain gliomas and skin fibromas in rats have been observed at varying exposures. Co-administration of entecavir with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of either entecavir or the co-administered drug.
INVESTIGATIONAL AGENTS
Compounds in advanced stages of clinical development for the treatment of patients with HBV infection include the nucleoside/nucleotide analogs clevudine, telbivudine, valtorcitabine, and alamifovir, as well as the immunologic modulator thymosin alpha-1, agents that facilitate uptake by the liver using conjugation to ligands, and RNA interference compounds.
TREATMENT OF HEPATITIS C INFECTION
INTRODUCTION
The primary goal of treatment in patients with HCV infection is viral eradication. In clinical trials, the primary efficacy end point is typically achievement of sustained viral response (SVR), defined as the absence of detectable viremia for 6 months after completion of therapy. SVR is associated with improvement in liver histology and reduction in risk of hepatocellular carcinoma and occasionally with regression of cirrhosis as well. Late relapse occurs in fewer than 5% of patients who achieve SVR.
In acute hepatitis C, the rate of clearance of the virus without therapy is estimated to be 15-30%. In one (uncontrolled) study, treatment of acute infection with interferon alfa-2b, in doses higher than those used for chronic hepatitis C (Table 49-6), resulted in a sustained rate of clearance of 95% at 6 months. Therefore, if HCV RNA testing documents persistent viremia 12 weeks after initial seroconversion, antiviral therapy is recommended.
The current standard of treatment in patients with chronic HCV infection is once-weekly pegylated interferon alfa in combination with daily oral ribavirin. Pegylated interferon alfa-2a and -2b have replaced their unmodified interferon alfa counterparts because of superior efficacy in combination with ribavirin, regardless of genotype. It is also clear that combination therapy with oral ribavirin is more effective than monotherapy with either interferon or ribavirin alone. Therefore, monotherapy with pegylated interferon alfa is recommended only in patients who cannot tolerate ribavirin. Factors associated with a favorable response to therapy include HCV genotype 2 or 3, absence of cirrhosis on liver biopsy, and low pretreatment HCV RNA levels.
RIBAVIRIN
Ribavirin is a guanosine analog that is phosphorylated intracellularly by host cell enzymes. Although its mechanism of action has not been fully elucidated, it appears to interfere with the synthesis of guanosine triphosphate, to inhibit capping of viral messenger RNA, and to inhibit the viral RNA-dependent polymerase of certain viruses. Ribavirin triphosphate inhibits the replication of a wide range of DNA and RNA viruses, including influenza A and B, parainfluenza, respiratory syncytial virus, paramyxoviruses, HCV, and HIV-1.
The absolute oral bioavailability of ribavirin is about 64%, increases with high-fat meals, and decreases with co-administration of antacids. Ribavirin elimination is primarily through the urine; therefore, clearance is decreased in patients with creatinine clearances less than 30 mL/min.
Evidence suggests that both a higher dose (ie, > 10.6 mg/kg) and a longer duration of ribavirin therapy, in combination with one of the interferon alfas, may improve response. This must be balanced with an increased likelihood of toxicity. A dose-dependent hemolytic anemia occurs in 10-20% of patients. Other potential adverse effects are depression, fatigue, irritability, rash, cough, insomnia, nausea, and pruritus. Contraindications to ribavirin therapy include uncorrected anemia, end-stage renal failure, ischemic vascular disease, and pregnancy. Ribavirin is teratogenic and embryotoxic in animals as well as mutagenic in mammalian cells. Patients exposed to the drug should not conceive children for at least 6 months thereafter.
INVESTIGATIONAL AGENTS
Investigational agents for the treatment of HCV infection include inhibitors of the HCV RNA polymerase such as valopicitabine, the nucleoside analogs isatoribine and viramidine, monoclonal antibodies against the glycoprotein, several new types of interferons, congeners of ribavirin, PIs, and the immunomodulator thymosin alpha-1.
V. ANTI-INFLUENZA AGENTS
INTRODUCTION
Influenza virus strains are classified by their core proteins (ie, A, B, or C), species of origin (eg, avian, swine), and geographic site of isolation. Influenza A, the only strain that causes pandemics, is classified into 16 H (hemagglutinin) and 9 N (neuraminidase) known subtypes based on surface proteins. Although influenza B viruses usually infect only people, influenza A viruses can infect a variety of animal hosts. Current influenza A subtypes that are circulating among people worldwide include H1N1, H1N2, and H3N2. Fifteen subtypes are known to infect birds, providing an extensive reservoir. Although avian influenza subtypes are typically highly species-specific, they have on rare occasions crossed the species barrier to infect humans and cats. Viruses of the H5 and H7 subtypes (eg, H5N1, H7N7, and H7N3) may rapidly mutate within poultry flocks from a low to high pathogenic form and have recently expanded their host range to cause both avian and human disease. Of particular concern is the H5N1 virus, which first caused human infection (including severe disease and death) in 1997 and has become endemic in Southeast Asia poultry since 2003. It is feared that the virus will become transmissible from person to person rather than solely from poultry to human, thus initiating the potential for a global outbreak (ie, pandemic).
Although both classes of antiviral drugs available for influenza have activity against influenza A, many or most of the circulating strains of avian H5N1, as well as the H1 and H3 strains causing seasonal influenza in the United States, are resistant to the adamantanamine agents.
AMANTADINE & RIMANTADINE
Amantadine (1-aminoadamantane hydrochloride) and its a-methyl derivative, rimantadine, are cyclic amines of the adamantine family that block the M2 proton ion channel of the virus particle and inhibit uncoating of the viral RNA within infected host cells, thus preventing its replication. They are active against influenza A only. Rimantadine is four to ten times more active than amantadine in vitro. Amantadine is excreted unchanged in the urine, whereas rimantadine undergoes extensive metabolism by hydroxylation, conjugation, and glucuronidation before urinary excretion. Dose reductions are required for both agents in the elderly and in patients with renal insufficiency and for rimantadine in patients with marked hepatic insufficiency.
In the absence of resistance, both amantadine and rimantadine, at 100 mg twice daily or 200 mg once daily, are 70-90% protective in the prevention of clinical illness when initiated before exposure. When begun within 1-2 days after the onset of illness, the duration of fever and systemic symptoms is reduced by 1-2 days.
The primary target for both agents is the M2 protein within the viral membrane, incurring both influenza A specificity and a mutation-prone site that results in the rapid development of resistance in up to 50% of treated individuals. Resistant isolates with single-point mutation are genetically stable, retain pathogenicity, can be transmitted to close contacts, and may be shed chronically by immunocompromised patients. The prevalence of resistance to both agents in clinical isolates in the United States increased from 2% in the 2003-2004 influenza season, to 12% in 2004-2005, to an alarming 91% in 2005-2006 (99% in H3N2, 1% in H1N1). Cross-resistance to zanamivir and oseltamivir does not occur.
The most common adverse effects are gastrointestinal (nausea, anorexia) and central nervous system (nervousness, difficulty in concentrating, insomnia, light-headedness). Central nervous system toxicity may be due to alteration of dopamine neurotransmission (see Chapter 28), is less frequent with rimantadine than with amantadine, tends to diminish after the first week of use, and may increase with concomitant antihistamines, anticholinergic drugs, hydrochlorothiazide, and trimethoprim-sulfamethoxazole. Serious neurotoxic reactions, occasionally fatal, may occur in association with high amantadine plasma concentrations and are more likely in the elderly or with renal insufficiency. Peripheral edema is another potential adverse effect. Both agents are teratogenic in rodents, and birth defects have been reported after exposure during pregnancy.
ZANAMIVIR & OSELTAMIVIR
The neuraminidase inhibitors zanamivir and oseltamivir, analogs of sialic acid, interfere with release of progeny influenza virus from infected to new host cells, thus halting the spread of infection within the respiratory tract. Unlike amantadine and rimantadine, zanamivir and oseltamivir have activity against both influenza A and influenza B viruses. Early administration is crucial because replication of influenza virus peaks at 24-72 hours after the onset of illness. When a 5-day course of therapy is initiated within 36-48 hours after the onset of symptoms, the duration of illness is decreased by 1-2 days compared with those on placebo, severity is diminished, and the incidence of secondary complications in children and adults decreases. Once-daily prophylaxis is 70-90% effective in preventing disease after exposure. Oseltamivir is FDA-approved for patients 1 year and older, whereas zanamivir is approved in patients 7 years or older.
Zanamivir is delivered directly to the respiratory tract via inhalation. Ten to twenty percent of the active compound reaches the lungs, and the remainder is deposited in the oropharynx. The concentration of the drug in the respiratory tract is estimated to be more than 1000 times the 50% inhibitory concentration for neuraminidase. Five to fifteen percent of the total dose (10 mg twice daily for treatment and 10 mg once daily for prevention) is absorbed and excreted in the urine with minimal metabolism. Potential side effects include cough, bronchospasm (occasionally severe), reversible decrease in pulmonary function, and transient nasal and throat discomfort.
Oseltamivir is an orally administered prodrug that is activated by hepatic esterases and widely distributed throughout the body. The dosage is 75 mg twice daily for treatment and 75 mg once daily for prevention; dosage must be modified in patients with renal insufficiency. The half-life of oseltamivir is 6-10 hours, and excretion is primarily in the urine. Potential side effects include nausea, vomiting, and abdominal pain, which occur in 5-10% of patients early in therapy but tend to resolve spontaneously. Taking oseltamivir with food does not interfere with absorption and may decrease nausea and vomiting. Headache, fatigue, and diarrhea have also been reported and appear to be more common with prophylactic use.
In adults, resistance during therapy is rare but may cause fatal disease. It is unknown whether resistant mutants retain pathogenicity or are spread between people. Worldwide resistance is rare and has not been documented in any clinical isolate from the 2005-2006 influenza season to date in the USA. Avian influenza is expected to retain susceptibility to the neuraminidase inhibitors.
VI. OTHER ANTIVIRAL AGENTS
INTERFERONS
Interferons have been studied for numerous clinical indications. In addition to HBV and HCV infections (see IV. Antihepatitis Agents), intralesional injection of interferon alfa-2b or alfa-n3 may be used for treatment of condylomata acuminata (see also Chapter 62).
RIBAVIRIN
In addition to oral administration for hepatitis C infection in combination with interferon alfa, aerosolized ribavirin is administered by nebulizer (20 mg/mL for 12-18 hours per day) to children and infants with severe respiratory syncytial virus (RSV) bronchiolitis or pneumonia to reduce the severity and duration of illness. Aerosolized ribavirin has also been used to treat influenza A and B infections but has not gained widespread use. Aerosolized ribavirin is generally well tolerated but may cause conjunctival or bronchial irritation. Health care workers should be protected against extended inhalation exposure.
Intravenous ribavirin decreases mortality in patients with Lassa fever and other viral hemorrhagic fevers if started early. High concentrations inhibit West Nile virus in vitro, but clinical data are lacking. Clinical benefit has been reported in cases of severe measles pneumonitis and certain encephalitides, and continuous infusion of ribavirin has decreased virus shedding in several patients with severe lower respiratory tract influenza or parainfluenza infections. At steady state, cerebrospinal fluid levels are about 70% of those in plasma.
PALIVIZUMAB
Palivizumab is a humanized monoclonal antibody directed against an epitope in the A antigen site on the F surface protein of RSV. It is licensed for the prevention of RSV infection in high-risk infants and children such as premature infants and those with bronchopulmonary dysplasia or congenital heart disease. A placebo-controlled trial using once-monthly intramuscular injections (15 mg/kg) for 5 months beginning at the start of the RSV season demonstrated a 55% reduction in the risk of hospitalization for RSV in treated patients, as well as decreases in the need for supplemental oxygen, illness severity score, and need for intensive care. Although resistant strains have been isolated in the laboratory, no resistant clinical isolates have yet been identified. Potential adverse effects include upper respiratory tract infection, fever, rhinitis, rash, diarrhea, vomiting, cough, otitis media, and elevation in serum aminotransferase levels.
IMIQUIMOD
Imiquimod is an immune response modifier shown to be effective in the topical treatment of external genital and perianal warts (ie, condyloma acuminatum; see Chapter 62). The mechanism of action against these human papillomavirus (HPV)-induced lesions is unknown. The 5% cream is applied three times weekly and washed off 6-10 hours after each application. Recurrences appear to be less common than after ablative therapies. Imiquimod is also effective against actinic keratoses. Local skin reactions are the most common side effect; these resolve within weeks after therapy. However, pigmentary skin changes may persist. Systemic adverse effects such as fatigue and influenza-like syndrome have occasionally been reported.
PREPARATIONS AVAILABLE
Abacavir
Oral (Ziagen): 300 mg tablets; 20 mg/mL solution
Oral (Epzicom): 600 mg plus 300 mg lamivudine
Oral (Trizivir): 300 mg tablets in combination with 150 mg lamivudine and 300 mg zidovudine
Acyclovir (generic, Zovirax)
Oral: 200 mg capsules; 400, 800 mg tablets; 200 mg/5 mL suspension
Parenteral: 50 mg/mL; powder to reconstitute for injection (500, 1000 mg/vial)
Topical: 5% ointment
Adefovir (Hepsera)
Oral: 10 mg tablets
Amantadine (generic, Symmetrel)
Oral: 100 mg capsules, tablets; 50 mg/5 mL syrup
Amprenavir (Agenerase)
Oral: 50 mg capsules; 15 mg/mL solution
Atazanavir (Reyataz)
Oral: 100, 150, 200 mg capsules
Cidofovir (Vistide)
Parenteral: 375 mg/vial (75 mg/mL) for IV injection
Darunavir (Prezista)
Oral: 300 mg tablets (must be taken with ritonavir)
Delavirdine (Rescriptor)
Oral: 100, 200 mg tablets
Didanosine (dideoxyinosine, ddI)
Oral (Videx): 25, 50, 100, 150, 200 mg tablets; 100, 167, 250 mg powder for oral solution; 2, 4 g powder for pediatric solution
Oral (Videx-EC): 125, 200, 250, 400 mg delayed-release capsules
Docosanol (Abreva) (over-the-counter)
Topical: 10% cream
Efavirenz (Sustiva)
Oral: 50, 100, 200 mg capsules; 600 mg tablets
Emtricitabine
Oral (Emtriva): 200 mg tablets
Oral (Truvada): 200 mg plus 300 mg tenofovir tablets
Enfuvirtide (Fuzeon)
Parenteral: 90 mg/mL for injection
Entacavir (Baraclude)
Oral: 0.5, 1 mg tablets; 0.05 mg/mL oral solution
Famciclovir (Famvir)
Oral: 125, 250, 500 mg tablets
Fosamprenavir (Lexiva)
Oral: 700 mg tablets
Fomivirsen (Vitravene)
Intraocular injection: 6.6 mg/mL
Foscarnet (Foscavir)
Parenteral: 24 mg/mL for IV injection
Ganciclovir (Cytovene)
Oral: 250, 500 mg capsules
Parenteral: 500 mg/vial for IV injection
Intraocular implant (Vitrasert): 4.5 mg ganciclovir/implant
Idoxuridine (Herplex)
Ophthalmic: 0.1% solution
Imiquimod (Aldera)
Topical: 5% cream
Indinavir (Crixivan)
Oral: 100, 200, 333, 400 mg capsules
Interferon alfa-2a (Roferon-A)
Parenteral: 3, 6, 9, 36 million IU vials
Interferon alfa-2b (Intron A)
Parenteral: 3, 5, 10, 18, 25, and 50 million IU vials
Interferon alfa-2b (Rebetron)
Parenteral: 3 million IU vials (supplied with oral ribavirin, 200 mg capsules)
Interferon alfa-n3 (Alferon N)
Parenteral: 5 million IU/vial
Interferon alfacon-1 (Infergen)
Parenteral: 9 and 15 mcg vials
Lamivudine
Oral (Epivir): 150, 300 mg tablets; 10 mg/mL oral solution
Oral (Epivir-HBV): 100 mg tablets; 5 mg/mL solution
Oral (Combivir): 150 mg tablets in combination with 300 mg zidovudine
Oral (Trizivir): 150 mg tablets in combination with 300 mg abacavir and 300 mg zidovudine
Lopinavir/ritonavir (Kaletra)
Oral: 133.3 mg/33.3 mg capsules; 80 mg/20 mg per mL solution
Nelfinavir (Viracept)
Oral: 250, 625 mg tablets; 50 mg/g powder
Nevirapine (Viramune)
Oral: 200 mg tablets; 50 mg/5 mL suspension
Oseltamivir (Tamiflu)
Oral: 75 mg capsules; powder to reconstitute as suspension (12 mg/mL)
Palivizumab (Synagis)
Parenteral: 50, 100 mg/vial
Peginterferon alfa-2a (pegylated interferon alfa-2a, Pegasys)
Parenteral: 180 mcg/mL
Peginterferon alfa-2b (pegylated interferon alfa-2b, PEG-Intron)
Parenteral: powder to reconstitute as 100, 160, 240, 300 mcg/mL injection
Penciclovir (Denavir)
Topical: 1% cream
Ribavirin
Aerosol (Virazole): powder to reconstitute for aerosol; 6 g/100 mL vial
Oral (Rebetol, generic): 200 mg capsules, tablets; 40 mg/mL oral solution
Oral (Rebetron): 200 mg in combination with 3 million units interferon alfa-2b (Intron-A)
Rimantadine (Flumadine)
Oral: 100 mg tablets; 50 mg/5 mL syrup
Ritonavir (Norvir)
Oral: 100 mg capsules; 80 mg/mL oral solution
Saquinavir
Oral (Invirase): 200 mg hard gel capsules, 500 mg tablets
Oral (Fortovase): 200 mg soft gel capsules
Stavudine
Oral (Zerit): 15, 20, 30, 40 mg capsules; powder for 1 mg/mL oral solution
Oral extended-release (Zerit XR): 37.5, 50, 75, 100 mg capsules
Tenofovir (Viread)
Oral: 300 mg tablets
Tipranavir (Aptivus)
Oral: 250 mg capsules
Trifluridine (Viroptic)
Topical: 1% ophthalmic solution
Valacyclovir (Valtrex)
Oral: 500, 1000 mg tablets
Valganciclovir (Valcyte)
Oral: 450 mg capsules
Vidarabine (Vira-A)
Topical: 3% ointment
Zalcitabine (dideoxycytidine, ddC) (Hivid)
Oral: 0.375, 0.75 mg tablets
Zanamivir (Relenza)
Inhalational: 5 mg/blister
Zidovudine (azidothymidine, AZT) (Retrovir)
Oral: 100 mg capsules, 300 mg tablets, 50 mg/5 mL syrup
Oral (Combivir): 300 mg tablets in combination with 150 mg lamivudine
Oral (Trizivir): 300 mg tablets in combination with 150 mg lamivudine and 300 mg zidovudine
Parenteral: 10 mg/mL
REFERENCES
Asmuth DM, Nguyen HH, Melcher GP et al: Treatments for hepatitis B. Clinical Infect Dis 2004;39:1353.
Drugs for non-HIV viral infections. Med Lett Drugs Ther 2005;23.
Gnann JW, Whitley RJ: Herpes zoster. N Engl J Med 2002;347:340.
Hammer SM et al: Treatment for Adult HIV Infection, 2006. Recommendations of the International AIDS Society-USA Panel. JAMA 2006;296:827.
Kimberlin DW, Rouse DJ: Genital herpes. N Engl J Med 2004;350:1970.
Moscona A: Neuraminidase inhibitors for influenza. N Engl J Med 2005;353:1363.
Panel on Clinical Practices for Treatment of HIV Infection convened by the Department of Health and Human Services (DHHS). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. April 7, 2005. Available at http://AIDSinfo.nih.gov
Piscitelli SC, Gallicano KD: Interactions among drugs for HIV and opportunistic infections. N Engl J Med 2001;344:984.
Public Health Service Task Force. Recommendations for Use of Antiretroviral Drugs in Pregnant HIV-1-Infected Women for Maternal Health and Interventions to Reduce Perinatal HIV-1 Transmission in the United States. February 24, 2005. Available at http://AIDSinfo.nih.gov
Thio CL, Sulkowski MS, Thomas DL: Treatment of chronic hepatitis B in HIV-infected persons: Thinking outside the black box. Clin Infect Dis 2005;41:1035.