Tobias Else, MD, & Gary D. Hammer, MD, PhD
The hypothalamus is the part of the brain where activity of the autonomic nervous system and endocrine glands, which directly control various systems of the body, is integrated with input from other centers that give rise to emotions and behavior. The hypothalamus thus serves to ensure that (1) the organism responds appropriately to deviations from various internal set points (including those for temperature, volume, osmolality, satiety, and body fat content), (2) the responses to such deviations from a set point include coordinated activity of the nervous and endocrine systems, and (3) the emotions and behavior being manifested are appropriate for reflex responses being triggered to correct the deviations from internal set points. The following description outlines the integrative function of the hypothalamus in regard to the coordination of endocrine and CNS responses.
Intravascular volume loss from any cause activates autonomic neural responses, mainly via the sympathetic nervous system to retain fluid and electrolytes, maintain blood pressure through vascular smooth muscle contraction, and maintain cardiac output by increasing heart rate. The effect of these immediate neural responses is reinforced by activation of several hormonal systems. In response to a decrease in intravascular volume, the renin-angiotensin-aldosteronesystem (RAAS) is activated and sodium is retained. Additionally increasing osmolarity triggers thirst and leads to release of vasopressin (antidiuretic hormone [ADH]) from hypothalamic neurons that end in the posterior pituitary, resulting in free water absorption in the kidney. In short, the body maintains intravascular volume by regulating sodium reabsorption through aldosterone, while it regulates osmolarity by increasing fluid intake (thirst) and free water retention by vasopressin.
Emotions interplay with these systems to coordinate appropriate behavioral and hormonal responses. Fear and pain activate limbic, hypothalamic and other centers to coordinate respective defensive (fight or flight) and recuperative stereotypic behaviors. These emotional responses to various stressors (eg, perceived threat to body; fear) also activate the sympathetic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis, which coordinate the mammalian stress response through preparing the body for “fight and flight” and through mobilization of energy stores. Any kind of stress (eg, physical, mental, metabolic stress) leads to the release of corticotropin-releasing hormone (CRH) from the hypothalamus and consequent adrenocorticotropin (ACTH; pituitary) and cortisol (adrenal cortex) secretion. For example, starvation leads to the activation of the HPA axis and ultimately cortisol-mediated increased gluconeogenesis to maintain basic physiologic functions.
The pituitary gland is the partner of the hypothalamus on the body side of the mind-body interface. Once viewed as the “master gland” in regulation of neuroendocrine systems, the pituitary is now known to be a “middle manager” responding to input from both the brain (via the hypothalamus) and the body (via the various peripheral endocrine glands).
The basic framework for hypothalamic-pituitary function is the neuroendocrine axis, a cascade of interacting hormonal products from various regions of the CNS to the hypothalamus, anterior pituitary gland, peripheral endocrine end organs, and peripheral target tissues. Some neuroendocrine axes involve hormones released by the hypothalamus that stimulate cells in the anterior pituitary to secrete other hormones into the systemic circulation. Each of these anterior pituitary hormones travels to a distant endocrine gland to stimulate secretion of yet other hormones that affect various target tissues. Thus, disorders of the hypothalamus and pituitary have important consequences for the pathophysiologic mechanisms of a wide range of disorders involving many different tissues and organs.
This chapter focuses on five clinical entities. The first four reflect the diversity of pituitary disease: pituitary adenomas, panhypopituitarism, vasopressin excess, and vasopressin deficiency. The last, obesity, is one in which the hypothalamus plays a crucial role and which has enormous implications for diseases involving many other organ systems.
NORMAL STRUCTURE & FUNCTION OF THE HYPOTHALAMUS & PITUITARY GLAND
ANATOMY, HISTOLOGY, & CELL BIOLOGY
The hypothalamus is located in the floor and lateral walls of the third ventricle below the hypothalamic sulcus and comprises about 1% of the mass of the brain (Figure 19-1). Hypothalamic nuclei are clusters of neurons whose cell bodies lie in discrete regions (Figure 19-2). From these nuclei, hypothalamic neurons send projections either directly or via neuronal relay to other parts of the central and peripheral nervous systems and secrete hormones that make possible the hierarchical control of various physiologic processes (Table 19-1).
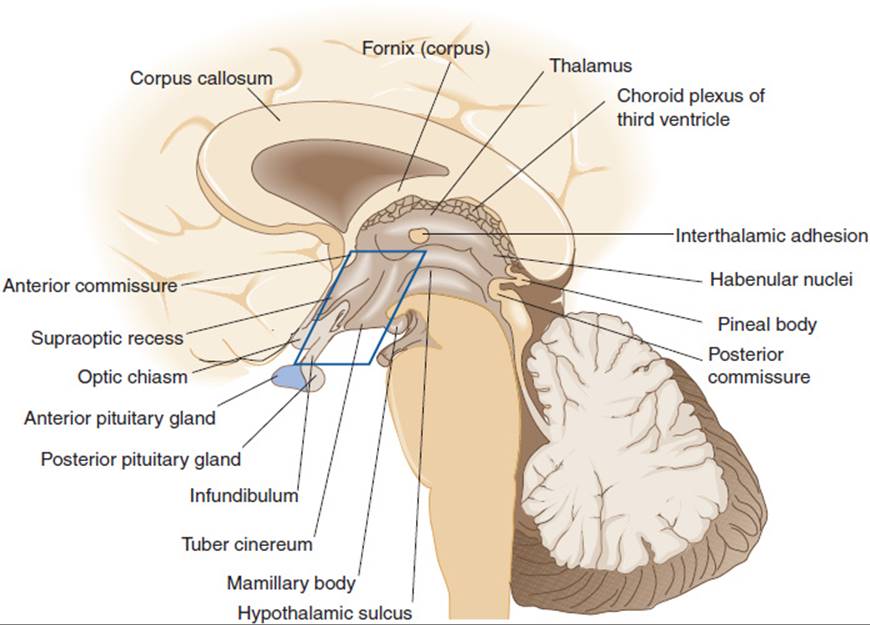
FIGURE 19-1 Sagittal section through the brain showing the diencephalon. (Redrawn, with permission, from Chusid JG. Correlative Neuroanatomy and Functional Neurology, 19th ed. Originally published by Lange Medical Publications. Copyright © 1985 by The McGraw-Hill Companies, Inc.)
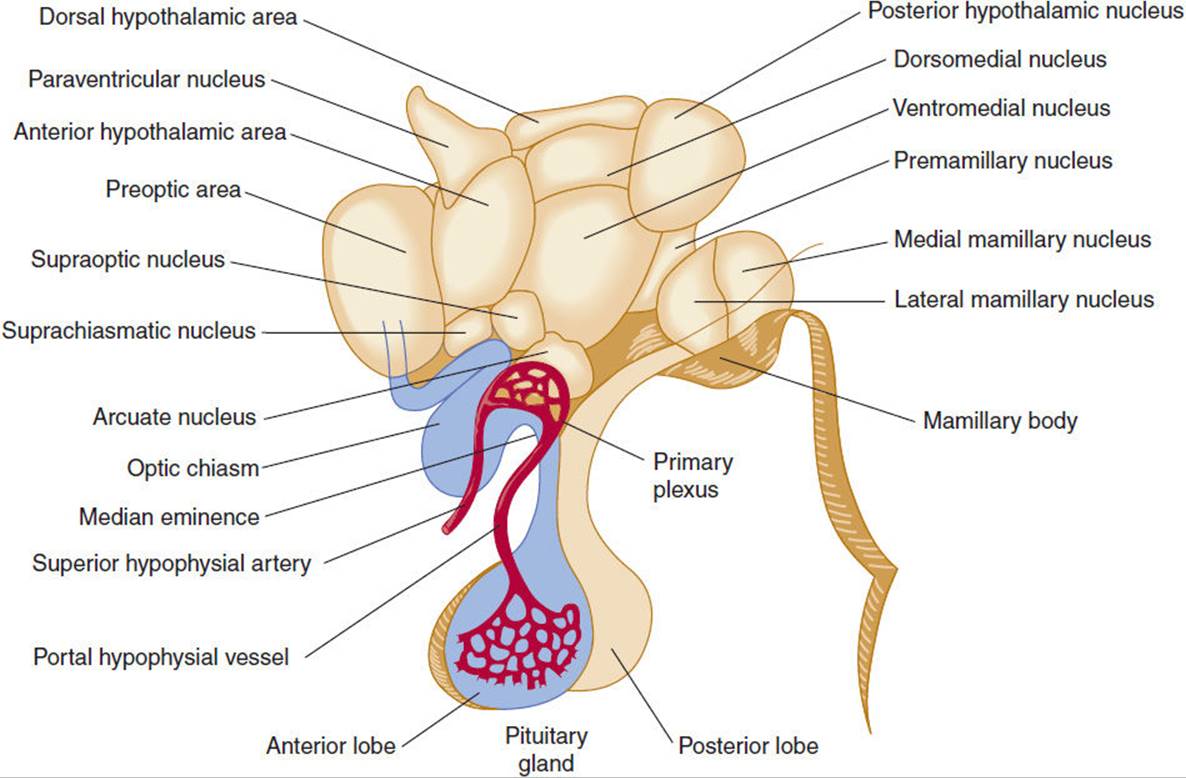
FIGURE 19-2 Human hypothalamus, with a superimposed diagrammatic representation of the portal hypophyseal vessels. (Redrawn, with permission, from Ganong WF. Review of Medical Physiology, 20th ed. Originally published by McGraw-Hill. Copyright © 2001 by The McGraw-Hill Companies, Inc.)
TABLE 19-1 The hypothalamic nuclei and their main functions.

The hypothalamus is connected to the pituitary gland by a stalk, which is composed of axons of some hypothalamic neurons with terminal boutons comprising the posterior pituitary gland (Figure 19-3). The posterior pituitary neurons secrete the peptide hormones oxytocin and vasopressin directly into the systemic circulation. The development of the anterior pituitary from the oral ectoderm is dictated by a tight program of consecutive activation of distinct transcription factors in the differentiating pituitary cell types (Figure 19-4). The pituitary gland is encased in a tough fibrous capsule, positioned in the bony sella turcica. The pituitary gland is bounded above by the optic chiasm and laterally by the cavernous sinus and the structures that traverse it (internal carotid artery, cranial nerves [CN] III and IV, first and second divisions of CN V and VI).
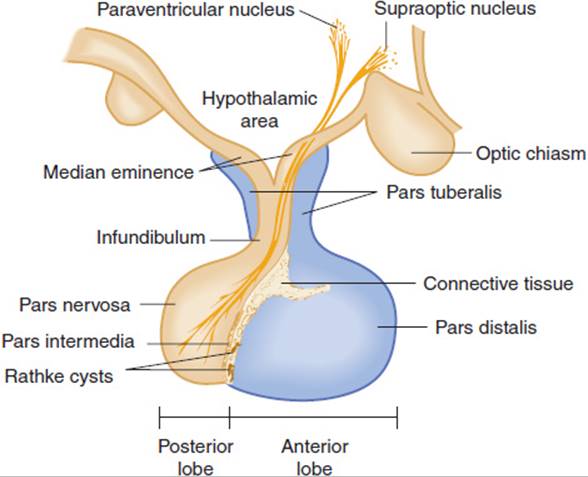
FIGURE 19-3 The component parts of the pituitary and their relationship to the hypothalamus. The pars tuberalis, pars distalis, and pars intermedia, which are rudimentary in humans, form the adenohypophysis. The infundibulum and pars nervosa form the neurohypophysis. (Redrawn and modified, with permission, from the Ciba Collection of Medical Illustrations, by Frank H. Netter, MD.)
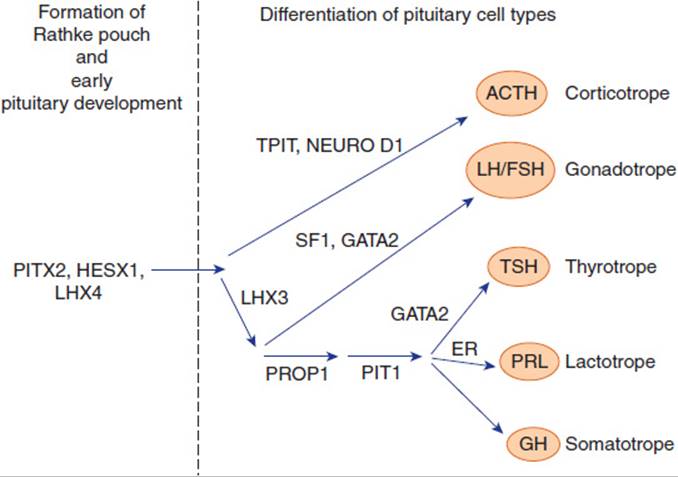
FIGURE 19-4 Diagram of the transcription factors involved in anterior pituitary development. Factors on the left are mainly responsible for Rathke pouch formation and early pituitary development. On the right side are factors inducing the differentiation into the major five pituitary cell types. Mutations of some of the genes encoding these transcription factors have been shown to result in hypopituitarism.
In circumventricular parts of the CNS, the capillaries are fenestrated, allowing neurons to sense various specific chemical stimuli in the bloodstream. These sensory neurons transmit the information regarding changes in stimuli (eg, change in osmolality) to other hypothalamic neurons involved in a variety of specific types of secretory activities.
Other hypothalamic neurons secrete peptide hormones into a specialized capillary bed termed the pituitary portal system. Blood in this capillary system flows directly from the median eminence to the anterior pituitary gland, where specific cells that display receptors for the various hypothalamic releasing hormones are found. Binding of hypothalamic hormones to their receptors on cells of the anterior pituitary in turn stimulates the secretion of specific anterior pituitary hormones into the systemic circulation. The portal system allows the cells of the anterior pituitary to be bathed in blood rich in hypothalamic hormones without the dilution that would have occurred in the systemic circulation. This intimate connection between hypothalamus and pituitary has important pathophysiologic consequences (see later).
Once secreted, the anterior pituitary hormones travel via the general bloodstream throughout the body and trigger the release of other hormones from particular endocrine glands. These hormones, in turn, have effects on target tissues that influence growth, reproduction, metabolism, and responses to stress. In addition to their effects on target tissues, hormones secreted in response to stimulation by pituitary hormones also feed back and inhibit secretion of the corresponding pituitary and hypothalamic hormones.
The posterior pituitary hormones are involved in a very different type of neuroendocrine axis, one that bypasses secondary endocrine glands and affects peripheral target tissues directly.
Although most peptide factors secreted by the hypothalamus cause release of a pituitary hormone, some are inhibitory factors that block or diminish secretion of particular hormones. There are five main cell types in the anterior pituitary, each of which produces and secretes one of five families of hormones: pro-opiomelanocortin and adrenocorticotropic hormone (ACTH), thyrotropin (TSH), growth hormone (GH), prolactin (PRL), and the gonadotropins, luteinizing hormone (LH), and follicle-stimulating hormone (FSH) (Table 19-2).
TABLE 19-2 Pituitary hormones.
In addition to their roles in regulation of neuroendocrine axes, some hypothalamic and pituitary hormones are important, but poorly understood, regulators of immune functions and the inflammatory response. Furthermore, secretion of hypothalamic and pituitary hormones can be significantly influenced by cytokines that regulate the immune response.
CHECKPOINT
1. What is the role of the hypothalamus?
2. What are the neuroendocrine axes, and how do they work?
3. What structures surround the pituitary?
4. Where do the neurons whose axons comprise the posterior pituitary originate?
PHYSIOLOGY OF THE HYPOTHALAMUS & PITUITARY GLAND
ANTERIOR PITUITARY HORMONES
Pro-opiomelanocortin & ACTH
The HPA axis is a major part of the physiologic stress system. A variety of stressors (eg, metabolic, physical, mental stress) result in activation of the HPA axis. The major hypothalamic regulator is the peptide CRH and to a lesser extent arginine vasopressin (AVP), which are produced in the paraventricular and supraoptic nuclei of the hypothalamus and are released into the hypothalamic-pituitary portal system. These hormones trigger synthesis and intracellular transport of a large protein termed pro-opiomelanocortin (POMC). POMC is further processed by proteases (prohormone convertases) to release smaller peptides, including a 39-amino-acid-residue peptide, ACTH (Figure 19-5). Although ACTH is the major pituitary hormone that stimulates adrenocortical endocrine function, the amino-terminal part of the POMC peptide (N-POMC) seems to harbor an adrenal growth-promoting function.

FIGURE 19-5 Schematic representation of the prepro-opiomelanocortin molecule formed in pituitary cells, neurons, and other tissues. The numbers in parentheses identify the amino acid sequences in each of the polypeptide fragments. For convenience, the amino acid sequences are numbered from the amino terminal of corticotrope (ACTH) and read toward the carboxyl terminal portion of the parent molecule, whereas the amino acid sequences in the other portion of the molecule read to the left to –131, the amino terminal of the parent molecule. The locations of Lys-Arg and other pairs of basic amino acid residues are also indicated; these are the sites of proteolytic cleavage in the formation of the smaller fragments of the parent molecule. AL, anterior lobe; IL, intermediate lobe. (Redrawn, with permission, from Barrett KE et al, eds. Ganong’s Review of Medical Physiology, 24th ed. McGraw-Hill, 2011.)
ACTH released into the systemic circulation triggers synthesis and secretion of corticosteroids and adrenal androgens. The effect of ACTH on mineralocorticoid synthesis and release is much less pronounced, as it is mainly regulated by the RAAS.
These steroid hormones, in turn, have complex effects on many tissues to protect the individual from stress: They raise blood pressure and blood glucose, alter responsiveness of the immune system, and so on. Glucocorticoids also feed back to the hypothalamus, where they inhibit CRH secretion, and to the pituitary, where they further inhibit ACTH secretion. In the absence of unusual stress, there is a daily diurnal rhythm of CRH, ACTH, and adrenal steroid release.
Pituitary factors (ie, N-POMC, ACTH) are involved in regulating proliferation of adrenal cells and growth of the adrenal layers involved in glucocorticoid and androgen secretion. As a result of chronic activation of the HPA axis, hypertrophy of the target organ (adrenal cortex) occurs. Conversely, conditions that downregulate the HPA axis (eg, exogenous glucocorticoids) result in atrophy of the adrenal cortex. On the other hand, the overall tone of the HPA axis has little or no effect on the growth of the mineralocorticoid-secreting tissues, despite the fact that acute ACTH stimulation triggers the release of mineralocorticoids.
The Glycoprotein Hormones
TSH and the gonadotropins belong to the family of glycoprotein hormones (Table 19-2). The classic glycoprotein hormone family members TSH and the gonadotropins, FSH and LH, as well as the placenta-derived pregnancy hormone human chorionic gonadotropin (hCG) are composed of a common α-glycoprotein subunit (α-GSU) and an individual β-subunit (eg, TSH-β, LH-β). The unique β-subunit of the glycoprotein hormones is responsible for the biologic differences of these hormones. Another member of this family is thyrostimulin, which shares the composition of an α- and β-subunit (α-2, β-5). The physiologic role of this hormone has yet to be determined.
A. Thyrotropin (Thyroid-Stimulating Hormone)
Thyrotropin (thyroid-stimulating hormone [TSH]) is released from specific cells in the pituitary on stimulation by thyrotropin-releasing hormone (TRH) from the hypothalamus. A hypothalamic factor negatively regulating TSH release is somatostatin. TSH, in turn, travels via the systemic bloodstream to the thyroid gland, where it stimulates synthesis and secretion of the thyroid hormones thyroxine and triiodothyronine. Thyroid hormones have effects on nearly every tissue in the body but especially the cardiovascular, respiratory, skeletal, and central nervous systems. Thyroid hormones are critical at key points in development, and their deficiency during development has effects (eg, severe mental retardation and short stature) that are not fully reversible by subsequent thyroid hormone administration (Chapter 20).
Besides its target tissue effects, thyroid hormone feeds back to the pituitary and hypothalamus to inhibit secretion of TSH and TRH. TSH also triggers growth of thyroid tissue, resulting in goiter under conditions of chronic TSH stimulation such as iodine deficiency (see Chapter 20).
B. Gonadotropin
The role of the gonadotropins is to regulate the reproductive system’s neuroendocrine axis. Thus, a releasing factor from the hypothalamus termed gonadotropin-releasing hormone (GnRH) stimulates LH and FSH secretion, which stimulates steroidogenesis within the ovaries and testes. Furthermore, the gonadotropins promote Sertoli and theca cell function and gametogenesis. The steroids produced by the ovaries (estrogens) and by the testes (testosterone) inhibit GnRH, LH, and FSH production and have target tissue effects on developing follicles within the ovary itself, on the uterus (controlling the menstrual cycle), on breast development, on spermatogenesis, and on many other tissues and physiologic processes (see Chapters 22 and 23).
As is the case with all neuroendocrine axes, the simple feedback loop is complicated by other inputs (eg, from the CNS) that modify responsiveness (Chapter 7). The discovery that KiSS1-derived peptides (eg, metastin) induce hypothalamic GnRH-release via signaling through a G protein–coupled receptor (GPR54) illustrates this point. Mutations in either of these can result in the failure of puberty to develop. A notable feature for many hypothalamic releasing factors, but particularly GnRH, is that secretion occurs in pulsatile fashion and that changes in the rate and amplitude of secretion result in altered pituitary responsiveness because of downregulation or upregulation of the receptors for the hypothalamic releasing factors found on the surface of the pituitary cells. Not only is the secretion of GnRH episodic, but the secretion of FSH and LH is as well, with a secretory burst every 60 minutes. The gonadotropins follow a typical secretion pattern during the estrus cycle with a midcycle LH surge initiating ovulation.
Growth Hormone & Prolactin
Growth hormone and prolactin are structurally related single-chain polypeptides with different spectrums of action.
A. Growth Hormone
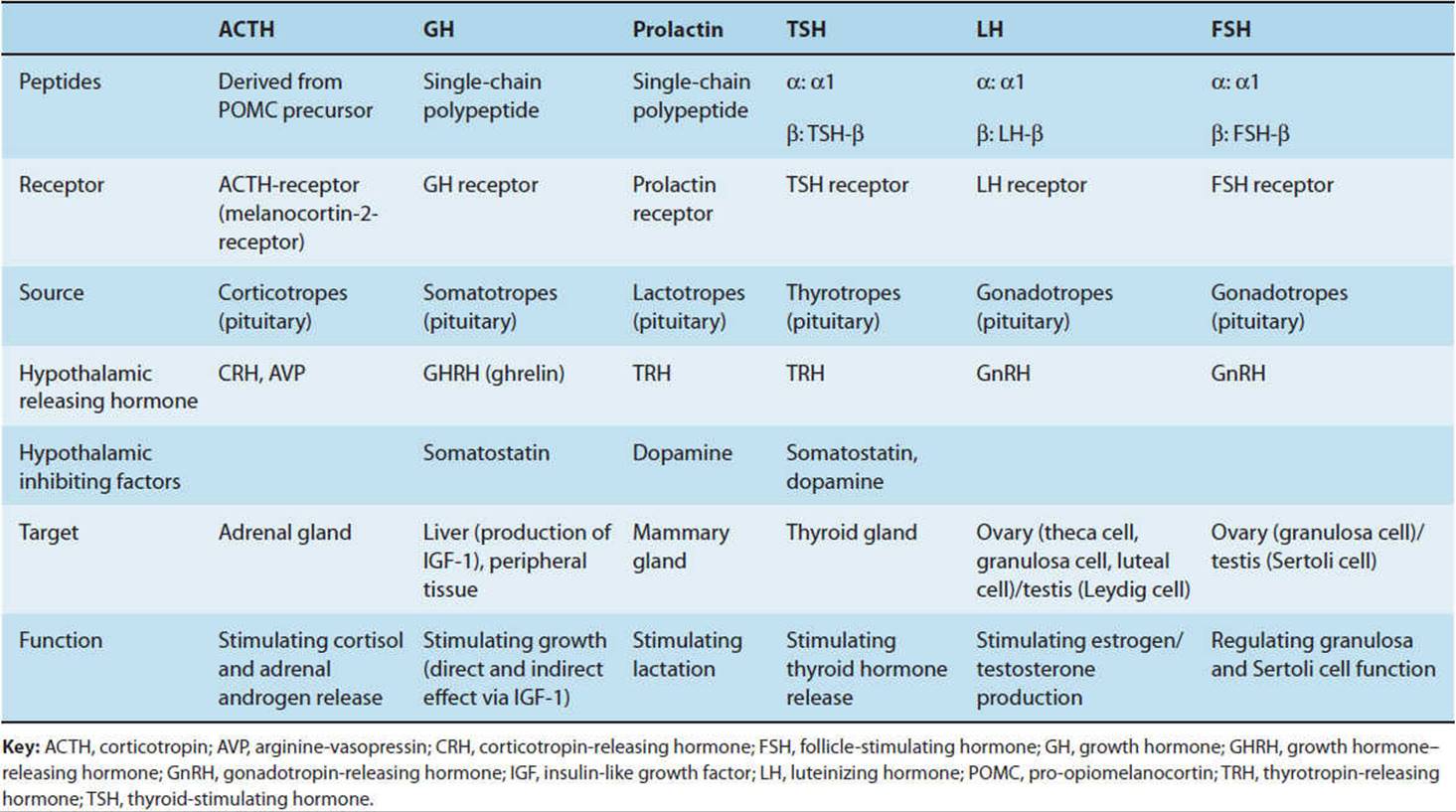
Growth hormone (GH), positively regulated by hypothalamic growth hormone–releasing hormone (GHRH) and inhibited by somatostatin, triggers growth-promoting effects in a wide range of tissues (Figure 19-6). GH has direct (eg, stimulating the growth of cartilage) as well as indirect (eg, via insulin-like growth factor-1 [IGF-1], a polypeptide secreted by the liver and other tissues) actions (Figure 19-7). IGF-1 has insulin-like effects of promoting fuel storage in various tissues. IGF-1 in turn inhibits GHRH and GH secretion. As in the other neuroendocrine feedback axes, the CNS and other factors can significantly influence the simple regulatory axis (Table 19-3). One of these factors is the gastrointestinal peptide hormone, ghrelin, which acts through the growth hormone–secretagogue receptor to induce GH release. The physiological significance of this process has yet to be determined. Somatostatin inhibits GH release and somatostatin analogs are therefore used to inhibit GH secretion from GH-secreting pituitary tumors.
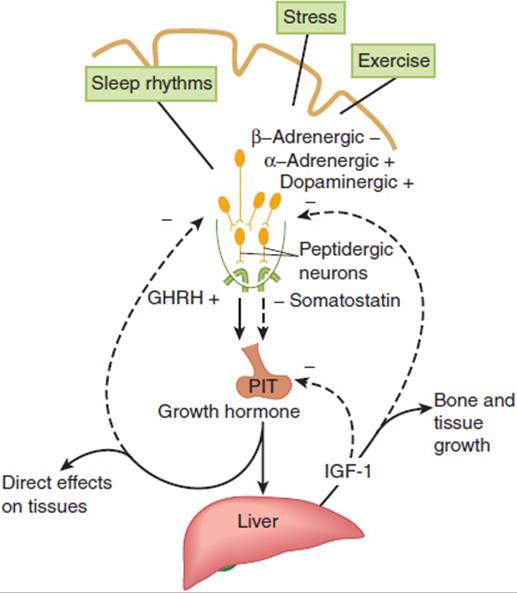
FIGURE 19-6 Schematic diagram of the hypothalamic control of growth hormone secretion. Inhibitory arrows are dashed; stimulating arrows are solid. GHRH, growth hormone–releasing hormone; IGF, insulin-like growth factor. (Redrawn from Reichlin S. Neuroendocrinology. In: Wilson JD et al, eds. Williams Textbook of Endocrinology, 9th ed. Saunders, 1998.)
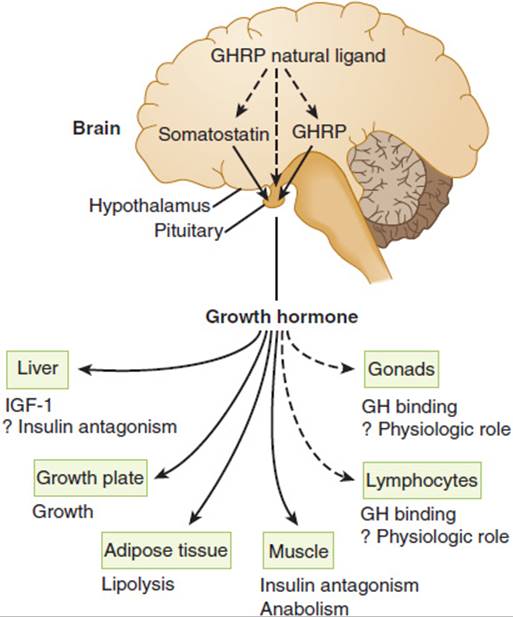
FIGURE 19-7 Schematic representation of multiple sites of growth hormone (GH) action. GHRH, growth hormone–releasing hormone; GHRP, growth hormone–releasing peptide; IGF-I, insulin-like growth factor I. (Redrawn, with permission, from Thorner MO et al. The anterior pituitary. In: Wilson JD et al, eds. Williams Textbook of Endocrinology, 9th ed. Saunders, 1998.)
TABLE 19-3 Factors influencing normal growth hormone secretion.
Some of the actions of GH appear to have a counter-regulatory character in that they raise blood glucose levels and antagonize the action of insulin. In contrast, other actions of GH via IGF-1 are insulin-like. This apparent contradiction makes sense when one considers that promoting growth requires first raising blood levels of substrates and then using them for synthesis. To do the latter without the former would simply make the individual hypoglycemic without promoting long-term growth.
B. Prolactin
The primary role of prolactin in humans is to stimulate breast development and milk synthesis. It is discussed in greater detail in Chapter 22. Prolactin secretion is mainly negatively regulated by the neurotransmitter dopamine from the hypothalamus rather than by a peptide. That is, dopamine acts to inhibit rather than stimulate prolactin secretion. Pathologic processes that result in separation of the pituitary gland from the hypothalamus cause loss of all pituitary hormones except prolactin (panhypopituitarism from lack of the hypothalamic releasing hormones). Loss of dopamine results instead in an increase in prolactin secretion from specific anterior pituitary cells now freed of inhibition by dopamine. Primary hypothyroidism is often accompanied by hyperprolactinemia because increased levels of TRH can exhibit prolactin-releasing factor properties.
POSTERIOR PITUITARY HORMONES
Vasopressin & Oxytocin
The peptide hormones vasopressin and oxytocin are synthesized in the supraoptic and paraventricular nuclei of the hypothalamus. The axons of the neurons in these nuclei form the posterior pituitary, where these peptide hormones are stored. Thus, there is no need for a separate set of hypothalamic releasing factors to trigger vasopressin or oxytocin release.
A. Vasopressin
In response to a small increase in blood osmolality, the hypothalamic “osmostat” responds by triggering the subjective sense of thirst and at the same time the release of vasopressin. Vasopressin increases the number of active water channels in the cell membranes of renal collecting duct cells, allowing conservation of free water. This increases the concentration of the urine. Conservation of free water and stimulation of thirst have the net effect of correcting the small change in blood osmolality.
Vasopressin binds to at least three classes of receptors. One of these classes of vasopressin receptors (V1A) is found on smooth muscle. Its major effect is to trigger vasoconstriction. V1B receptors are found on corticotropes, and they contribute to increased ACTH secretion. The other class of receptors (V2) is found in the distal nephrons in the kidneys; its major action is to mediate vasopressin’s effects on osmolality. Because of its V2-mediated actions, vasopressin is also known as antidiuretic hormone (ADH). The relationship among osmotic forces, volume, and vasopressin secretion is illustrated in Figure 19-8. Although the minute-to-minute function of vasopressin is to maintain blood osmolality, its secretion is also increased by large decreases in intravascular volume. This assists aldosterone in raising intravascular volume, albeit at the expense of lowered osmolality. The combination of ADH-mediated peripheral vasoconstriction and water retention (in the setting of hypotension even with lower or normal osmolarity) can be understood as a way of helping to maintain perfusion in the face of major intravascular volume deficits (eg, with hemorrhage), even if the volume and osmolar composition of the perfusing blood are not ideal. In pharmacologic doses, vasopressin can be used as an adjunct in the treatment of severe hypotensive crises.
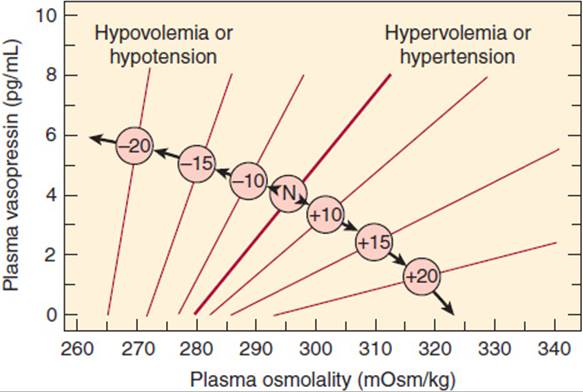
FIGURE 19-8 The influence of hemodynamic status on the osmoregulation of vasopressin in otherwise healthy humans. The numbers in the center circles refer to the percentage change in volume or pressure; N refers to the normovolemic normotensive subject. Note that the hemodynamic status affects both the slope of the relationship between the plasma vasopressin and osmolality and the osmotic threshold for vasopressin release. (Redrawn and adapted from Robertson GL et al. The osmoregulation of vasopressin. Kidney Int. 1976;10:25. Adapted by Rose BD in: Clinical Physiology of Acid-Base and Electrolyte Disorders, 3rd ed. McGraw-Hill, 1989. Reprinted, with permission, from Kidney International.)
B. Oxytocin
Like vasopressin, this peptide is stored in nerve terminals of hypothalamic neurons in the posterior pituitary. It plays an important role in breast and uterine smooth muscle contraction both on a minute-to-minute basis during breast-feeding and in contraction of the uterus during parturition. Besides its function in parturition and lactation, recent research suggests a significant role for oxytocin in the neuropsychological regulation of behavior, such as trust formation and interpersonal bonding (eg, pair and parental attachment).
CHECKPOINT
5. How do neuroendocrine feedback loops of the anterior and posterior pituitary differ?
6. How can two polypeptide hormones whose mature forms have no sequence in common be derived from the same precursor?
7. Describe the distinguishing features of each pituitary neuroendocrine feedback axis.
8. What is the significance of receptor downregulation for hypothalamic control of pituitary function?
PHYSIOLOGY OF THE NEUROENDOCRINE AXIS
A number of features of neuroendocrine axis physiology have important implications for the pathophysiology of disease.
First, the hypothalamic hormones that traverse the pituitary portal system are short-lived. They also have relatively low affinities for their receptors. These properties are generally more characteristic of neurotransmitters in the nervous system than of hormones in the bloodstream. Some of these hormones, and the receptor systems with which they interact, have evolved in ways that take advantage of the unique features of a neuroendocrine axis. For example, in the case of GnRH, secretion is markedly pulsatile in character; a particular rate and amplitude of hypothalamic hormone secretion are crucial for a proper response by the receptor-bearing gonadotropes. If the pulse rate or amplitude is too high, the receptors are downregulated.
Second, for some of the neuroendocrine axes, measurement of a random blood level of the end-organ hormone is not generally clinically useful. A more reliable approach to assessment of neuroendocrine axis function is often to assess the secretory response to a provocative stimulus, or challenge test. Thus, an adequate increase in blood cortisol 1 hour after an intravenous injection of ACTH provides far more compelling evidence for an intact adrenal gland than does a randomly drawn, unprovoked normal blood level of cortisol.
Finally, besides stimulating end-organ hormone secretion, most of the pituitary hormones exert trophic effects on the hormone-secreting cells of the end organ. Thus, excess of pituitary hormone results in end-organ hypertrophy, and lack of the pituitary hormone results in end-organ atrophy.
PHYSIOLOGY OF BODY WEIGHT CONTROL
Various physiologic control mechanisms integrated by the hypothalamus work to maintain body weight over the short and the long term (Figure 19-9).
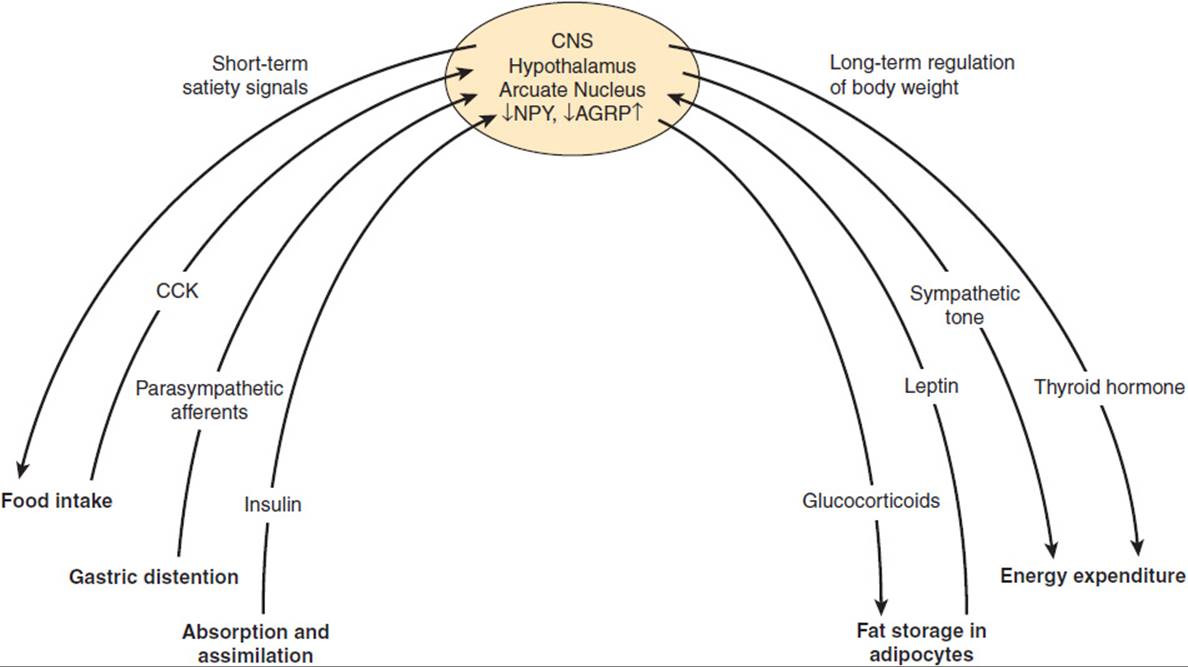
FIGURE 19-9 Physiologic control mechanisms regulating body weight. NPY, neuropeptide Y; AGRP, agouti-related peptide; CCK, cholecystokinin.
The key parameters of short-term regulation of body weight are (1) the amount and composition of food, (2) nutrient absorption and assimilation, and (3) satiety, the sense of having eaten enough food. Satiety is a complex response to food intake that has mechanical, neural, and hormonal components.
A main mechanism by which short-term food intake and satiety are regulated is the communication via the “gut-brain axis.” The gut-brain crosstalk uses two main routes of communication, including both neural components, mainly afferent vagal fibers, and hormonal components. Thus, we feel a sense of fullness in response to mechanical distention of the stomach, which triggers afferent neural pathways to the hypothalamus or via brainstem centers (eg, nucleus of the solitary tract). In addition, hormones are secreted in response to food ingestion and absorption and have direct effects on the hypothalamus to induce satiety. These hormonal signals mainly include anorexigenic satiety signals, such as cholecystokinin (CCK) and glucagon-like peptide-1 (GLP-1), which are released in the gut and directly impact on gastrointestinal mobility and function, but also stimulate gastrointestinal neural signaling to the hypothalamus. Some of these hormones travel directly to the brain and bind to receptors in the hypothalamus or in areas of regulated “open” blood-brain barrier. The only known orexigenic signal arising from the gut is the peptide hormone, ghrelin, suggesting that satiety is more abundantly regulated by the gastrointestinal system than hunger.
In contrast to short-term control of body weight, long-term regulation is largely influenced by the degree of obesity. Fat cells secrete the hormone leptin in proportion to the amount of triglyceride they have stored. Thus, over the long term, excess ingestion of calories resulting in increased fat deposition triggers an increase in leptin secretion. Leptin impinges on its receptors in the hypothalamus so that the individual eats less and, therefore, assimilates fewer calories. Another response to leptin is to increase sympathetic nervous system activity so that more calories are burned.
Conversely, when caloric intake is insufficient to maintain body weight, fat is mobilized, leptin secretion decreases, and set points in the hypothalamus are changed in ways that promote food-seeking behavior, diminish sympathetic neural activity, and generally conserve calories to offset the tendency toward weight loss. As a result of this feedback loop, further decrease in body weight is resisted. It is likely that this system evolved primarily as a defense against starvation, but it also serves to defend against obesity.
How these signals are normally integrated in the hypothalamus to achieve satiety in the short term and maintain normal body weight in the long term is less clear. The arcuate nucleus of the hypothalamus is the best understood integrator of the regulation of food intake. However, several other hypothalamic nuclei appear to be involved in the control of energy homeostasis and food intake. For example, lesions in the ventromedial region result in obesity, and lesions in the lateral hypothalamus result in weight loss. One hypothesis attempting to integrate current information on the regulation of fuel homeostasis proposes different responses by the body to falling versus rising leptin concentrations, as would be seen in weight loss versus weight gain, respectively. Thus, in response to falling leptin levels, neuropeptide Y is secreted from leptin receptor–bearing cells of the arcuate nucleus in the ventromedial hypothalamus. Neuropeptide Y is believed to mediate hypothalamic responses to starvation.
Another well-described system regulating satiety and food intake within the arcuate nucleus of the hypothalamus is the POMC system. Though the hypothalamic POMC system uses the same peptides for signaling mediators as the pituitary POMC system, they are very different in POMC expression, processing, and receptors. In particular, the main receptor mediating satiety and food intake is a special subtype of melanocortin receptors (MC4-R). In the state of caloric excess, hypothalamic-derived POMC peptides such as melanocyte-stimulating hormone (α-MSH) keep these MC4-R in a tonic activated state. In addition, hypothalamic POMC neurons are leptin responsive; therefore, these neurons represent an interface between the leptin and the POMC system. As circulating leptin levels parallel the total quantity of fat storage, it makes sense that activation of POMC neurons leads to inhibition of food intake. In the event of caloric restriction, the tonic activation of the MC4-R is reduced by two mechanisms: a decrease in agonists (MSH) and, even more important, an increase in availability of antagonists, namely agouti-related peptide (AGRP). These antagonists downregulate not only MSH (agonist driven) but also an intrinsic constitutive activity of the MC4-R. The mode of action of the MC4-R antagonism by AGRP has been termed “inverse agonism.” Furthermore, it is believed that many other neuropeptides, including bombesin, insulin, and a group of peptides termed orexins, have complex effects on the hypothalamus that affect feeding, satiety, energy balance, and other parameters relevant for weight control (Table 19-4). The orexins appear to be ligands for previously “orphan” G protein–coupled receptors in the brain. How the effects of these peptides are integrated with those of leptin and neuropeptide Y is a current focus of research. Finally, research strongly implicates leptin in other physiologic functions such as regulating reproductive and immune function as well as bone density.
TABLE 19-4 Peptides regulating food intake (mainly on the level of the hypothalamus).

CHECKPOINT
9. What are the short- and long-term factors involved in normal control of body weight?
10. What is the significance of the short half-life, low affinity, and restricted circulation of most hypothalamic hormones?
11. Why are challenge tests particularly important in assessing function of a neuroendocrine axis?
12. What happens to an end organ in the absence of the pituitary hormone that normally triggers its secretion?
PATHOPHYSIOLOGY OF SELECTED HYPOTHALAMIC & PITUITARY DISEASES
The hypothalamus and pituitary are implicated in the pathophysiology of a variety of complex diseases. These include anxiety disorders, in which abnormalities of the hypothalamic-pituitary-growth hormone axis appear to be a specific pathologic marker; alcoholism, in which neuropeptide Y has been implicated in mouse models of this condition; and obesity, in which a host of hypothalamic neuropeptides are affected and, in turn, affect parameters of fuel homeostasis. In most of these disorders, it remains unclear whether hypothalamic and endocrine dysregulation are important causative factors in pathogenesis or epiphenomena mirroring central nervous dysfunction.
OBESITY
Changes in body weight can occur through alteration of several variables, including (1) amount and type of food ingested, (2) central control of satiety, (3) hormonal control of assimilation or storage, and (4) physical activity or metabolic rate.
Clinical Presentation & Etiology
Obesity can be defined as excess body weight sufficient to increase overall morbidity and mortality. Although extreme obesity is associated with dramatically increased mortality, the risks of mild to moderate obesity are less clear. An index of “fatness” is the body mass index (BMI), which equals the weight (in kilograms) divided by height (in meters squared). The normal range is 18.5–25 kg/m2, and clinically significant obesity is a BMI > 30 kg/m2. More than 20% of the U.S. population is obese by this criterion. Individuals with a BMI of 150% of normal have an overall twofold risk of premature death, whereas those who are 200% of normal BMI have a 10-fold risk. Table 19-5 lists some important causes of morbidity and mortality associated with obesity, and Figure 19-10 shows possible pathophysiologic mechanisms involved in their production.
TABLE 19-5 Some disorders associated with obesity.
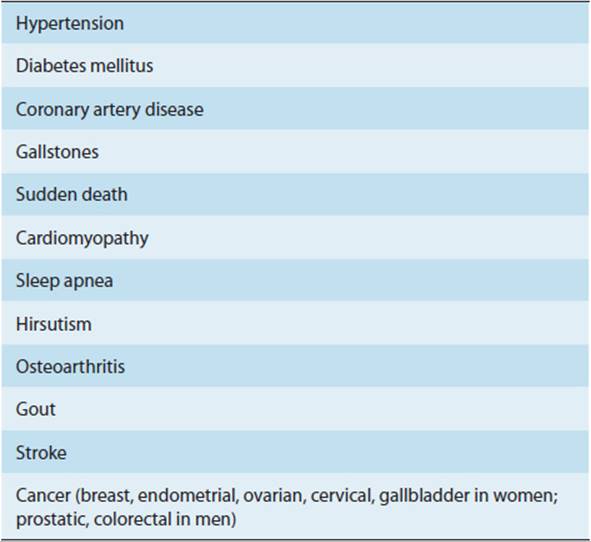
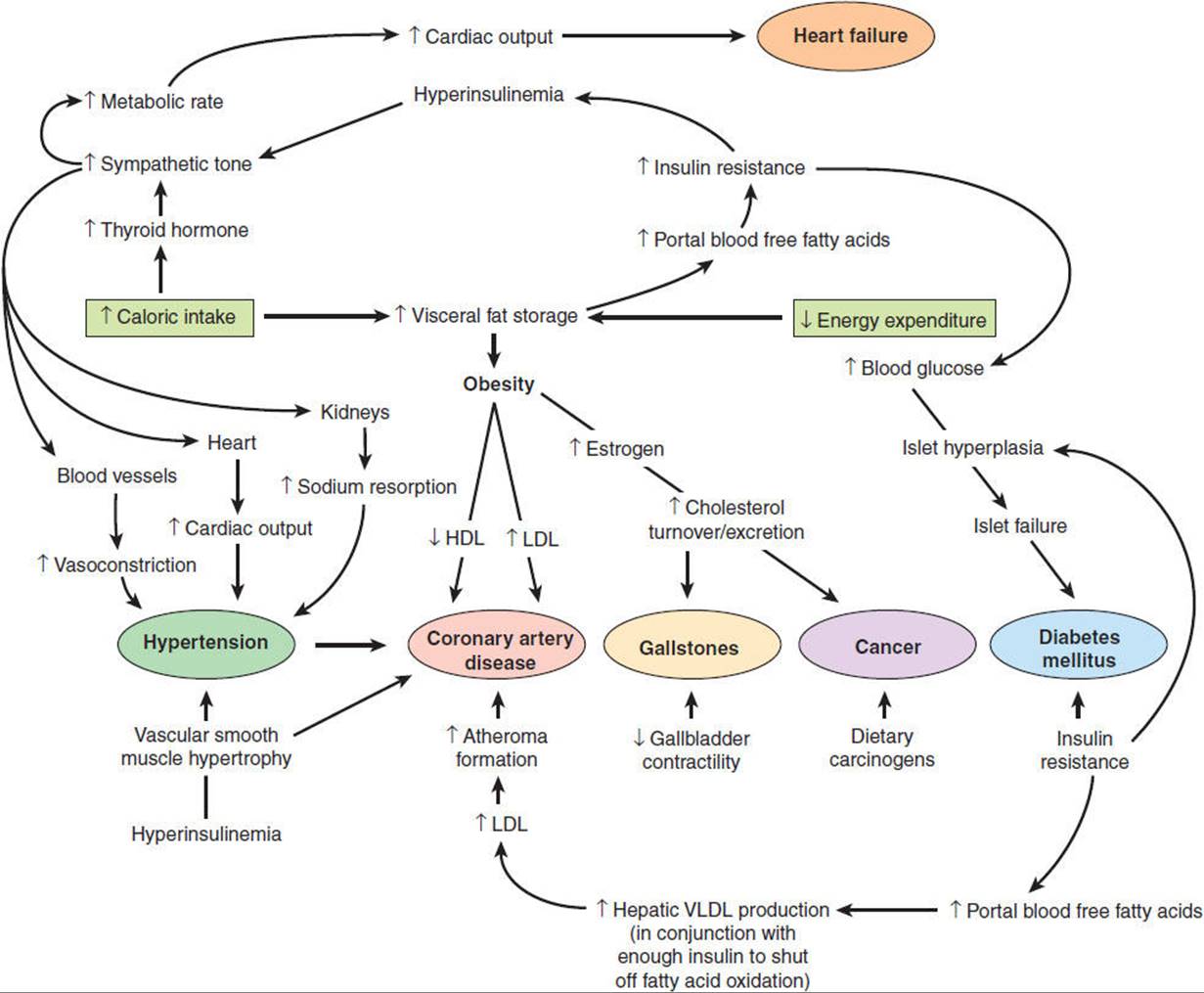
FIGURE 19-10 Role of obesity in the pathophysiology of disease. Some ways by which obesity contributes to disease. Short arrows refer to a change in the indicated parameter, and long arrows indicate a consequence of that change. In some cases, evidence is epidemiologic; in others, it is experimental. HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoproteins. (Redrawn, with permission, from Bray GA. Pathophysiology of obesity. Am J Clin Nutr. 1992;55:488S.)
Pathophysiology
Recognition that obesity plays a role in the pathophysiology of disease comes from epidemiologic studies identifying obesity as a risk factor without providing insight into the mechanism of the risk.
Although growing, only a very small number of cases of monogenetic disorders result in obesity in humans. Those syndromes highlight the importance of the aforementioned hypothalamic regulatory systems of body weight control. Several mutations in leptin or the leptin receptor, both resulting in the lack of sufficient leptin effect on the hypothalamus, have been described as a cause of both human and murine obesity. Most strikingly, leptin replacement therapy in cases of leptin deficiency leads to complete normalization of body weight. Other mutations have been described in the hypothalamic POMC system. Mutations in the MC4-R as well as mutations in the POMC gene or in POMC-processing proteases, both resulting in reduced MSH levels, lead to severe childhood obesity. Consistent with data describing the involvement of the POMC system in hypothalamic body weight regulation, all mutations within this system result in decreased signaling through the MC4-R and, therefore, increased food intake.
Aside from the monogenic disorders mentioned previously, obesity appears to be the result of multiple mechanisms and many studies have established an imbalance in the neuroendocrine hypothalamic and brain-gut systems. Thus, obesity may be either a cause or a consequence of disease, depending on the disorder. For example, type 2 diabetes mellitus is sometimes first manifested clinically by sudden weight gain, and this disorder can be difficult to control without weight loss, reflecting the insulin-resistant character of the obese state. Moreover, if the weight can be lost, the diabetes may once again become latent, controlled by diet and exercise alone. In such cases, obesity seems clearly to be an etiologic factor in the development of diabetes mellitus. Yet insulin injections, which may be necessary to control the symptoms of diabetes in such a patient, further exacerbate the weight gain that precipitated the disorder in the first place. Such “chicken-or-egg” relationships make the pathophysiology of obesity particularly difficult to dissect. Nevertheless, important progress has been made toward developing a coherent framework in which to view obesity as both cause and consequence of disease. Some of these observations are noted next.
The number of fat cells in the body is probably established during infancy. One hypothesis is that obesity appearing during adulthood results from enlargement of individual fat cells (hypertrophy) rather than an increased number of fat cells (hyperplasia). Obesity from fat cell hypertrophy appears to be much more easily controlled than obesity from fat cell hyperplasia. Perhaps feedback signals in response to the degree of fat cell hypertrophy are important to the hypothalamic “lipostat.”
It now appears that where fat is deposited is more important than how much is deposited. Thus, so-called visceral or central obesity (omental fat in the distribution of blood flow draining into the portal vein) seems far more important as a risk factor for obesity-related morbidity and mortality than so-called subcutaneous (gynecoid, lower body) or peripheral fat. It appears that visceral fat is more sensitive to catecholamines and less sensitive to insulin, making it a marker of insulin resistance. Consistent with these findings is the observation that obese individuals who engage in vigorous physical activity and whose obesity is largely due to high caloric intake (eg, sumo wrestlers) have subcutaneous rather than visceral fat and do not demonstrate substantial increased insulin resistance. In contrast, the obesity associated with a sedentary lifestyle is believed to be largely visceral obesity and is associated with a greater degree of insulin resistance in patients both with and without a diagnosis of diabetes mellitus. A parameter reflecting the different kinds of fat distribution is the waist-to-hip ratio, which has been shown to correlate with morbidity.
As mentioned, mutated leptin genes are also associated with obesity in some humans. However, in the vast majority of obese humans, excessive rather than deficient leptin levels are observed. Thus, it appears that the most common form of human obesity involves leptin resistance in the face of high endogenous leptin levels rather than defective leptin secretion as observed in ob/ob mice. An animal model for this condition is the obese db/db mouse, in which there is a defective leptin receptor. A variety of mechanisms, including diminished signaling through the leptin receptor and diminished transport across the blood-brain barrier, could account for leptin resistance in different individuals.
Psychologic factors also make an important contribution to the development of obesity. For example, obese individuals appear to regulate their desire for food by greater reliance on external cues (eg, time of day, appeal of the food) rather than endogenous signals (eg, feeling hungry).
Last, there is great interest in the development of drugs that alter these pathways (eg, neuropeptide Y and endocannabinoid antagonists) in ways that would promote weight loss as a treatment for obesity. On the contrary, endocannabinoid agonists are used to promote appetite and weight gain in the setting of severe wasting syndrome.
CHECKPOINT
13. Define obesity.
14. What diseases are associated with obesity?
15. Outline several pathophysiologic mechanisms by which obesity contributes to disease.
PITUITARY ADENOMA
An adenoma is a benign tumor of epithelial cell origin. Pituitary adenomas are of particular significance because (1) the pituitary is in an enclosed space with very limited capacity to accommodate an expanding mass and (2) pituitary adenomas may arise from cells that secrete hormones, giving rise to hormone overproduction syndromes.
Clinical Presentation
Pituitary adenomas are extremely common and are observed in about one in six autopsies. The majority of pituitary adenomas are clinically inapparent, either because they are nonfunctional or because hormone production does not reach the critical threshold to elicit clinical symptoms. If pituitary adenomas come to medical attention, symptoms and signs are related either to an expanding intracranial mass (headaches, diabetes insipidus, vision changes) or to manifestations of excess or deficiency of one or more pituitary hormones. Hormone deficiency results from destruction of the normal pituitary by the expanding adenoma. Hormone excess occurs when the adenoma secretes a particular hormone. Microadenomas (<10 mm in diameter) are more likely to present with complaints related to hormone excess than to local mass effects because they are small. Conversely, whether or not they secrete hormones, macroadenomas (>10 mm in diameter) can impinge on the optic chiasm above the sella turcica or the cavernous sinuses laterally.
Etiology
Any cell type in the pituitary gland can undergo hyperplasia or give rise to a tumor. Whether the patient with a pituitary tumor presents with a mass effect or symptoms referable to pituitary hormones depends on the size, growth rate, and secretory characteristics of the tumor. Which, if any, hormones the tumor secretes is generally a reflection of the cell type from which the tumor originated. Gigantism and acromegaly are due to oversecretion of growth hormone. Cushing disease is a syndrome of glucocorticoid excess resulting from oversecretion of ACTH. Galactorrhea occurs in patients with prolactin-secreting tumors. Tumors secreting TSH, LH, and FSH are extremely rare and (in accordance with their physiological function) can cause secondary hyperthyroidism, precocious puberty, or ovarian hyperstimulation.
Pathophysiology
Most pituitary adenomas are clonal in origin: A single cell with altered growth control and feedback regulation gives rise to the adenoma. Evidence for the involvement of genetic mutations in the cause of pituitary adenomas comes from the occurrence of familial pituitary tumor syndromes. At least four different syndromes caused by defined genetic mutations are known to raise significantly the incidence of pituitary tumor formation: multiple endocrine neoplasia type 1 (MEN-1), Carney complex (CNC), McCune-Albright syndrome, and AIP (aryl hydrocarbon receptor–interacting protein)-related predisposition to pituitary adenoma. Mutation of the MENIN tumor suppressor gene is the underlying cause of the multiple endocrine neoplasia syndrome type 1 (MEN-1). As is typical for tumor suppressor genes, loss of heterozygosity results in tumor formation. Pituitary tumors as well as tumors of the pancreas and hyperplasia of the parathyroid glands are typical manifestations in MEN-1 patients. Pituitary hyperplasia and microadenomas are also part of CNC. A subgroup of these patients harbor a mutation in the gene encoding for a protein A kinase subunit, resulting in an altered response to growth regulatory factors. In McCune-Albright syndrome, the GNAS1 gene, which encodes a G-protein stimulatory subunit, is mutated and renders the protein product constitutively active. Thus, cyclic adenosine monophosphate levels are chronically elevated in these cells, resulting in constitutive hormone gene activation and cell hyperplasia. Patients with AIP mutations are mainly predisposed to the development of growth hormone–secreting tumors.
Aside from these rare syndromes, the pathogenesis of pituitary adenomas is believed to be a multistep process analogous to the well-described consecutive mutations necessary for the induction of colon carcinomas. Several known or proposed factors have been shown to be part of transformation of pituitary cells (eg, GNAS1, PTTG). Other factors promoting pituitary tumor formation include chromosomal instability, presumably because of an unknown gene mutation, that results in further gene mutations and aneuploidy, altered hypothalamic signaling, and other endocrine and paracrine factors (eg, estrogens, growth factors).
Clinical Manifestations
Clinical manifestations related to mass effects are summarized in Figure 19-11. Bitemporal hemianopia is the classic visual field defect in a patient with an expanding pituitary mass (see Figure 19-11, panel C). It occurs because the crossing fibers of the optic tract, which lie directly above the pituitary gland and innervate the part of the retina responsible for temporal vision, are compressed by the tumor. However, in practice, a wide variety of visual field defects is seen, reflecting the unpredictable nature of the direction and extent of tumor growth as well as anatomic variability. The clinical manifestations of hormone excess are discussed under specific syndromes next.
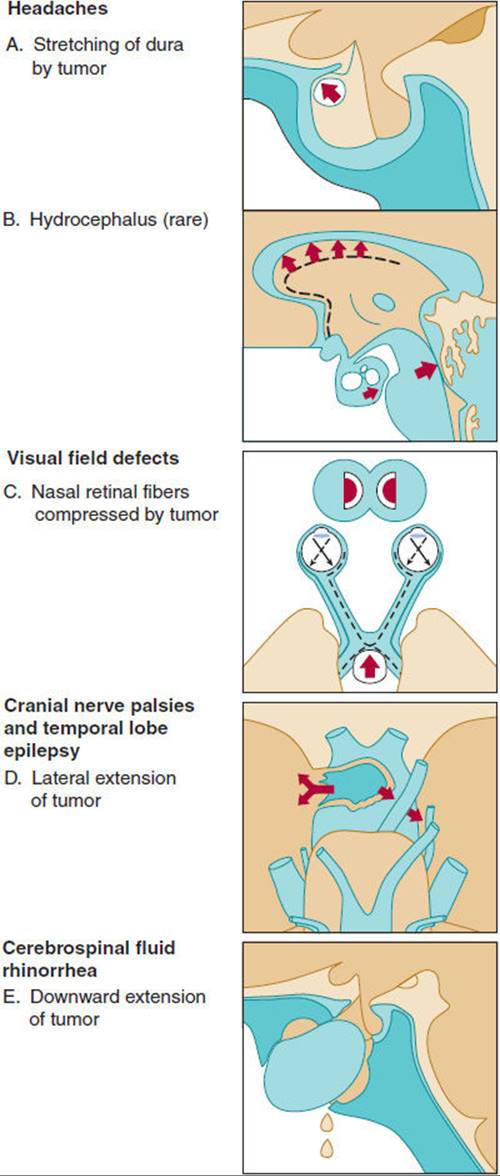
FIGURE 19-11 Various symptoms of pituitary tumor. Headaches are rarely caused by hydrocephalus. Visual field defects caused by extension of the tumor are plotted with the Goldmann perimeter. (Redrawn from Wass JAH. Hypopituitarism. In: Besser GM et al, eds. Clinical Endocrinology: An Illustrated Text. Gower, 1987.)
Regardless of whether a pituitary tumor is producing hormones or not, infarction of or hemorrhage into the expanding mass can destroy the normal pituitary gland. This leaves the patient without one or more of the pituitary hormones. The resulting clinical manifestations are considered later in the discussion of panhypopituitarism.
A. Prolactinoma
Hyperprolactinemia is the most common anterior pituitary disorder and has many causes (Table 19-6). Pathologic hyperprolactinemia, caused by prolactin-secreting adenomas (prolactinomas) or other clinical states that result in elevated prolactin levels such as primary hypothyroidism or dopamine-receptor blocking drug therapy, must be distinguished from the physiologic hyperprolactinemia of pregnancy and lactation. Roughly 40% of pituitary adenomas found in autopsies are prolactinomas. Most of the patients had no symptoms from microadenomas and died of unrelated causes.
TABLE 19-6 Causes of hyperprolactinemia.
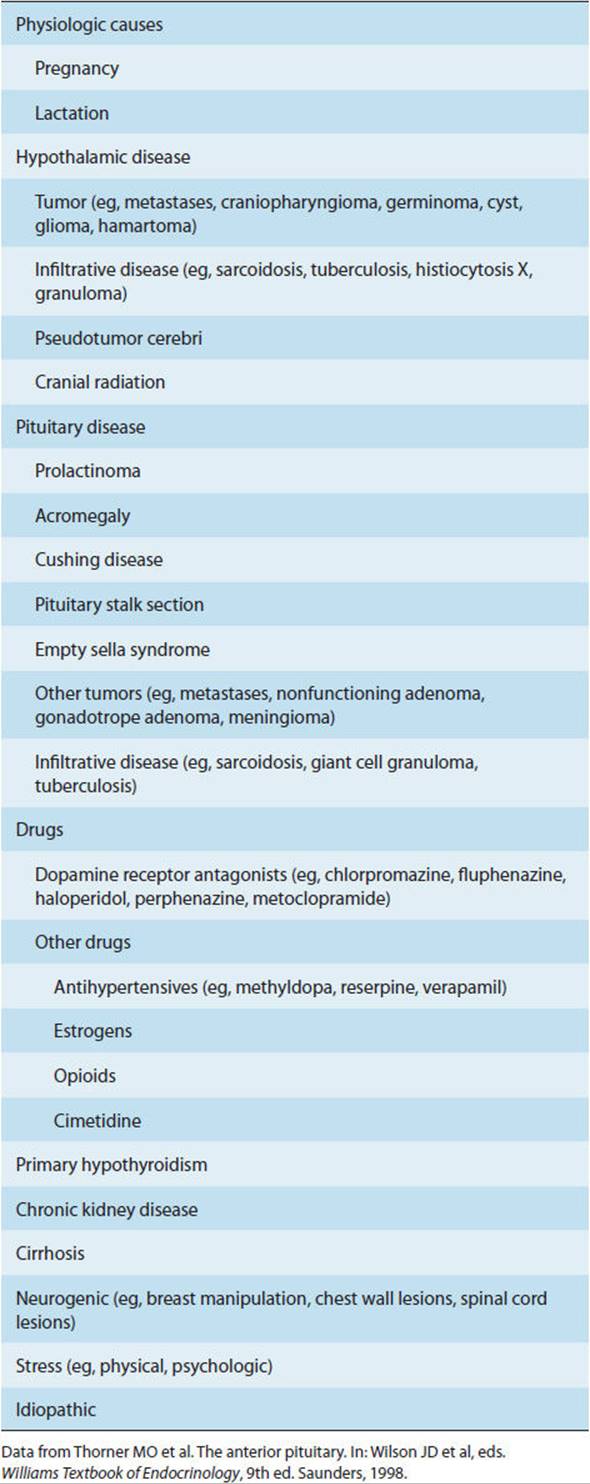
Patients with prolactin-secreting macroadenomas generally present with mass effect symptoms, whereas those with microadenomas may develop symptoms related to hormonal effects, from either the direct actions of prolactin (galactorrhea in 30–80% of women and up to 33% of men) or prolactin’s inhibitory effects on the hypothalamic-pituitary-gonadal axis. The resulting reproductive dysfunction presents variably: amenorrhea, irregular menses, or menses with infertility in women and decreased libido and partial or complete impotence or infertility in men.
Decreased bone density is another common consequence of hyperprolactinemia resulting from hypogonadism and perhaps also poorly understood direct effects of prolactin on bone.
B. Growth Hormone–Secreting Adenoma
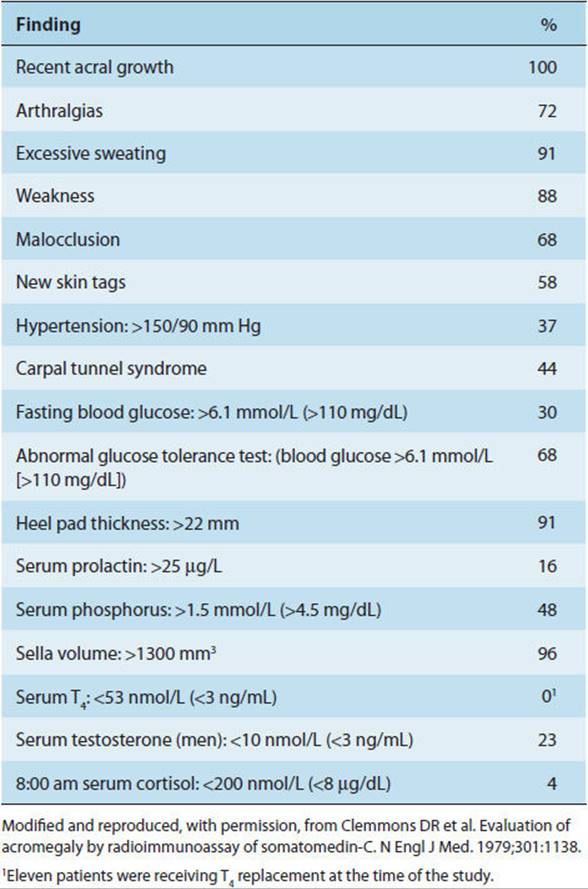
GH-secreting tumors give rise to the syndromes of gigantism or acromegaly depending on whether they develop before or after closure of the epiphyses. Clinical findings in gigantism and acromegaly are summarized in Table 19-7and reflect a combination of the insulin-like effects of the hormone, promoting visceromegaly, and the counterregulatory effects, promoting glucose intolerance.
TABLE 19-7 Clinical and laboratory findings in 57 patients with acromegaly.
C. ACTH-Secreting Pituitary Adenoma (Cushing Syndrome)
Secretion of excess cortisol as a result of overproduction of ACTH by a pituitary adenoma is the most common cause of spontaneous Cushing syndrome (Chapter 21). ACTH-secreting pituitary adenomas are eight times more common in women than in men and must be distinguished from the effects caused by CRH or ACTH arising from outside the hypothalamus and pituitary gland, respectively, and from hypercortisolism due to adrenal adenomas and carcinomas.
The symptoms and signs of ACTH-secreting pituitary adenomas are a consequence of both local mass effects, similar to those discussed previously for other types of pituitary tumors, and effects from overproduction of cortisol by the adrenal gland, as discussed in Chapter 21. Nelson syndrome is the rapid progression of an ACTH-secreting pituitary adenoma, which is often observed after bilateral adrenalectomy to control the symptoms of cortisol excess. With the advent of vigorous glucocorticoid substitution regimens, transsphenoidal pituitary surgery, and radiation therapy, the incidence of this complication has greatly diminished.
CHECKPOINT
16. What is a pituitary adenoma?
17. What brings patients with pituitary adenomas to medical attention?
18. What are the most common forms of pituitary adenoma?
19. How does a pituitary adenoma develop?
HYPOPITUITARISM
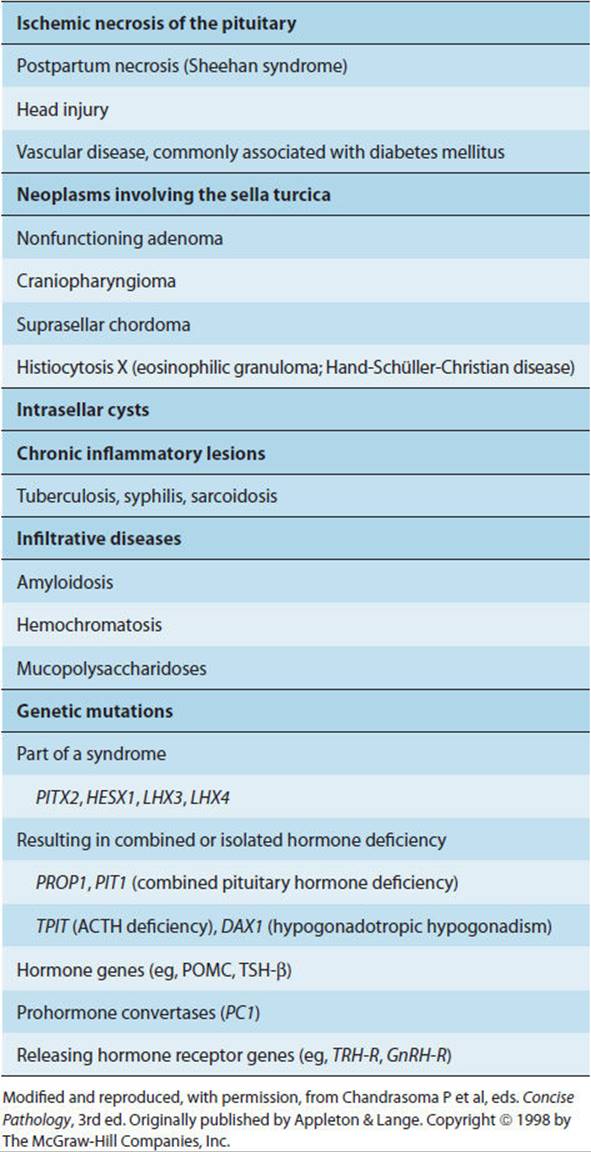
Panhypopituitarism is the syndrome resulting from complete loss of all of the hormones secreted by the pituitary gland. Hypopituitarism refers to the loss of one or more pituitary hormones. Causes of hypopituitarism are listed in Table 19-8.
TABLE 19-8 Causes of hypopituitarism.
Clinical Presentation
The complex of symptoms in hypopituitarism varies depending on the extent and duration of disease. Regardless of the underlying cause, in non-congenital forms of hypopituitarism, GH deficiency occurs as the earliest hormonal deviance, followed by ACTH and gonadotropin (LH and FSH) deficiencies, and finally, TSH deficiency. In some cases, panhypopituitarism is of sudden onset (eg, caused by pituitary infarction or trauma). These patients may rapidly develop two potentially life-threatening situations as a consequence of loss of ACTH and vasopressin. First, since the patient is unable to mount a stress response because of a lack of ACTH-stimulated glucocorticoid secretion, even relatively mild stress may be lethal. Second, a patient unable to maintain water intake will be unable to compensate for the massive diuresis associated with vasopressin deficiency (diabetes insipidus). Thus, the patient will quickly become comatose as a result of profound water loss and the complications of dehydration and hyperosmolarity.
In other cases, pituitary insufficiency develops more insidiously (eg, from progressive destruction of the pituitary gland by a nonsecreting tumor or subsequent to pituitary radiation therapy). In many of these slowly developing cases of panhypopituitarism, the patient comes to medical attention with complaints related to reproductive functions (amenorrhea in women; infertility or erectile dysfunction in men) caused by LH and FSH deficiency. Other patients have nonspecific complaints (eg, lethargy or altered bowel habits), perhaps related to the gradual development of hypothyroidism (from TSH deficiency). Panhypopituitarism may be unmasked only when the patient does poorly during some other unrelated medical emergency because of an inability to mount a protective stress response because of an ACTH and consequent glucocorticoid deficiency.
Etiology
Panhypopituitarism of sudden onset is usually due to traumatic disruption of the pituitary stalk, infarction and hemorrhage into a pituitary tumor, or ischemic destruction of the pituitary after systemic hypotension (eg, Sheehan syndrome or postpartum hypopituitarism after massive blood loss in childbirth). A number of rare genetic causes have also been reported (Table 19-8, Figure 19-4). Gradually acquired hypopituitarism is most often due to extension of pituitary tumors or occurs as a complication of radiation therapy for brain tumors.
Pathophysiology
The biochemical hallmark of hypopituitarism is low levels of pituitary hormones in the face of low end-organ products of one or more components of the neuroendocrine axes involving the pituitary. By contrast, primary end-organ failure results in compensatory high levels of the relevant pituitary hormones.
Another biochemical difference between primary end-organ failure and end-organ failure secondary to hypopituitarism is that not all end-organ functions are equally controlled by the pituitary. In the case of the adrenal cortex, for example, although mineralocorticoid secretion can be stimulated by ACTH, it is not dependent on it.
Both of the biochemical distinctions between primary end-organ failure and pituitary failure have important clinical implications. For example, hyperpigmentation occurs in primary adrenal insufficiency because several POMC-derived peptides (MSHs, ACTH) stimulate skin pigmentation via binding to the melanocortin-1 receptor (MC1-R). Because levels of POMC-derived peptides are not elevated in pituitary and hypothalamic insufficiency, hyperpigmentation does not occur. Similarly, the symptoms of adrenal insufficiency secondary to pituitary disease may be more subtle than in the case of primary adrenal failure, because a significant fraction of mineralocorticoid production is preserved even in the absence of ACTH (Chapter 21).
In the case of trauma and pituitary stalk transection, it is notable that hypopituitarism may improve over time as local edema diminishes and some degree of integrity of the pituitary stalk with its connection to the hypothalamus is reestablished. Sometimes, however, these symptoms and signs may worsen over time as the few residual intact cells or connections are lost.
Notably, injuries disconnecting the pituitary from the hypothalamus result in deficiencies of most of the anterior pituitary hormones except prolactin. Indeed, prolactin secretion is usually preserved or elevated because it is the only pituitary hormone regulated by tonic hypothalamic inhibition.
Clinical Manifestations
The symptoms and signs of hypopituitarism depend on the extent and duration of specific pituitary hormone deficiencies and the patient’s overall clinical status. Thus, a relative deficiency of vasopressin can be compensated for by increasing water intake; adrenal insufficiency may not be manifest until the patient needs to mount a stress response. Hypothyroidism may become manifest gradually over months because of the relatively long half-life and large reservoir of thyroid hormone normally available in the gland.
The clinical manifestations of hypopituitarism are those of the end-organ deficiency syndromes. Most important are adrenal insufficiency, hypothyroidism, and diabetes insipidus. Less crucial but often the most sensitive clues to the presence of pituitary disease are amenorrhea in women and infertility or impotence in men.
CHECKPOINT
20. What are the most common causes of panhypopituitarism?
21. How do patients with panhypopituitarism come to medical attention?
22. How would you determine what replacement therapy is required for a patient with panhypopituitarism?
DIABETES INSIPIDUS
Diabetes insipidus is a syndrome of polyuria resulting from the inability to concentrate urine and, therefore, to conserve water as a result of lack of vasopressin action.
Clinical Presentation
The initial clinical presentation of diabetes insipidus is polyuria that persists in circumstances that would normally lead to diminished urine output (eg, dehydration), accompanied by thirst. Adults may complain of frequent urination at night (nocturia), and children may present with bed-wetting (enuresis). No further symptoms develop if the patient is able to maintain a water intake commensurate with water loss. The volume of urine produced in the total absence of vasopressin may reach 10–20 L/d. Thus, should the patient’s ability to maintain this degree of fluid intake be compromised (eg, damage to hypothalamic thirst regulating centers), dehydration can develop and may rapidly progress to coma.
Etiology
Diabetes insipidus can be due to (1) diseases of the CNS (central diabetes insipidus), affecting the synthesis or secretion of vasopressin; (2) diseases of the kidney (nephrogenic diabetes insipidus), with loss of the kidney’s ability to respond to circulating vasopressin by retaining water; or (3) pregnancy, with probable increased metabolic clearance of vasopressin. In both central and nephrogenic diabetes insipidus, urine is hypotonic. The most common central causes are accidental head trauma, intracranial tumor (eg, craniopharyngioma), and the postintracranial surgery state. Less common causes are listed in Table 19-9. Nephrogenic diabetes insipidus may be familial or caused by renal damage from a variety of drugs. Diabetes insipidus–like syndromes may result from mineralocorticoid excess, pregnancy, and other causes. True nephrogenic diabetes insipidus must be distinguished from an osmotic (and hence vasopressin-resistant) diuresis. Likewise, washout of the medullary interstitial osmotic gradient, which is necessary for the concentration of urine, may occur with prolonged diuresis resulting from any cause and may be confused with true diabetes insipidus. In both cases (osmotic diuresis and medullary washout), the urine is hypertonic or isotonic rather than hypotonic. Finally, extreme primary polydipsia (drinking excessive amounts of water, often because of a psychiatric disorder) results in an appropriately large volume of dilute urine and a low plasma vasopressin level, thus mimicking true central diabetes insipidus.
TABLE 19-9 Causes of central and nephrogenic diabetes insipidus.
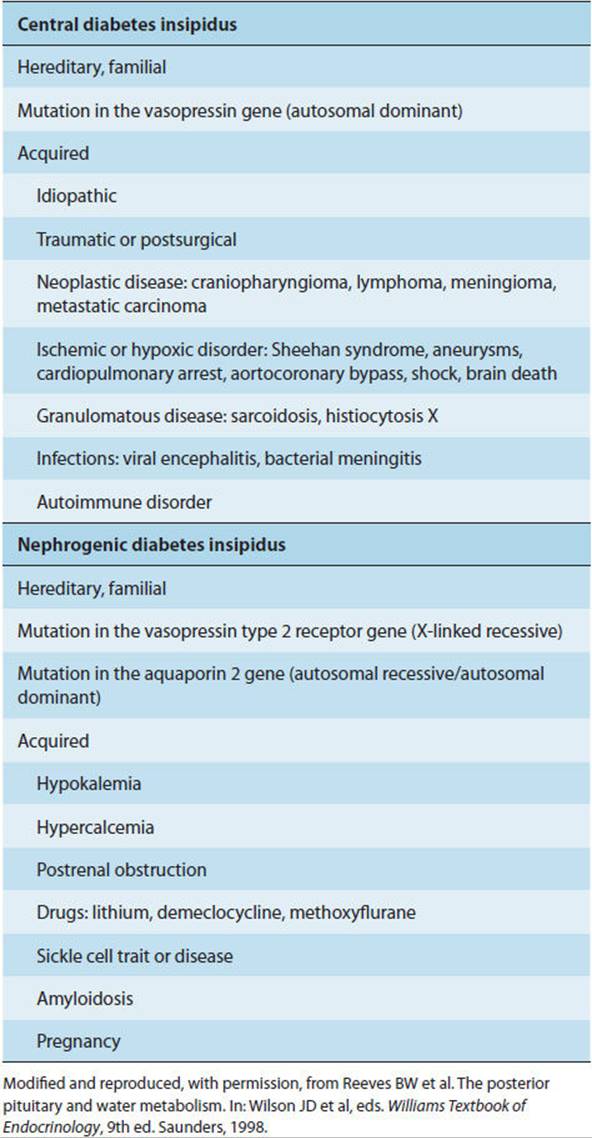
Pathophysiology
A. Central Diabetes Insipidus
Central diabetes insipidus can be either permanent or transient, reflecting the natural history of the underlying disorder (Table 19-9). Only about 15% of the vasopressin-secreting cells of the hypothalamus need to be intact to maintain fluid balance under normal conditions. Simple destruction of the posterior pituitary does not cause sufficient neuronal loss to result in permanent diabetes insipidus. Rather, destruction of the hypothalamus or at least some of the supraoptic-hypophysial tract must also occur.
A more common finding is transient disease resulting from acute injury with neuronal shock and edema (eg, post-infarction or post-trauma), leading to cessation of vasopressin secretion with subsequent resumption of sufficient vasopressin secretion to resolve symptoms, because of either neuronal recovery or resolution of edema with reestablishment of hypothalamic-pituitary neurovascular integrity.
B. Nephrogenic Diabetes Insipidus
Familial nephrogenic diabetes insipidus is the result of a generalized defect in either the V2 class of vasopressin receptors or the aquaporin-2 water channel of the renal collecting ducts.
Drug-induced nephrogenic diabetes insipidus appears to result from sensitivity of the vasopressin receptor to lithium, fluoride, and other salts. This occurs in 12–30% of patients treated with these drugs. It is generally reversible on termination of exposure to the offending drug (Table 19-9).
C. Diabetes Insipidus–Like Syndromes
There are several diabetes insipidus–like syndromes. As an example, diabetes insipidus is a rare complication of pregnancy. It appears to be due to excessive vasopressinase in plasma. This enzyme, which selectively degrades vasopressin, is presumably released from the placenta. A hallmark of this entity is that it is reversed by administration of the vasopressin analog desmopressin acetate, which is resistant to degradation by the enzyme.
Clinical Manifestations
Diabetes insipidus must be distinguished from other causes of polyuria and hypernatremia (Table 19-10). The hallmark of diabetes insipidus is dilute urine, even in the face of hypernatremia. Dipstick testing of the urine for glucose distinguishes diabetes mellitus. Conditions in which osmotic diuresis is responsible for polyuria can be distinguished from diabetes insipidus by their normal or elevated urine osmolality. Primary polydipsia is distinguished by the presence of hyponatremia, whereas in diabetes insipidus the serum sodium should be normal or elevated. In primary polydipsia, uncontrolled excess water ingestion drives the polyuria, whereas in diabetes insipidus, hypertonicity stimulates thirst.
TABLE 19-10 Major causes of hypernatremia.
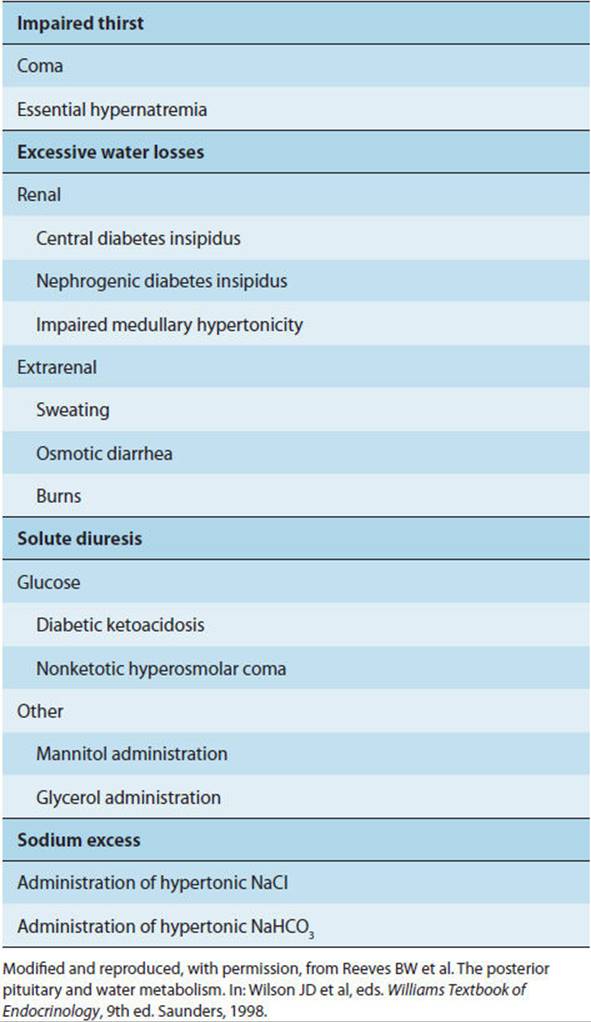
Distinguishing central from nephrogenic diabetes insipidus depends ultimately on a determination of responsiveness to injected vasopressin, with a dramatic decrease in urine volume and increase in urine osmolality in the former and little or no change in the latter. In central diabetes insipidus, circulating vasopressin levels are low for a given plasma osmolality, whereas in nephrogenic diabetes insipidus they are high.
Polyuria in nephrogenic diabetes insipidus results from an inability to conserve water in the distal nephron because of a lack of vasopressin-dependent water channels. These channels, which reside within vesicles in the cytoplasm of collecting duct cells, are normally inserted into the apical plasma membrane in response to vasopressin stimulation, permitting increased reabsorption of water. Up to 13% of the volume of the glomerular filtrate can be reclaimed in this manner.
In diabetes insipidus of either central or nephrogenic origin, if the patient is unable to maintain sufficient water intake to offset polyuria, dehydration with consequent hypernatremia develops. Hypernatremia leads to a number of neurologic manifestations, including progressive obtundation (decreased responsiveness to verbal and physical stimuli), myoclonus, seizures, focal deficits, and coma. These neurologic manifestations result from cell shrinkage and volume loss as a result of osmotic forces, sometimes complicated by intracranial hemorrhage because of stretching and rupture of small blood vessels. Barring structural changes such as those leading to hemorrhage, the neurologic consequences of hypernatremia are reversible on resolution of the underlying metabolic disorder.
The time course of hypernatremia is an important variable in the development of neurologic symptoms in that, over time, neurons generate “idiogenic osmoles” (ie, amino acids and other metabolites that serve to raise intracellular osmolality to the level in the blood and thereby minimize fluid shifts out of the cells of the brain). Thus, the more slowly hypernatremia develops, the less likely are neurologic complications resulting from fluid shifts in the brain or from a vascular catastrophe.
CHECKPOINT
23. What clues would suggest diabetes insipidus in a new patient?
24. How would you make a definitive diagnosis of diabetes insipidus?
25. What are the pathophysiologic differences between central and nephrogenic diabetes insipidus?
SYNDROME OF INAPPROPRIATE VASOPRESSIN SECRETION (SIADH)
The syndrome of inappropriate ADH (vasopressin) secretion (SIADH) is one of several causes of a hypotonic state (Table 19-11). SIADH is due to the secretion of vasopressin in excess of what is appropriate for hyperosmolality or intravascular volume depletion.
TABLE 19-11 The hypotonic syndromes.
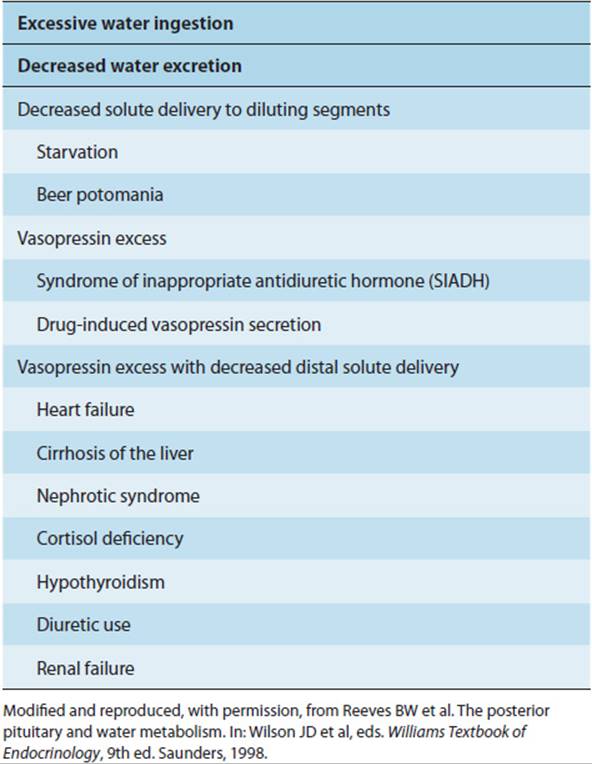
Clinical Presentation
The cardinal clinical presentation of SIADH is hyponatremia without edema. Depending on the rapidity of onset and the severity, the neurologic consequences of hyponatremia include confusion, lethargy and weakness, myoclonus, asterixis, generalized seizures, and coma.
Etiology
A variety of vasopressin-secreting tumors, CNS disorders, pulmonary disorders, and drugs have been associated with SIADH (Table 19-12). Therefore, it is worth mentioning that the hypothalamic neurons and the posterior pituitary are not always the source of vasopressin secretion. In fact, the hypothalamus and pituitary account for elevated vasopressin levels in only one-third of patients with SIADH, and it is important to regard SIADH as not necessarily a disorder of the hypothalamic-pituitary system. Several metabolic disorders can produce hyponatremia and must be investigated and ruled out before the diagnosis of true SIADH is made. In particular, adrenal insufficiency and hypothyroidism are often associated with hyponatremia. In these conditions, sodium deficiency and subsequent volume depletion trigger vasopressin secretion. Hyponatremia accompanying CNS disorders is caused either by SIADH or by cerebral salt wasting (CSW) with an increased release of natriuretic peptides (eg, BNP, ANP). A major difference between these two disorders is in the total extracellular volume, which is increased in SIADH and reduced in CSW.
TABLE 19-12 Causes of SIADH.
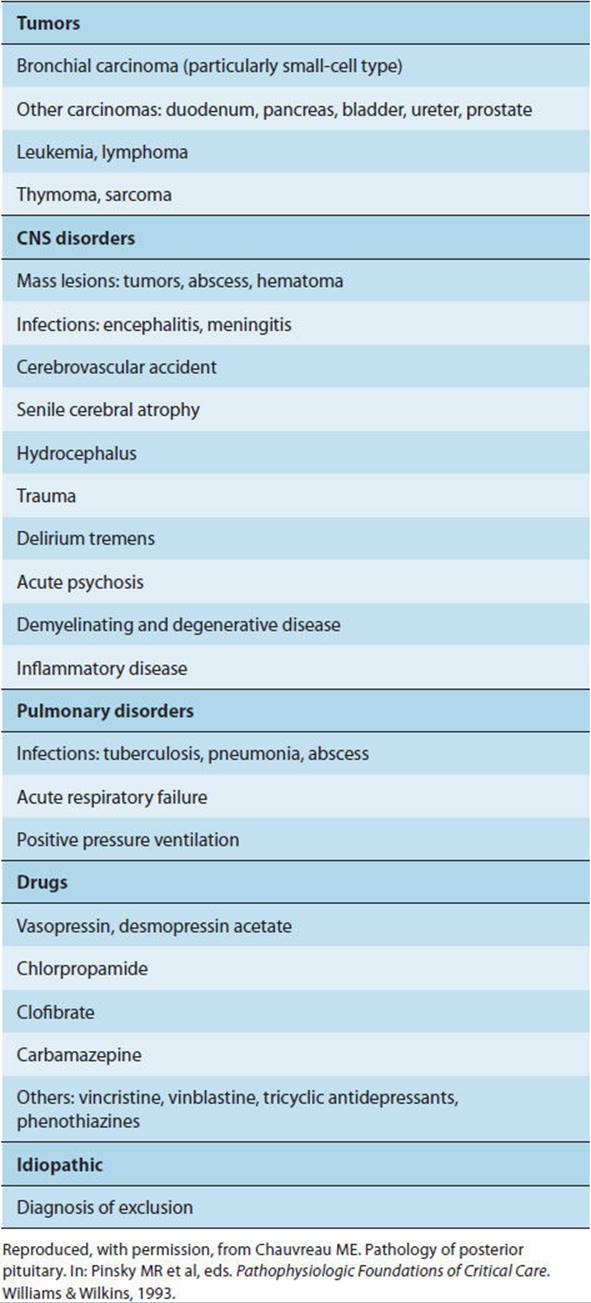
Pathophysiology
The serum sodium concentration (and hence osmolarity) is normally determined by the balance of water intake, renal solute delivery (a necessary step in water excretion), and vasopressin-mediated distal renal tubular water retention. Disorders in any one of these features of normal sodium balance, or factors controlling them, can result in hyponatremia. Hyponatremia occurs when the magnitude of the disorder exceeds the capacity of homeostatic mechanisms to compensate for dysfunction. Thus, simple excess water ingestion is generally compensated for by renal water diuresis. The exceptions are (1) when water ingestion is extreme (greater than the approximately 18 L daily that can be excreted via the kidney) or (2) when renal solute delivery is limited (eg, in salt depletion), thereby limiting the ability of the kidney to excrete free water.
In hypoadrenal states, renal sodium loss resulting from lack of aldosterone has two consequences. Most importantly, volume depletion as a consequence of renal sodium loss results in the release of vasopressin; although the primary stimulus for ADH secretion is an elevated plasma osmolarity, ADH release is stimulated by low intravascular volume as well. Second, diminished renal solute delivery impairs the ability of the kidney to excrete a water load, in the case in which ingestion of water exceeds nonrenal water loss.
In hypothyroidism, both renal solute delivery and function of the osmostat to which vasopressin secretion is coupled appear to be impaired, resulting in hyponatremia.
True causes of hyponatremia, including SIADH, must also be distinguished from so-called pseudohyponatremia. Pseudohyponatremia occurs in two groups of conditions (Table 19-13). First, there are those in which infusion of hyperosmolar solutions (eg, glucose) pulls water out of cells, thereby diluting the sodium. The key feature of these conditions is hyponatremia without hypo-osmolality. Second, pseudohyponatremia occurs when the nonaqueous fraction of plasma is larger than normal. Sodium only equilibrates with, and is regulated in, the aqueous fraction of plasma, and calculations of serum sodium concentration typically correct for total plasma volume because the nonaqueous fraction of plasma volume is normally negligible. In those relatively rare conditions in which the nonaqueous fraction is significant (eg, severe hyperlipidemic states, multiple myeloma, and other conditions with higher than normal serum lipid or protein concentrations), the calculated sodium concentration will, therefore, be misleadingly low.
TABLE 19-13 Causes of pseudohyponatremia.
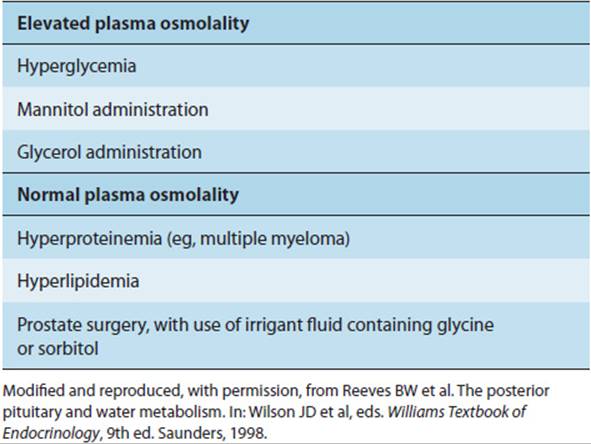
The pathophysiologic mechanisms behind most cases of SIADH are not well understood. It has been proposed that baroreceptor input from the lung is impaired in those pulmonary disorders that result in SIADH. CNS lesions causing SIADH are presumed to interrupt the vasopressin-inhibiting neural pathways. Regardless of the mechanism, in most cases the hyponatremia of SIADH is partially limited by secretion of atrial natriuretic peptide. Thus, severe hyponatremia develops only when water intake is relatively increased, and edema formation is rare. The simplest therapy is restriction of free water intake and, in the case of CNS or pulmonary lesions, treatment of the underlying disease.
Clinical Manifestations
The clinical manifestations of SIADH are in part determined by the nature and course of any underlying disorder (eg, CNS or pulmonary disease), by the severity of hyponatremia, and by the rapidity with which hyponatremia develops. Regardless of its cause, SIADH can have neurologic manifestations, including confusion, asterixis, myoclonus, generalized seizures, and coma. These occur as a result of osmotic fluid shifts and resulting brain edema and elevated intracranial pressures; brain swelling is limited by the size of the skull. Physiologic mechanisms to counter this swelling include depletion of intracellular osmoles, especially potassium ions. The more rapid the progression of hyponatremia, the more likely it is that brain edema and increased intracranial pressure will develop and that the neurologic complications and herniation will lead to permanent damage. However, even when hyponatremia develops slowly, it can in extreme cases (eg, serum sodium <110 mEq/L) result in seizures and altered mental status. Central pontine myelinolysis can develop and cause permanent neurologic damage in patients whose hyponatremia is corrected too rapidly.
CHECKPOINT
26. What conditions are associated with SIADH?
27. How would you distinguish SIADH from other causes of hyponatremia?
28. What are the neurologic consequences of SIADH, and how may they be prevented?
CASE STUDIES
Yeong Kwok, MD
(See Chapter 25, p. 733 for Answers)
CASE 94
A 53-year-old woman came to the clinic to get help managing her weight. She has been overweight since childhood and has continued to gain weight throughout her adult life. She has tried numerous diets without lasting success. She initially loses weight, but then regains it after a few months. She is otherwise healthy and is not taking any medications. Other family members are also overweight or obese. She does not do any regular exercise and has a sedentary office job. On examination, she is 5 feet 3 inches tall and weighs 260 pounds, with a body mass index (BMI) of 46.2 (normal <25).
Questions
A. How is body weight controlled?
B. How is obesity defined?
C. What medical conditions is she at increased risk for due to her obesity?
CASE 95
A 30-year-old woman presents to the emergency department after sideswiping a parked car. She reports that she never saw the car until after she hit it. She denies any trauma to herself but does complain of headache. She states that she has had headaches every day for the past 3 months, and this one is similar to her other headaches. She describes the headache as a frontal throbbing pain that is worse when she lies down and occasionally it wakes her from sleep. She has no significant medical history, takes no medications, and denies alcohol, tobacco, or drug use. On review of systems, she notes irregular menses but denies having other complaints. On examination she appears to be well, with normal vital signs. Her neurologic examination is notable for bitemporal hemianopia. On breast examination, galactorrhea is present but no masses. The remainder of the examination is unremarkable.
Questions
A. What is the likely diagnosis?
B. How did this condition arise?
C. What is the pathogenetic mechanism of her bitemporal hemianopia? Her headaches?
D. What is the cause of her irregular menses? Her galactorrhea?
CASE 96
A 31-year-old woman with a medical history significant for pituitary macroadenoma status post-radiation therapy presents to the clinic with a complaint of amenorrhea. Before the diagnosis of pituitary adenoma, she had irregular menses. This irregularity had persisted, with menses lasting approximately 3 days, and occurring about once every 1.5–2 months. However, for the past 4 months, she has had no menses. She denies sexual activity. On review of systems, she notes progressive fatigue and 10 pounds of weight gain over several months.
The pituitary macroadenoma was treated with radiation therapy 1 year ago. She has been without medical care since completing therapy because she moved and has not yet found a physician. She is taking no medications. On examination her blood pressure is 100/60 mm Hg and heart rate is 80 bpm. Neurologic examination is normal except for a slight delay in the relaxation phase of her deep tendon reflexes. On head-neck examination, she has somewhat coarse, brittle brown hair. Neck examination discloses no goiter or masses. Lung, cardiac, and abdominal examinations show no abnormalities. Pelvic examination reveals normal female genitalia without uterine or ovarian masses. Urine pregnancy test is negative.
Questions
A. What is the likely cause of this and why?
B. On the basis of her history and physical examination, do you suspect any other hormone deficiencies? Why do you think so?
C. What other hormonal deficiencies should you be concerned about in this patient? Why might they be asymptomatic currently?
CASE 97
A 54-year-old man with a medical history significant for bipolar disease presents to his physician with complaints of polyuria. He states that he must get up three or four times each night to urinate. He also notes frequent thirst. He denies polyphagia, urinary urgency, difficulty initiating urination, and postvoid dribbling. His medical history is notable only for bipolar disease. He has a long-standing history of noncompliance with medications for this disease, with frequent hospitalizations for both mania and depression, but has been stable on lithium for the past 6 months. He denies any symptoms of mania or depression at this time. He takes no other medications. Family history is notable for depression and substance abuse but is otherwise negative. The patient has a history of polysubstance abuse but has been “clean and sober” for the past 6 months.
On examination, the patient’s vital signs are within normal limits. Head-neck examination reveals slightly dry mucous membranes. Rectal examination reveals a normal prostate without masses. The remainder of his examination is unremarkable. Urinalysis reveals dilute urine without glucose or other abnormality. Serum electrolytes reveal a mildly increased sodium level. A diagnosis of diabetes insipidus is entertained.
Questions
A. Do you suspect central or nephrogenic diabetes insipidus? Why? How would you confirm the diagnosis?
B. How does lithium cause diabetes insipidus?
C. What is the cause of this patient’s polyuria? His thirst?
D. What might occur if this patient were unable to maintain sufficient water intake?
CASE 98
A 75-year-old man with terminal small cell carcinoma of the lung presents to the emergency department with altered mental status. The patient’s wife, who cares for him at home, states that he is quite weak at baseline, requiring assistance with all activities of daily living. Over the past few days, he has become progressively more lethargic. She has been careful to adequately hydrate him, waking him every 2 hours to give him water to drink. His appetite has been poor, but he willingly ingests the water, consuming 2–3 quarts per day. He is taking morphine for pain and dyspnea.
On examination, the patient is a cachectic white man in mild respiratory distress. He is lethargic but arousable. He is oriented to person only. Vital signs reveal a temperature of 38 °C, blood pressure of 110/60 mm Hg, heart rate of 88 bpm, respiratory rate of 18/min, and oxygen saturation of 96% on 3 L of oxygen. On head-neck examination, pupils are 3 mm and reactive, scleras are anicteric, and conjunctivas are pink. Mucous membranes are moist. Neck is supple. There are decreased breath sounds in the left lower posterior lung field and rales in the upper half. Cardiac examination shows a regular heartbeat without murmur, gallop, or rub. Abdomen is benign without masses. Extremities are without edema, cyanosis, or clubbing. Neurologic examination shows only bilateral positive Babinski reflexes and asterixis. Laboratory studies reveal a serum sodium level of 118 mEq/L.
Questions
A. What conditions are associated with SIADH? Which are present in this patient?
B. What pathophysiologic mechanism produces SIADH?
C. What is the cause of this patient’s lethargy, confusion, and asterixis?
D. How would you treat this patient’s hyponatremia?
REFERENCES
General
Javorsky BR et al. Hypothalamus and pituitary gland. In: Gardner DG et al, eds. Greenspan’s Basic & Clinical Endocrinology, 9th ed. McGraw-Hill, 2011.
Obesity
Berthoud HR et al. Neural and metabolic regulation of macronutrient intake and selection. Proc Nutr Soc. 2012 Aug;71(3):390–400. [PMID: 22617310]
Farooqi IS. Genetic, molecular and physiological insights into human obesity. Eur J Clin Invest. 2011 Apr;41(4):451–5. [PMID: 21391993]
Suzuki K et al. The role of gut hormones and the hypothalamus in appetite regulation. Endocr J. 2010;57(5):359–72. [PMID: 20424341]
Pituitary Adenoma
Colao A. Pituitary tumours: the prolactinoma. Best Pract Res Clin Endocrinol Metab. 2009 Oct;23(5):575–96. [PMID: 19945024]
Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011 May;7(5):257–66. [PMID: 21423242]
Melmed S et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011 Feb;96(2):273–88. [PMID: 21296991]
Hypopituitarism
Fernandez-Rodriguez E et al. Subclinical hypopituitarism. Best Pract Res Clin Endocrinol Metab. 2012 Aug;26(4):461–9. [PMID: 22863388]
Romero CJ et al. The molecular basis of hypopituitarism. Trends Endocrinol Metab. 2009 Dec;20(10):506–16. [PMID: 19854060]
Diabetes Insipidus/Oxytocin
Devin JK. Hypopituitarism and central diabetes insipidus: perioperative diagnosis and management. Neurosurg Clin N Am. 2012 Oct;23(4):679–89. [PMID: 23040752]
Devuyst O. Physiopathology and diagnosis of nephrogenic diabetes insipidus. Ann Endocrinol (Paris). 2012 Apr;73(2):128–9. [PMID: 22503803]
Fenske W et al. Clinical review: current state and future perspectives in the diagnosis of diabetes insipidus: a clinical review. J Clin Endocrinol Metab. 2012 Oct;97(10):3426–37. [PMID: 22855338]
Syndrome of Inappropriate ADH Secretion
Adrogué HJ et al. The challenge of hyponatremia. J Am Soc Nephrol. 2012 Jul;23(7):1140–8. [PMID: 22626822]
Esposito P et al. The syndrome of inappropriate antidiuresis: pathophysiology, clinical management and new therapeutic options. Nephron Clin Pract. 2011;119(1):c62–73. [PMID: 21677440]
Hannon MJ et al. The syndrome of inappropriate antidiuretic hormone: prevalence, causes and consequences. Eur J Endocrinol. 2010 Jun;162(Suppl 1):S5–12. [PMID: 20164214]
Maesaka JK et al. Is it cerebral or renal salt wasting? Kidney Int. 2009 Nov;76(9):934–8. [PMID: 19641485]
Peri A et al. Hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). J Endocrinol Invest. 2010 Oct;33(9):671–82. [PMID: 20935451]
Thompson C et al. Hyponatraemia: an overview of frequency, clinical presentation and complications. Best Pract Res Clin Endocrinol Metab. 2012 Mar;26(Suppl 1):S1–6. [PMID: 22469246]