Diana M. Juriloff
Orofacial clefting has been observed sporadically in a variety of vertebrates, including monkeys (Stills and Bullock, 1981), gorillas (Siebert et al., 1998), cattle (Swartz et al., 1982), cats (Loevy and Feynes, 1968), dogs (Dreyer and Preston, 1974), and chickens (Juriloff and Roberts, 1975), but genetic studies have usually been limited. The most advanced animal models for genetic analysis and linkage mapping of orofacial defects are laboratory mice, where the extensive resources of specialized strains and crosses, spontaneous and targeted mutations, polymorphic markers, and welldeveloped linkage and physical maps can be used to identify genes and their effects on orofacial development. Conservation of genes and linkage relationships between mice and humans is well documented, and the chromosomal location of a gene in humans can often be predicted from its genetic map position in mice. Development of the orofacial complex is very similar between mouse and human embryos, and much of the understanding of developmental mechanisms in humans has been inferred from mice (Diewert and Wang, 1992).
Highly penetrant Mendelian defects of craniofacial development are relatively easily mapped in both mice and humans. The most common orofacial clefting in humans does not follow Mendelian transmission ratios, however, and is clearly genetically complex and not well understood (Mitchell and Risch, 1992; Schutte and Murray, 1999). Mapping and identification of mouse genes that participate in genetically complex (“multifactorial”) causes of orofacial clefting will lead to knowledge of the gene-regulatory pathways important to nonsyndromic orofacial clefting and will identify candidate pathways, as well as candidate genes, for examination in studies of human genetic risk factors. The emphasis of this chapter is on the results of approaches to mapping the genetic components of the multifactorial nonsyndromic class of orofacial defects in mice.
Developmental and Genetic Independence of Types of Orofacial Clefts in Mice
Cleft lip with or without cleft palate (CL/P), isolated cleft palate (CP), and median cleft lip (MCL) are developmentally distinct malformations in mice. Cleft lip with or without cleft palate arises on embryonic days 10 to 11 of gestation, from the failure of a mesenchymal bridge to fuse the embryonic maxillary prominence with the lateral nasal prominence internally and with the lateral and medial prominences on the external surface of the prospective upper lip (Diewert and Wang, 1992; Millicovsky et al., 1982). The result is a unilateral or bilateral cleft between the maxilla and the premaxilla, extending into the nostril (see Figs. 1.3,1.6,1.7, and 1.12 in this volume). Cleft palate arises later, on days 14 to 16 of gestation in mice, from a failure of outgrowth of the palatal shelves from the medial aspect of the maxillary prominence or from a failure of the palatal shelves to elevate to a horizontal orientation to contact in the midline and fuse, resulting in a failure to form the roof of the mouth (see Fig. 2.1 in this volume). The cleft palate that usually accompanies cleft lip (CL) has been observed to originate in mechanical obstruction of palatal shelf elevation by the tongue, due to alterations in the shape of the oral cavity caused by the CL, and thus is secondary to the CL (Trasler and Fraser, 1963; Pourtois, 1967). Median cleft lip arises on gestational days 9 to 11 in mice, from a midline defect of outgrowth the mesenchyme between and merging with the medial prominences, leading to a midline cleft of the premaxilla.
|
TABLE 22.1. Genetic Causes of Cleft Lip with or without Cleft Palate in Mice |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Median CL and CL/P are different traits genetically as well as developmentally. Median CL occurs in syndromic mutants that do not have CL/P, such as patch (Ph) (Gruneberg and Truslove, 1960), doublefoot (Dbf) (Lyon et al., 1996), the Kara dominant-negative mutation (Damm et al., 1993), and the MSX2 transgene (Winograd et al., 1997). Also, CL/P is usually genetically distinct from CP; they are caused by mutations at different loci, the exception being the twirler (Tw) syndromic mutant, where CL/P and CP can occur as alternative phenotypes (Lyon, 1958). Only five Mendelian mutations are known that cause CL/P, and all are syndromic (Table 22.1). Only a few Mendelian mutations cause nonsyndromic CP (Table 22.2), but numerous spontaneous and gene-knockout mutations cause syndromes that include CP; these will not be reviewed in detail. This pattern parallels the genetics of human orofacial clefting syndromes, and it is likely that the syndromic mouse mutants are homologs of human syndromes.
Nonsyndromic CL/P occurs spontaneously in some mouse strains, and nonsyndromic CP occurs spontaneously at unusually high frequencies in other strains (Table 22.3). This pattern indicates that they are genetically independent traits. The genetics and linkage mapping of CL/P and CP therefore will be discussed separately.
Cleft Lip with or without Cleft Palate
Nonsyndromic
Occurrence Only in A-Strctin Lineages
Spontaneous CL/P, i.e., not induced by teratogens, is very rare in most strains of mice. All stocks with frequent occurrence of spontaneous nonsyndromic CL/P trace to a single genetic origin, a closely related set of strains, referred to as the “A” strains. These strains share at least 30 generations of brother-sister inbreeding before their dispersion to different laboratories about 70 years ago and subsequent designation as substrains, and they are expected to be identical at nearly all of their loci.
All A strains examined produce CL/P at relatively high frequencies in their fetuses or newborns (Kalter, 1979; Juriloff, 1982). The frequency of CL/P varies among these strains. The A/J strain, with about 10% CL/P, was used in most genetic and developmental studies of CL/P until about 1980, when the A/WySnJ strain was discovered to have a higher risk of CL/P, 20% to 30% (Juriloff, 1982), and became the A strain of choice for CL/P studies. In the 1970s, outcrosses of the A/J strain, with subsequent selection for production of CL/P during inbreeding of descendants, led to two new stocks that also produced CL/P, the CL/Fr strain, with 20% to 25% CL/P in the offspring (Juriloff and Fraser, 1980), and the “L line,” with about 10% CL/P (Trasler and Trasler, 1984).
An outcross of the A/J strain, to the normal C57BL/6J strain, was also the origin of a new set of re combinant inbred (RI) strains, descended from a series of closed lineages, each tracing back to a separate F2 breeding pair. These strains are called the AXB-1, AXB-2…and the BXA-1, BXA-2…strains; the AXB set has A/J cytoplasm and the BXA set has C57BL/6J cytoplasm. Each RI strain is expected to have a unique combination of pieces of its genome derived equally and randomly from the two parental strains, and half of the strains are expected to be homozygous for the A strain allele at any given locus (Sampson et al., 1998). Among these strains, only AXB-6/Pgn and BXA-8/Pgn have been reported to have spontaneous CL/P (Diehl and Erickson, 1997; Juriloff et al., 2001).
|
TABLE 22.2. Genetic Causes of Liability to Nonsyndromic Cleft Palate (CP) in Mice |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
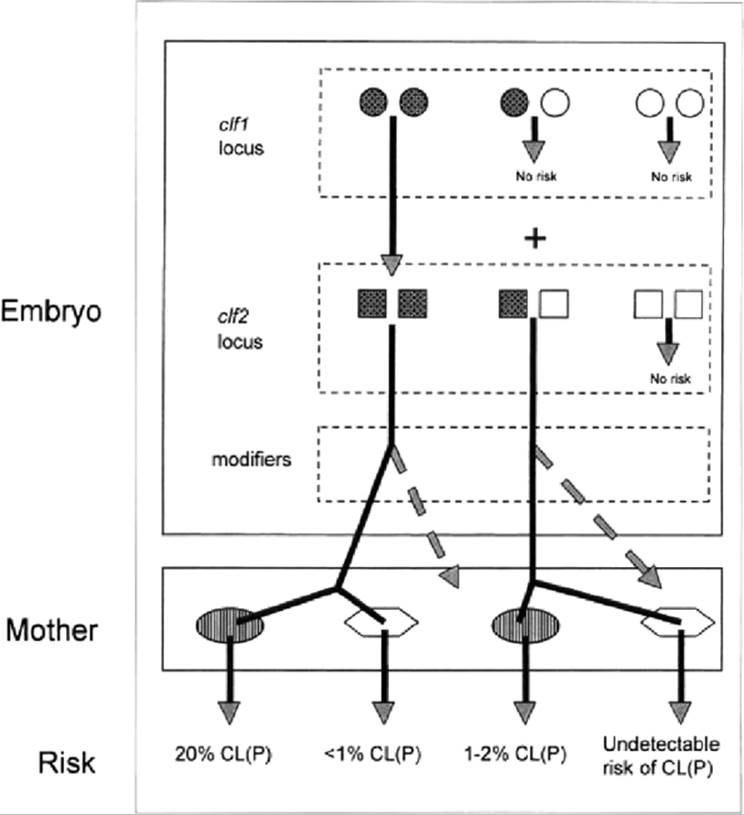
The cumulative understanding of the genetics and mapping of the liability to spontaneous CL/P of Astrain mice has been deduced in genetic contrasts with the normal C57BL/6J strain and the C57BL-related normal AEJ/GnRk strain. In the A strains, CL/P is a genetically complex trait but does not appear to have the polygenic additive basis often assumed of multifactorial threshold traits. Historical assumptions about the number and nature of the CL/P genes acting directly in the embryos strongly influenced the approaches taken to linkage mapping, as discussed below. The genetic architecture of the trait has genetic components in two layers (Fig. 22.1): a layer of genes acting directly in the embryo to cause risk of CL/P and to influence expression of that risk and a layer of genes acting in the mother to indirectly modify the embryos' risk of CL/P.
|
TABLE 22.3. Mouse Strains with High Frequencies of Spontaneous Nonsyndromic Orofacial Clefts |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||
|
FIG. 22.1. Genetic architecture of risk cleft lip with or without cleft palate (CL/P) derived from the A strains mice. Two compartments influence risk: genes acting in the embryo and genes mother. Filled circles and squares denote alleles from the A strain; open circles and squares denote normal-strain alleles. Only a combination of A alleles across the clf1 and clf2 loci can cause risk of CL/P. The variety of specific risk levels created by subsequent modifiers is not shown; only the effect of A-strain modifiers is shown as the embryo risks are carried forward into the maternal effect compartment. Two types of maternal effect are shown: A/WySnJ mothers (dark ovals) and Fl mothers from A/WySnJ x C57BL/6J (light polygons). |
||||||||||||||||||||||||||||||||||||||||
Developmental Threshold
The developmental defect in A/J, CL/Fr, L Line, and A/WySnJ embryos is a minor quantitative change in the dynamic spatial relationships between the facial prominences (Trasler, 1968; Juriloff and Trasler, 1976). In particular, a delay in the forward growth of the maxillary prominence leads to its inadequate contact and fusion with the lateral and medial prominences (Diewert and Wang, 1992; Wang et al., 1995). By a critical time in the development of the face, the fusion (mesenchymal bridge) must reach a critical size or fail. Variation between embryos, stochastic or micro-environmental, distributes some embryos to the “failure” side of this threshold. Thus, among genetically identical embryos, some fail to successfully form one or both sides of the upper lip. The genes that cause risk of CL/P in the A strains therefore cause a quantitative change in the growth of embryonic facial prominences, which is not a severe abnormality in itself, although the potential consequence, CL/P, is severe. Embryos that successfully form the mesenchymal bridges go on to become normal adults. The genes causing risk of CL/P in the A strains are expected to be expressed in the cells that populate the maxillary prominence, and candidate loci identified in linkage studies can be evaluated by this criterion.
Maternal Effects
Every study of CL/P that has used reciprocal crosses has demonstrated that the genotype of the mother influences the frequency of CL/P in genetically liable embryos. The maternal effects do not cause CL/P; crosses of A-strain females to normal-strain males do not produce CL/P in the F1; the effect of the maternal genotype is to influence the risk of CL/P in embryos that have the CL/P-causing genes. Reciprocal crosses between A/HeJ and A/WySnJ, between A/J and CL/Fr, and between A/WySnJ and AXB-6/Pgn each indicated that the strain of the mother, not the genotype of the embryo, causes the differences in CL/P frequency between strains. The frequency of CL/P obtained in each cross was that of the maternal strain (Juriloff and Fraser, 1980; Juriloff, 1982; Juriloff et al., 2001). Maternal effects are not confined to strains that have CL/P liability; the standard normal C57BL/6J strain used in genetic studies of CL/P also differs from A/J, A/WySnJ, and CL/Fr by genetic maternal effects, detected in reciprocal backcrosses, where the frequency of CL/P is lower in progeny from F1 females than from F1 males (Davidson et al., 1969; Bornstein et al., 1970; Juriloff et al., 2001). The variety of strain combinations that demonstrate maternal effects indicates that multiple loci and/or at least three alleles are responsible for the maternal effects. None has been identified or mapped, and the mechanism is not known.
A Major Gene, clf1
Nonsyndromic spontaneous CL/P in mice was for many years thought to be a polygenic trait with low penetrance (Gruneberg, 1952; Davidson et al., 1969), leading to the assumption that linkage mapping of the genes involved would require impractically large sample sizes. However, in the 1980s, two separate studies showed that a major recessive gene in A/J, clf1, is necessary for risk of CL/P (Juriloff, 1980; Biddle and Fraser, 1986). In each study, after a cross between A/J and C57BL/6J and backcross of Fl to A/J, the resultant BC1 males were individually test-crossed with A/J and the frequency of CL/P in their progeny was observed. Two approximately equal groups of BC1 males were discerned, one producing CL/P in the progeny at about half the frequency of the other. This pattern is diagnostic of the presence of a major recessive variant necessary to risk of CL/P. If a set of closely spaced, highly polymorphic mapped linkage markers had been available at that time, it would have been possible to do a genome screen of marker genotypes in the sires or in BC1 segregants with CL/P to map clf1.
With evidence of a major gene, clf1, in hand, it was clearly feasible to construct a congenic strain pair, i.e., inbred strains differing only by the chromosomal segment containing clf1. This entailed transferring the CL/P liability trait from A/WySnJ by repeated backcrossing into the normal AEJ/GnRk strain background; segregants in each generation were testcrossed and the CL/P producers were used for further backcrossing. Simultaneously, assuming clf1 to be a recessive essential mutation and using a variety of available markers in standard linkage crosses and other approaches, a significant part of the genome was excluded as the site of clf1 (Juriloff, 1993). Later, when the ideal mapped polymorphic linkage markers, simple sequence length polymorphisms (SSLPs), became available, the A-strain alleles at passenger SSLP loci on the selected differential CL/P-causing chromosomal segment in the congenic strain were used to map clf1 to mid-distal chromosome 11 near the marker, DllMitlO (Juriloff and Mah, 1995; Juriloff et al., 1996).
A Second Epistatic Gene, clf2
In addition to the evidence of a major recessive causative locus, clf1., historical genetic studies of CL/P indicated the presence of at least one other important contributing locus in A-strain mice because the frequencies of CL/P were consistently significantly lower in segregants after outcrosses than expected for segregation of a single locus (Davidson et al., 1969; Juriloff, 1980; Juriloff, 1995). Analysis of the CL/P frequencies recovered across all of the generations of testcrosses during construction of the congenic strain and comparison with expected patterns under various multigenic hypotheses excluded all additive genetic models and indicated that an allele from A strains at a second locus, clf2, was necessary to permit expression of CL/P caused by clf1 homozygosity; heterozygotes at clf1 permitted some CL/P at low frequencies, and homozygotes allowed high frequencies, similar to those of the A strains. With the map position of clf1 in hand, a genome screen in a new independent genetic study was done to map elf2. It was based on a cross of A/WySnJ with C57BL/6J, followed by a backcross to A/WySnJ females to produce “BC1” embryos; 2.4% of the 1485 BC1 embryos had CL/P. The first 29 embryos with CL/P were used in a genome screen of informative mapped SSLP loci spaced 10 to 25 cM across the genome (Juriloff et al., 2001). For loci not linked to CL/P risk loci, half of the embryos were expected to be homozygous for the A allele; loci with an excess of A homozygotes with an uncorrected p < 0.02 were further typed in an additional seven CL/P BC1 embryos obtained later in the study. All 36 CL/P embryos were homozygous A at DllMitlO near clf1, confirming this locus. Of the 36, 32 were also homozygous A at D13Mitl3, mapping elf2 to midproximal chromosome 13 (Juriloff et al., 2001). Both were significant at p < 0.001 as individual tests and, when corrected for the multiple tests of a genome screen, at p < 0.05. No other regions were significant. The hypothesis (Juriloff, 1995) that the A strain-like high risk of CL/P requires both clf1 and clf2 simultaneously in homozygous A state and that homozygosity for the clf1 causative gene in clf2 heterozygotes leads to a detectable but lower risk of CL/P was supported by the data.
Epistatic Interaction between clf1 and clf2 and Finer Mapping of el/2 in Testcrosses of the AXB/BXA RI Strain Set
Gene mapping based on RI strains requires an observable phenotypic variation distributed among the strains, with several strains of each phenotype, to be correlated with the strain distribution pattern (SDP) of allelic alternatives at mapped marker loci throughout the genome. Although several strains have the clf1 region from the A/J parent, CL/P occurs in only two of the several AXB/BXA RI strains examined, AXB-6/Pgn and BXA-8/Pgn; and their risk is similar to that of the A/J parental strain (Table 22.3). This SDP for CL/P has little statistical power for linkage mapping by a direct approach. The pattern, fairly high risk of CL/P in two RI strains and no detectable risk in all others examined, is not consistent with a polygenie additive genetic architecture, where a variety of levels of risk of CL/P would be expected to occur among the strains, but it is consistent with an epistatic genetic architecture, where A-strain alleles at a combination of particular loci must be reconstituted for the risk of CL/P to be present.
An indirect measure of the CL/P genotype of 10 of the RI strains was obtained by testcrossing them with A/WySnJ females. Testcross embryos are heterozygous for all CL/P risk loci not already present in the homozygous A allelic state in the RI strain itself and are within the most permissive maternal environment available. Consistent with the recessive epistatic model, only strains with the A allele at DllMitlO near clf1 produced CL/P in testcrosses, and these strains produced CL/P at two distinct levels, one rate near 20% (two RI strains) and similar to that of the A/WySnJ strain, as expected for homozygotes for A alleles at both clf1 and clf2, and one rate low, 0% to 2% (four RI strains), as expected for homozygotes for the A allele at clf1 with heterozygosity at clf2 (Fig. 22.1).
The A-strain derived chromosomal segments around DllMitlO in the RI strains that produced CL/P in testcrosses are relatively large (SDP database, http://www.informatics.jax.org/searches/riset_form.shtml) and cannot refine the map position of clf1. However, these strains have a variety of recombination breakpoints in the broad region previously defined for elf2; and on the assumption that only the strains with the A allele at clf2 produce high risk of CL/P in the testcross, they were used to refine the clf2 candidate region as the subregion common to the two high risk-producing strains (Juriloff et al., 2001), between D13Mitl3 and D13Mit231 (Fig. 22.2).
Modifiers
There are other modifier loci that influence risk levels for CL/P. The frequency of CL/P expected in the BC1 from A/WySnJ and C57BL/6J (in A/WySnJ mothers), if simultaneous homozygosity at two loci is required, is one-quarter of the A/WySnJ frequency, or about 6%, and the observed frequency of 2.4% was significantly lower (Juriloff et al., 2001). Furthermore, some RI strains whose genotypes at the clf1 and clf2 loci predicted 1% to 2% CL/P in testcrosses, produced none in samples of 130 to 225 embryos (Juriloff et al., 2001). The most promising candidate locations of modifier loci are near D7Mitl58 and D18Mit4, two genetic regions that had suggestive nonsignificant association with CL/P in the genome screen, with >75% probability of linkage after correction for multiple tests by a Bayesian approach (Silver, 1995; Juriloff et al., 2001). Both regions were homozygous for the A allele in the four BC1 CL/P embryos heterozygous at elf2 and in the two RI strains with high risk of CL/P in testcrosses but not in strains with lower risk. The random probability of each genotype at each locus in each individual or strain is 0.5, and the random probability of the combination of the AA genotypes at the two loci across all six observations is <0.001. The human homologs of these two regions are on 19q and 18q.
Candidate Loci for clf1
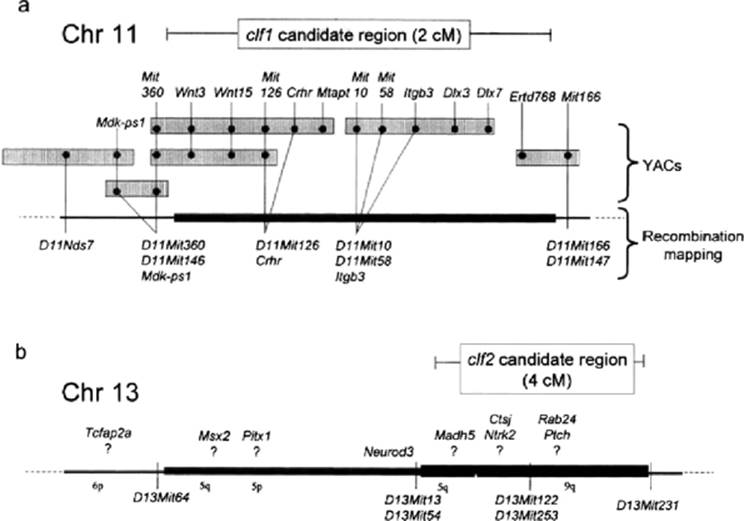
The recombination and physical map for the region of clf1 on chromosome 11 is summarized in Figure 22.2. The recombination map position of clf1 is defined by an A-strain haplotype between DllMit360 and DllMitl66 (Juriloff et al., 2001) held in common between congenic strain carriers of CL/P liability and BC1 embryos with CL/P from the genome screen. These markers flank an approximately 2 cM region, within which the genes Crhr and Itgb3 have been located by recombination mapping in the same materials (Juriloff et al., 2001). Yeast artificial chromosome (YAC) contigs, marked by SSLP loci, indicate that the loci WntlS, Wnt3, Crhr, and Mtapt are within the candidate region. The YAC contig is incomplete. One YAC that is well within the candidate region contains the loci Itgb3, Dlx3, and Dlx7 (Juriloff et al., 2001). It is expected that a bacterial artificial chromosome (BAG) contig will extend through this region as part of the current Mouse Genome Project (Graham et al., 2001) and that more candidate genes for the clf1 mutation may be identified. From the perspective that in knockout studies genes defined by a function in the maintenance of adult homeostasis have surprisingly demonstrated roles in embryogenesis, all of the loci in the candidate region can be considered to be candidate genes for clfl. Among these genes, embryonic expression of Crhr (corticotropin-releasing hormone receptor) and Mtapt (microtubule-associated protein tau) has not been described. Wnt3 and WntlS are members of a family of genes that encode secreted glycoproteins that act in signaling pathways to modulate cell behavior and fate in embryos (Moon et al., 1997). Expression domains have not been reported for WntlS. In the embryonic period just before development of the facial prominences, Wnt3 is expressed specifically in dorsal forebrain neuroepithelium, with a sharp rostal boundary between the prospective diencephalon and telencephalon, caudal to the part of the forebrain that will underlie the facial prominences, and not in the neural crest cells migrating to the facial prominences (Roelink and Nusse, 1991; Parr et al., 1993).
|
|
|
FIG. 22.2. Locations of clfl (a) and clf2 (b) on the mouse linkage map. All known genes that map into the physical interval defined for clf1 are shown. The order of genes mapped to yeast artificial chromosomes (YACs) but not by recombination mapping is arbitrary. For elf2, some potential candidate genes are shown; however, they have not been mapped against the interval defined for clf2, and their specific location is uncertain. For clf2, the thickest bar denotes the location defined by testcrosses of recombinant inbred strains; the less thick bar denotes the location from BC1 segregants (see text). |
Dlx3 and Dlx7 are members of the distal-less homeobox transcription factor gene family. Dlx7 is said to be expressed in the branchial arches and molar and incisor teeth (Nakamura et al., 1996), but details have not been published. As the maxillary prominence, the site of the primary defect in CL/P, is derived from the first branchial arch, Dlx7 appears to be a good candidate for clfl. Another good candidate, Dlx3, is expressed in the distal tips of the first branchial arch; its knockout mutant dies during embryogenesis before lip formation due to placental defects (Kraus and Lufkin, 1999; Morasso et al., 1999); a frameshift mutation in humans causes trichodento-osseous (TDO) syndrome (Price et al., 1998).
Integrin (β3 (Itgb3, also known as Cd61) is a member of a large family of genes coding for components of heterodimer transmembrane receptors for extracellular matrix proteins, and some are involved in cell migration during morphogenesis. The functional relationships among integrins are complex; each ft protein can form functionally different heterodimers with different a proteins; each integrin locus produces multiple different proteins through alternative mRNA splicing sites. One of the heterodimers of β3 (αllb, β3) is expressed on blood platelets, and human loss of function of the β3 gene causes Glanzmann's thrombasthenia, a bleeding disorder (Hynes, 1994). The embryonic expression domains of Itgb3 have not been described in detail; one of the heterodimers, αV/β3, is expressed in the blastocyst and appears necessary for implantation (Illera et al., 2000). There is an interrelationship between αV/β3 and αV/β1 functions (Blystone et al., 1995), and it is a way that Itgb3 could lead to CL/P: disruption of β1 function leads to deficient maxillary prominences (Baudoin et al., 1998), and aV/β1 with an expression domain in developing eyelids is associated with the defect open-eyelids-at-birth when deficient (Carroll et al., 1998). It is particularly interesting, therefore, that the CL-liable A strains and the CL/Fr strain occasionally have open-eyelids-at-birth (Kalter, 1979; Kadowaki et al., 1997), suggesting that they may have disruption of integrin signaling.
In summary, among the loci currently believed to be within the clf1 region, the best candidates to be examined for mutations are Dlx3, Dlx7, and Itgb3. The human homologs of these loci are on 17q, which has been associated with CL/P in some studies (Chenevix-Trench et al., 1992; Huie et al., 1999).
Candidate Loci for cl/f2
The identification of candidate loci for clf1 is at an earlier stage than that clf1 (Fig. 22.2). Many of the gene loci around clf2 have not been finely mapped or mapped against SSLPs, and their placement on the composite reference map MGD 2001 (Mouse Genome Database, http://www.informatics.jax.org), considered to be a work in progress, is expected to contain significant errors in the relative placement of loci. Consequently, loci shown on the current MGD map to be several centimorgans outside of the SSLP-defined clf1 candidate interval are not necessarily excluded as candidate genes, and the number of potential candidates is large. An approach to identifying good candidate loci for clf2 is based on the paralogy phenomenon. In mammals, a cluster of linked loci extending over several centimorgans and comprising several Hox genes, a pair of Dlx genes, and Rar, Neurod, Evx, Crhr, Mdk, Itga, Wnt, Krt, and Sen genes, is present in four copies, i.e., one on each of four mouse chromosomes (Chromosomes 2, 6, 11, 15), although not all loci are present in each of the four copies. It is thought that these conserved duplicated paralogous linkage groups originated in two sequential genome duplication events in the evolutionary path to mammals, with subsequent loss or sequence diversion of the duplicate loci, clf1 maps into one of these paralogous linkage groups on chromosome 11 and interacts epistatically with clf2 in the manner of duplicated loci. Although in mapping to chromosome 13, clf1 is not located in one of the recognized paralogous gene clusters, the approach of paralogy need not be rejected. It appears that one of the clusters has broken up in the evolutionary lineage to mice, and a piece of it, containing a Neurod locus, is on chromosome 13. clf1 is linked to Neurod2, and clf1 is linked to NeurodS. Thus, the possibility remains that clf1 and clf2 are functionally redundant paralogs. No pair of candidate paralogous loci have been recognized in the pair of candidate intervals, but more gene loci are expected to be identified in both.
Among the many loci currently known to map in the general region encompassing the clf1 locus, some of the most interesting potential candidate loci are Tcfap2a (AP-2) and Msx2, although both appear to be outside of the segment most likely to contain clf2 (Fig. 22.2). Locus AP-2 demonstrates a role in lip development by CL/P in mutants, as discussed below (Nottoli et al., 1998). Msx2 is a transcription factor expressed in the maxillary prominence (MacKenzie et al., 1992; Wang et al., 1996). Knockout mutants have defects of skull ossification and other abnormalities but not CL/P (Satokata et al., 2000). As the Msx2 knockout mutant has not been examined in the context of homozygosity for the requisite A-strain allele at clf1, it has not been examined in a context where it would be expected to cause CL/P. The homolog of a related gene, Msx1, has been associated with CL/P in humans (van den Boogaard et al., 2000). Msx gene products interact with candidates for clfl, Dlx products (Zhang et al., 1997), consistent with the epistatic relationship between clf2 and clfl.
Other interesting possible candidate loci are within the clf2 candidate region. These include Madh5 (formerly Smad5), which codes for a protein involved in bone morphogenetic protein signaling and whose knockout mutant has forebrain and branchial arch deficiencies within a severe multifaceted embryonic lethal syndrome (Chang et al., 1999).
The clf2 candidate interval is fragmented relative to human homologous linkage groups. Tcfap2a is on 6p, Msx2 and Madh5 are on 5q, and other loci in the region are reported to have homologs on 5p and 9q (MGD 2001).
Syndromic Mutants
Dancer
A semidominant mutation on proximal chromosome 19, Dancer (Dc) is characterized in heterozygotes by a circling behavior due to defects in the inner ear (Wenngren and Anniko, 1989). Homozygotes die at birth and often have CL/P due to deficiencies of the facial prominences (Trasler and Leong, 1982; Trasler et al., 1984). The human homologous region is on llq.
Twirler
A semidominant mutation on proximal chromosome 18, Twirler (Tw) is characterized in heterozygotes by circling behavior due to defects of the inner ear and by obesity. Homozygotes die at birth and have CL/P (Lyon, 1958; Gong et al., 2000). The human homologous region is on 18q (Griffith et al., 1996).
AP-2 Null Chimeras
A widely expressed transcription factor, AP-2 (or AP-2α, Tcfap2a), has a strong expression domain in the facial prominences (Mitchell et al., 1991). Knockout homozygotes have multiple early, severe embryonic defects (Schorle et al., 1996; Zhang et al., 1996) that preclude observation of later defects such as CL/P. Embryos chimeric for AP-2 null cells have regions lacking in AP-2 activity; in many, CL/P was observed (Nottoli et al., 1998). As AP-2 has multiple transcripts (Meier et al., 1995), one can speculate that point mutations in a facial prominence-specific transcript could lead to nonsyndromic CL/P. The human homolog is on 6p.
Brachyphalangy (Gli3Xt–Bph)
A radiation-induced mutation, brachyphalangy, causes homozygotes to have abnormal digits and various other defects, including CL/P (Johnson, 1969). It is allelic to mutations at the GU3 locus, which is mapped to proximal chromosome 13 (Perou et al., 1997), clearly outside the candidate region for df2. Only the Bph allele has been reported to cause CL/P, raising the possibility that the mutational event that caused this allele has also damaged clf2 and is in linkage disequilibrium. The human homolog is on 7p.
Legless
A 600 kb deletion on distal chromosome 12 that probably spans multiple genes (Supp et al., 1999), the legless (Igl) mutation causes a recessive syndrome of skeletal, craniofacial, and visceral malformations, including absence of distal limb structures, and a variety of severe facial and brain defects, such as frontonasal encephalocele with severe midline clefts. As part of the phenotypic spectrum, some specimens have CL/P; illustrations of this mutant (McNeish et al., 1990) suggest that the premaxilla is abnormally small. The human homologous region is on 7q.
Genetic Liability to Teratogen-Induced Cleft Lip with or without Cleft Palate
Role of Genes that Influence Risk of Teratogen Induced Cleft Lip with or without Cleft Palate
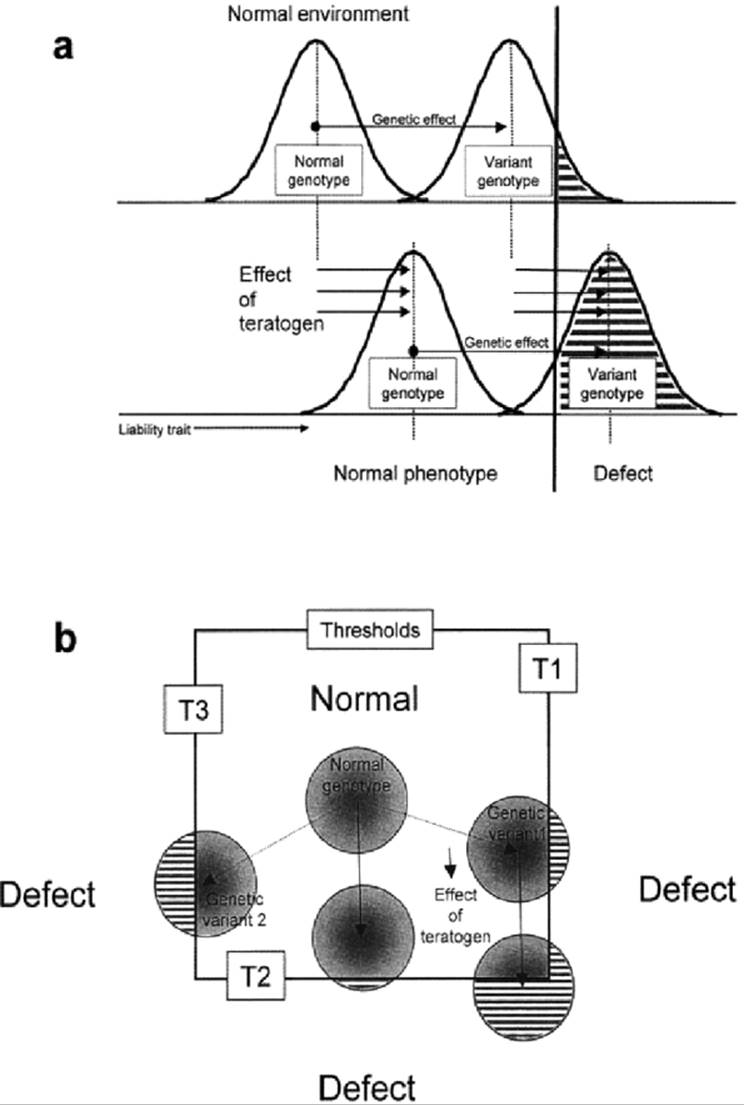
Heterozygotes for the CL/P liability gene DC have greater risk of CL/P than genetically normal embryos when treated with 6-aminonicotinamide (Trasler et al., 1984). The CL/Fr strain, which has about 20% spontaneous CL/P, is easily induced to near 100% CL/P at dosages of 6-aminonicotinamide that cause less than 10% CL/P in the normal C57BL/6J strain (Juriloff, 1980). These differences are often explained on the basis of the multifactorial threshold model, where the effect of the teratogen adds to the initial genetic effect on risk (Fig. 22.3a). However, normal mouse strains that do not have risk of spontaneous CL/P also differ in the frequency of CL/P induced by treatment with a variety of teratogens (Biddle, 1988). Interpretation of the genetic basis of risk of teratogen-induced CL/P is complex; not only loci that affect craniofacial development but also loci influencing maternal, placental, and embryonic metabolism of the teratogen are expected to influence the risk of CL/P (Atlas et al., 1980; Lankas et. al., 1998). Further complexity of interpretation derives from heterogeneity of defective developmental mechanisms; e.g., small lateral prominences (Sulik et al., 1979) vs. small maxillary prominences (Diewert and Wang, 1992; Wang et al., 1995) can result in CL/P. The developmental mechanism underlying the induced defect may differ from that underlying the spontaneous trait (Fig. 22.3b). A study of the genetic co-transmission of spontaneous CL/P risk and 6-aminonicotmamideinducible CL/P indicated that the two traits are not controlled by the same genes (Juriloff, 1980).
Phenytoin-Induced Cleft Lip with or without Cleft Palate
The anticonvulsant phenytoin induces CL/P if administered to the pregnant mother in the 2 days prior to embryonic lip formation. The A strains are more liable to phenytoin-induced CL/P than the C57BL/10 and C57BL/6J strains (Goldman et al., 1983b; Karolyi et al., 1990). Differences in liability to phenytoin-induced CL/P in reciprocal congenic strains differing by the genomic region containing the major histocompatibility locus H2 are present in the context of an A-strain background but not in a C57BL/10 background (Goldman et al., 1983b). Similarly, in other reciprocal congenic strains, a region that includes an N-acetyltransferase variant now called Nat1 alters liability to phenytoin-induced CL/P in the context of the A/J strain background but not in the C57BL/6J background (Karolyi et al., 1990). There is another N-acetyltransferase locus, Aanat, linked to clf1 about 20 cM distal (MGD 2001). The variant attributed to Nat1 on chromosome 8 was transferred between backgrounds based on biochemical phenotype (Karolyi et al., 1990), raising the possibility that Aanat and clf1 on chromosome 11 were also transferred in the congenic strains.
Another analysis to map loci influencing the risk of phenytoin-induced CL/P was done in the AXB and BXA RI strains (Diehl and Erickson, 1997). The parental strain C57BL/6J (B) had about 1% phenytoin-induced CL/P at the dosage used, whereas the A/J strain had about 43% CL/P. Nine RI strains with CL/P responses ranging from 0% to 34 % were used. A novel analytical method contrasted the set of risk values for strains with the A allele at a marker locus with the risk values for the strains with the B allele at the locus. The threshold value for type I error for linkage (false-positives) was estimated from the values obtained from all possible random distributions of marker alleles at that locus. By this strategy, a genome screen for linkage to closely spaced marker loci was done. Eight chromosomal regions were significant by the criterion of p < 0.05 for each test; among these regions, the B allele from the low-risk strain, C57BL/6J, was associated with high risk of CL/P in the RI strains at four of these eight loci (Table 22.1). None of the eight regions coincided with the spontaneous CL/P loci clf1 and clf2 or with the DC, Tcfap2a, or G//3 gene loci; however, one with an A-susceptibility allele coincided with the vicinity of the Tw locus on chromosome 19, one with a B-susceptibility allele coincided with the region of the lgl mutation, and one with a B-susceptibility allele coincided with the Nat1 region for which B in the previous congenic study conferred resistance.
|
|
|
FIG. 22.3. Conceptual models for the interpretation of genetic liability to teratogen-induced orofacial clefts, (a) Threshold model assumes additivity of genotypes and teratogens for risk of clefting. Gaussian curves represent relative frequencies of individuals, within a given genotype, having each value of a quantitative liability trait, shown on the x axis. The liability trait impinges on a critical threshold value, denoted by a bold vertical line, beyond which malformation occurs. Both the effect of genetic variants that increase liability and the effect of teratogens move the distributions toward the same threshold, and the effects of genes and teratogens act by the same developmental mechanism, (b) Threshold model assumes that there is mechanistic heterogeneity of various types of genetic and teratogen-induced orofacial cleft. Each circle represents a distribution of individuals within a given genotype. Normal development occupies a range of values for several liability traits, and the borders of “normal” space represent three different mechanistic thresholds (Tl, T2, T3; e.g., small lateral prominences vs. late growth of maxillary prominences). In this model, genetic liability may be created by mechanisms at least partly different from the mechanism of effect of the teratogen. |
The four regions (Table 22.1) with B alleles (from the low-risk strain) that contribute to high risk are difficult to interpret. If both parental strains have alleles conferring relative risk at various loci, it is expected that the frequencies of induced CL/P for some RI strains will fall beyond the parental strain range because of new allelic combinations across loci; but this expectation is not met. Each of the eight regions has a high likelihood of being a false “hit.” Genome screens for linkage involve many statistical analyses of one data set, and there is a high risk of false-positives at the standard p < 0.05 threshold for type I error. For example, if 100 independent markers are used, at p < 0.05, five false-positive linkages are expected; therefore, in this study using 342 markers at p < 0.05, many of the eight loci “mapped” may well be false-positives. Various approaches to identify the P value for each marker locus that corresponds to a functional value of p < 0.05 per locus in genome screens have been described (Silver, 1995; Belknap et al., 1996); these values tend to be around p < 0.001 or less, a criterion not met by any of the linkages indicated. On the basis of balancing the risk of false-positives against overlooking loci of moderate effect, Diehl and Erickson (1997) endorse a twostep mapping strategy to confirm these suggested linkages in new intercross or backcross data. In the meantime, it is premature to attempt to further interpret the genetic architecture implied by participation of several low-risk strain alleles in causing high risk, as well as the very large number of potential candidate loci within 15 cM of each of the eight marker loci.
Cleft Palate
A wide variety of perturbations of the developing craniofacial complex can, singly or in combinations, lead to CP (Eraser, 1976, 1980; Diewert, 1986). Some of these factors, e.g., small size of the palatal shelves, delayed timing of elevation of the shelves, and lack of fusion of apposed shelves, are intrinsic to the palate. Other factors, such as excessive head width, abnormal head posture, lack of tongue movement, lack of jaw movement, and deficient size of the lower jaw, are extrinsic to the palate but also contribute to risk of CP. This multifactorial and heterogeneous set of developmental factors predicts that the genetic factors involved also will be heterogeneous.
Nonsyndromic
Some genetically normal inbred strains of mice have a fairly high risk of spontaneous nonsyndromic CP at birth (Table 22.3): the SW/Fr strain was reported to have 6% CP (Vekemans and Fraser, 1979); several stocks of CF1 mice in Japan have 3% CP (Matsushima et al., 1992); and the J/Glw strain was said to have a “high” risk of CP (Staats, 1972), estimated to be up to 25% (S. Gluecksohn-Waelsch, personal communication, 1978). Generally, sporadic CP is not rare in genetically normal stocks of laboratory mice, and it is occasionally documented (Gasser et al., 1981; Proetzel et al., 1995). The genetic cause of a relatively high risk of spontaneous CP in mouse strains has not been studied, and there appear to be no mapping studies of sporadic, probably multifactorial, nonsyndromic CP in mice.
A few mutations that cause nonsyndromic CP in mice are known (Table 22.2). These are promising models for definition of the gene-regulatory pathways involved in human nonsyndromic multifactorial CP. Loss of function of the a transcription factor on chromosome 3, Lhx8, leads to CP in about half of homozygotes, through failure of the elevated palatal shelves to make contact (Zhao et al., 1999); the moderate penetrance suggests that genetic and environmental modifiers could influence risk, as would be expected in its human counterpart. Similarly, loss of function the γ- aminobutyric acid (GABA)-synthesizing enzyme Gad1 (also Gad67) on chromosome 2 leads to failure of fusion of elevated shelves in most homozygotes (Condie et al., 1997); loss of the β3 subunit for a GABA receptor (Gabrb3) on chromosome 7 also causes CP (Culiat et al., 1995). Lack of transforming growth factor-β3 (Tgfb3) on chromosome 12 also leads to CP, through failure of fusion of the apposed palatal shelves (Proetzel et al., 1995; Kaartinen et al. 1995, Taya et al., 1999). The human homologous regions are listed in Table 22.2.
Syndromic Mutants
More than 50 mapped mutations in mice cause syndromes of malformations that include CP. Many probably correspond to human syndromes caused by mutations at homologous loci. Searches of the TBASE (tbase.jax.org) and DHMHD (http://www.hgmp.mrc.ac.uk/DHMHD/dysmorph.html) electronic databases for “cleft palate” lead to lists of many of these mutants, and their human linkage homologies can be found in the MGD 2001 database; they will not be reviewed in detail here. In many of these syndromic mutants, the CP appears to be secondary to other defects. For example, defects in cartilage growth that cause short limbs and short snouts probably lead to mechanical obstruction of palatal closure by the tongue because of inadequate space in the oral cavity for the tongue, as in and (Rittenhouse et al., 1978), cho (Seegmiller and Fraser, 1977), and Col2a1 (Maddox et al., 1998) mutants. A similar obstructive mechanism may be present in mutants with a small mandible, such as the SEF1 transcription factor knockout (Takagi et al., 1998), the activin receptor II knockout (Matzuk et al., 1995a), and the Hoxa2 knockout with abnormal tongue movement (Barrow and Capecchi, 1999). In several other mutants, the cause of CP seems to be an independent pleiotropic effect of loss of function of genes with key roles in other developmental pathways. Among these are activin-βA (Inhba) (Matzuk et al., 1995b), follistatin (Fst) (Matzuk et al., 1995c), Tgfb2 (Sanford et al., 1997), Egfr (Miettinen et al., 1995, 1999), the regulator of Hox gene expression Edr1 (formerly rae28) (Takihara et al., 1997), and the transcription factors Titf2 (De Felice et al., 1998), Msx1 (Satokata and Maas, 1994), and Pax9 (Peters et al., 1998). In some syndromic mutants, the effect on palatal shelves appears to be direct; severe deficiency of the palatal shelves is the cause of CP in the Dlx2 transcription factor knockout (Qiu et al., 1997) and the Far mutant (Juriloff and Harris, 1983). A genetic complexity of CP risk may be introduced by the participation of imprinted loci from the genomic region on chromosome 7 homologous to Beckwith-Wiedemann syndrome loci (Eggenschwiler et al., 1997; Wise and Pravatcheva, 1997; Caspary et al., 1999).
Generic Liability to Terafogen-Induced Cleft Palate
Complexities of Interpretation of the Genetics of Liability to Teratogen-Induced Cleft Palate
Palatal closure is one of the last major morphogenetic events during development. The critical period for inducing CP by teratogens is usually after most other morphogenetic events are completed, and the CP is therefore usually nonsyndromic. Genetic liability to spontaneous CP expressed as late palatal closure can cause elevated liability to teratogen-induced CP, as demonstrated by the SW/Fr strain response to cortisone (Vekemans and Fraser, 1979). A/J, a strain with spontaneous CL/P, is more susceptible than C57BL/6J to isolated CP induced by various teratogens such as glucocorticoids, 6-aminonicotinamide, and phenytoin. A common preconception has been that liability to teratogen-induced CP is caused by a weakness in craniofacial development in A/J arising from the same loci as spontaneous CL/P. The available evidence indicates that the apparent convergence of these traits in A/J may be a misinterpretation. There are several avenues of evidence. A/J has neither an unusually high risk of spontaneous isolated CP, with less than 0.5% (Hackman and Brown, 1972; Brown et al., 1974; Gasser et al., 1981) (Table 22.3), nor a uniquely high risk of either cortisone- or 6-aminonicotinamide-induced CP. Another strain, SWV/Bc, has a genetic liability to cortisone-induced CP as high as that of A/J and a high liability to 6-aminonicotinamide-induced CP but never has CL/P (Biddle, 1978; Juriloff, 1987). The mechanisms leading to CP in A/J differ between cortisone and 6-aminonicotinamide, as demonstrated by their different critical periods and by their different dose-response slopes (Biddle, 1978), indicating that these two types of CP are different traits. In backcrosses descended from a cross of A/J and C57BL/6J, liability to CL/P and liability to either type of induced CP segregate independently (my analysis of combined data from Biddle and Fraser, 1979, 1986). Among the 12 RI strains derived from A/J and C57BL/6J that were tested for liability to 6-aminonicotinamide-induced CP, the only one that produced spontaneous CL/P, AXB-6, ranked ninth for CP, also indicating the genetic independence of spontaneous CL/P and 6-aminonicotinamide-induced CP (Diehl and Erickson, 1997).
Few genetic analyses have attempted to detect the number of differential loci responsible for a strain difference in liability to teratogen-induced CP. The A/J-C57BL/6J difference in liability to cortisone-induced CP may be due to two loci acting additively (Biddle and Fraser, 1977), that for 6-aminonicotinamide-induced CP may be due to three loci interacting epistatically (Biddle, 1977). and Another strain difference in susceptibility to cortisone-induced CP, between DBA/2J and C57BL/6J, where DBA/2J is the more susceptible strain, may be largely due to one gene (Vekemans et al., 1981).
Mapping studies (Table 22.2) for loci controlling liability to teratogen-induced CP are expected to detect a heterogeneity of types of locus, some involved in a variety of craniofacial mechanisms that lead to CP and some in teratogen binding or metabolism (Lankas et al., 1998; Atlas et al., 1980).
Glucocorticoid-Induced Cleft Palate
Several genomic regions, identified in different strain comparisons, influence liability to glucocorticoid-induced CP. The regions include at least three loci within the major histocompatibility gene (H2) complex, Dcp1, Dcp2, and Acp (Gasser et al., 1988, 1991; Tyan and Tyan, 1993), which were detected in congenic strains on the C57BL/10 background. On this background, in other congenic strains, another immune response gene region, H3, also influences susceptibility (Gasser et al., 1981). The H2-region loci do not appear to affect palatal development directly (Vekemans and Fraser, 1982), they seem to cause differences in palate binding of glucocorticoids (Katsumata et al., 1981), and biochemical studies indicate that the mechanism of teratogenesis is through the anti-inflammatory activity of glucocorticoids and consequent differences in inhibition of arachidonic acid release (Gupta et al., 1984). In contrast to the congenic strains, the DBA/2J-C57BL/6J strain difference in susceptibility to glucocorticoid-induced CP is not detectably influenced by the genes in the H2 complex, and a major susceptibility locus difference was mapped in the BXD RI strains to chromosome 5 near the Pgm1 marker locus (Vekemans et al., 1981). Another strain difference in susceptibility to glucocorticoid-induced CP, between A/J and C57BL/6J, where A/J is the relatively susceptible strain, was shown by reciprocal congenic strains to be strongly influenced by functional variants of N-acetyltransferase, attributed to Nat1 on chromosome 8 or closely linked loci (Karolyi et al., 1990). An effect of genes on the X chromosome also was demonstrated for the susceptibility differences between A/J, C57BL/6J, and C3H/HeJ, using comparisons of risk in specific categories of reciprocal backcrosses that differ only in X chromosome genes (Francis, 1973).
Phenytoin-Induced Cleft Palate
In many strains of mice, CP is induced when the anticonvulsant phenytoin is given to the mother during the 3 days before palatal closure in the fetus. Although phenytoin is not chemically similar to glucocorticoids, at least some of the same genes appear to influence strain susceptibilities to CP induced by both teratogens. On the C57BL/10 background, at least two loci in the H2 complex and a gene at or near the H3 locus strongly influence susceptibility to phenytoin-induced CP (Goldman et al., 1983a,b). The biochemical mechanism of the H2 genetic effect on susceptibility to phenytoin-induced CP has been observed to be the same as for glucocorticoids, involving an H2 effect on the degree of inhibition of arachidonic acid release (Gupta et al., 1984). Other loci are also implicated: the A/J background confers a higher degree of inhibition of arachidonic acid by phenytoin than the C57BL/10 background (Gupta et al., 1984). One of these loci may be the Nat1 region, also involved in glucocorticoid susceptibility, as shown by a significant increase in phenytoin-induced CP when the A/J N-acetyl-transferase trait was substituted into a congenic C57BL/6J background (Karolyi et al., 1990).
6-Aminonicotinamide-Induced Cleft Palate
Liability to 6-aminonicotinamide-induced CP is much higher in A/J than C57BL/6J mice and is in part attributable to the variant in the Nat1 region. Substitution of the C57BL/6J variant into A/J in a congenic strain significantly reduced the frequency of CP induced; however, this effect was limited to the context of the A/J background, and the reverse substitution into the C57BL/6J background had no detectable effect (Karolyi et al., 1990). This epistatic phenomenon is consistent with the independent genetic analysis that estimated that a combination of three recessive epistatic loci in A/J cause its high response to 6-aminonicotinamide (Biddle, 1977) as the epistasis would enable the dominant allele from C57BL/6J at any one of the three loci to suppress the effects of the other two loci, whereas the homozygous recessive state at one of the three loci would have no effect, as demonstrated by the reverse congenic on C57BL/6J.
In another approach (Diehl and Erickson, 1997) to mapping loci responsible for the A/J-C57BL/6J strain difference (54% CP vs. 9% CP at the 6-aminonicotinamide dosage used), the CP response and genome screen of markers from a set of nine AXB/BXA RI strains were analyzed by a novel method, as described above for the parallel study done on phenytoin-induced CL/P. Ten genomic regions were identified at p<0.05; however, as discussed above, the lack of correction for the multiple tests of a genome screen increases the actual risk of false linkages greatly, and it is likely that as many as half of the “hits” could be false-positives. Among the 10 potential linkages found, in three the higher-susceptibility allele was from the A/J strain (Table 22.2); in the rest, it was the B (low-risk strain) allele. Although the RI strains do exceed the parental range in CP response, predicting some B-susceptibility alleles, it is difficult to reconcile the very low risk of the C57BL/6J strain with its contributing most of the high-risk alleles to the cross, and this argues that many are false linkages. In keeping with the recommended two-step mapping strategy for genome surveys in RI strains (Belknap et al., 1996; Diehl and Erickson, 1997), which relaxes the criterion for significance on the first pass on condition that the suggested linkages be tested in independent segregating crosses, it is premature to extrapolate to genetic architecture or to candidate loci from these data.
Potential Future Roles for Quantitative Trait Locus Approaches
Measurable characteristics that normally vary among individuals on a continuous scale are termed quantitative traits. The normal variation in agriculturally important traits such as milk production, egg production, fruit yield, and growth rate appears to be due to the additive effects of numerous polymorphic gene loci. A theoretical base and statistical methods were developed to study and manipulate these traits (Falconer, 1981). An individual locus that contributes to the variation in a quantitative trait is termed a quantitative trait locus (QTL). Quantitative genetic methodology has been developed to use highly polymorphic mapped molecular markers in a genome screen of genetic segregants to identify chromosomal regions that contain QTLs (Lander and Botstein, 1989; Tanksley, 1993). In principle, the core of these methods is concerned with statistical detection of the impacts on variation in the quantitative trait by the QTL variants present in marked chromosomal regions. Several statistical approaches to QTL mapping have been developed and the available software packages reviewed (Manly and Olson, 1999). Generally, BC1 and F2 generations are more efficient for detection of QTLs than are RI strains; BC1 segregants are more efficient than F2s for mapping major QTLs, but F2s give a more accurate picture of genetic architecture (Darvasi, 1998).
Approaches to QTL mapping have been applied to diseases with complex inheritance that are expressed as observable extremes of quantitative physiological variables, such as hypertension (Jacob et al., 1991) or frequency of seizures induced by specific environments (Frankel et al., 1994). Application of the statistical methods for QTL mapping both to normal variation and to disease states tends to obscure some fundamental genetic questions. Whether there is a systematic difference in the genetic architecture and type of genetic variants or loci that contribute to the two situations is not known. It has become conventional to refer to the loci involved in genetically complex traits as QTLs, although their role may not be quantitative.
A longstanding quantitative genetic hypothesis (Fraser, 1976; Falconer, 1981) explains the complexity of heredity of some “all-or-none” (qualitative) birth defects such as CL/P and CP. It proposes that an underlying polygenic quantitative trait in a developmental process is translated into the observed binary trait by a critical threshold value, such as timing or size (Fig. 22.3a). This threshold hypothesis has been supported by observations of embryos. The polygenic aspect of the hypothesis has not been supported by mouse models; the genetic complexity originates in nonadditive interactions among a few loci rather than segregation of a large number of QTL's.
The QTL mapping programs that are based on statistical assumptions for quantitative variables are generally not directly applicable to the low-penetrance binary data for orofacial clefting in segregants. A function of the MAPMAKER/QTL program, termed a penetrance scan, can be used with binary data to map QTLs of large effect, as demonstrated by Cadfar in cadmium-induced forelimb malformations (Hovland et al., 2000). However, if a small number of loci of relatively large effect are involved, they can be mapped by direct analysis of marker segregation ratios in a genome screen of affected segregants, as demonstrated by clf1 and clf2 (Juriloff et al., 2001).
The goal of mapping QTLs is to identify the genes. Mapping is the first step. Because there is not a direct relationship between the genotype at a particular QTL and the phenotype, it is not possible, among segregants, to distinguish the rare recombinants that refine the definition of the QTL map position. Therefore, QTL map positions begin as large chromosomal segments (e.g., 10 cM) containing dozens of genes. A prerequisite to gene identification is more precise mapping of the QTL (e.g., a 1-2 cM segment). Precise mapping requires additional mapping strategies (reviewed in Darvasi, 1998). Some approaches that are applicable to binary threshold traits are as follows. In the recombinant progeny testing method, segregants are screened for the flanking markers surrounding the QTL interval, recombinants within the QTL interval are testcrossed, and the phenotypes of their progeny indicate which part of the interval contains the QTL. For example, if high risk of CL/P were associated with the A allele at the QTL, the recombinants retaining the A haplotype through the part of the interval containing the QTL would transmit a higher risk of CL/P to their testcross progeny than the recombinants that had been changed by recombination to B in this region. In the interval-specific congenic strain approach, the flanking markers are used to assist in transferring the QTL interval from one parental strain to the other, by repeated backcrossing. Then, individuals with recombinant haplotypes between the flanking markers are used as founders of homozygous strains, each containing a different section of the transferred interval, identified by a series of closely spaced markers. Comparison of the phenotypes of these strains is used to identify the fraction of the original interval that contains the QTL. Other, more complex approaches may also be taken (Darvasi, 1998).
With a precisely defined map position in hand, the gene at the QTL can be identified by examination of the sequence of candidate genes already known in the region.
Summary
Mouse models offer some insight into the gene regulatory pathways involved in nonsyndromic noninduced CL/P and CP. For CL/P, these are clf1 and clf2, which are naturally occurring variants that need to be identified at the molecular genetic level by examination of candidate genes or by positional cloning and sequencing. For CP, the genes are knockout mutations of known genes: Lhx8, Gad1, Gabrb3, and Tgfb3. Their reduced penetrance offers an access point to identification of genetic modifiers.
As there likely is a role of environment in risk of orofacial clefting (Schutte and Murray, 1999), the loci important to susceptibility to environmentally induced CL/P and CP offer a clue to other mechanisms of susceptibility. The loci identified for susceptibility to CL/P or CP induced by the anticonvulsant phenytoin, particularly those in the major histocompatibility complex and the N-acetyltransferase candidates, are attractive, clinically relevant entry points.
One of the very strong themes emerging from studies of genetics and mapping of genes for nonsyndromic orofacial clefting in mice is genetic complexity due to epistatic interaction between loci. Whether a particular allelic substitution at a locus important to the risk of orofacial clefting will have a detectable effect depends on the genetic context of the substitution. Although this is sometimes called the “strain background,” genetic analyses have demonstrated that large strain differences that appear genetically complex can be due to a small number of loci with epistatic interactions, as for the A/J-C57BL/6J difference for spontaneous CL/P or 6-aminonicotinamide-induced CP. The contextual background effect therefore may be due to the effect of a specific allele at a speciic locus, rather than the amorphous situation implied by background. It will be interesting to explore the mechanistic nature of these interactions using targeted alterations of known genes.
Mouse models provide a powerful tool that can be of immense value in unraveling the genetic complexity of human CL/P and CP. It remains to be seen whether the particular mutant gene loci identified in mice have human mutant counterparts responsible for a significant fraction of human orofacial clefts. More important is the identification in mice of gene-regulatory pathways important to orofacial clefting as these are expected to be highly conserved and will identify a series of candidate genes that may contribute to risk of human orofacial clefting. Future candidate genes can be targeted for mutagenesis in mice and the specific molecular, cellular, and developmental effects of the candidate gene isolated in congenic strains. The molecular developmental mechanisms and genetic mechanisms of interaction that lead to complexity of transmission patterns of orofacial clefts will likely be identified first in mice.
References
Atlas, SA, Zweier, JL, Nebert, DW (1980). Genetic differences in phenytoin pharmacokinetics. In vivo clearance and in vitro metabolism among inbred strains of mice. Dev Pharm Ther 1: 281–304.
Barrow, JR, Capecchi, MR (1999). Compensatory defects associated with mutations in Hoxa1 restore normal palatogenesis to Hoxa2 mutants. Development 126: 5011–5026.
Baudoin, C, Goumans, MJ, Mummery, C, Sonnenberg, A (1998). Knockout and knockin of the beta 1 exon D define distinct roles for integrin splice variants in heart function and embryonic development. Genes Dev 12: 1202–1216.
Belknap, JK, Mitchell, SR, O'Toole, LA, et al. (1996). Type I and type II error rates for quantitative trait loci (QTL) mapping studies using recombinant inbred mouse strains. Behav Genet 26: 149–160.
Biddle, FG (1977). 6-Aminonicotinamide-induced cleft palate in the mouse: the nature of the difference between the A/J and C57BL/6J strains in frequency of response and its genetic basis. Teratology 16: 301–312.
Biddle, FG (1978). Use of dose-response relationships to discriminate between the mechanisms of cleft-palate induction by different teratogens: an argument for discussion. Teratology 18: 247–252.
Biddle, FG (1988). Genetic differences in the frequency of acetazolamide-induced ectrodactyly in the mouse exhibit directional dominance of relative embryonic resistance. Teratology 37: 375–388.
Biddle, FG, Eraser, FC (1977). Cortisone-induced cleft palate in the mouse. A search for the genetic control of the embryonic response trait. Genetics 85: 289–302.
Biddle, FG, Eraser, FC (1979). Genetic independence of the embryonic reactivity difference to cortisone- and 6-aminonicotinamide-induced cleft palate in the mouse. Teratology 19: 207–211.
Biddle, FG, Eraser, FC (1986). Major gene determination of liability to spontaneous cleft lip in the mouse. J Craniofac Genet Dev Biol (Suppl) 2: 67–88.
Blystone, SD, Lindberg, FP, LaFlamme, SE, Brown, EJ (1995). Integrin beta3 cytoplasmic tail is necessary and sufficient for regulation of alpha5beta1 phagocytosis by alphavbeta3 and integrin associated protein. J Cell Biol 130: 745–754.
Bornstein, S, Trasler, DG, Eraser, FC (1970). Effect of the uterine environment on the frequency of spontaneous cleft lip in CL/Fr mice. Teratology 3: 295–298.
Brown, KS, Johnston, MC, Murphy, PF (1974). Isolated cleft palate in A/J mice after transitory exposure to drinking water deprivation and low humidity in pregnancy. Teratology 9: 151–158.
Carroll, JM, Luetteke, NC, Lee, DC, Watt, FM (1998). Role of integrins in mouse eyelid development: studies in normal embryos and embryos in which there is a failure of eyelid fusion. Mech Dev 78: 37–45.
Caspary, T, Cleary, MA, Perlman, EJ, et al. (1999). Oppositely imprinted genes p57(Kip2) and igf2 interact in a mouse model for Beckwith-Wiedemann syndrome. Genes Dev 13: 3115–3124.
Chang, H, Huylebroeck, D, Verschueren, K, et al. (1999). Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 126: 1631–1642.
Chenevix-Trench, G, Jones, K, Green, AC, et al. (1992). Cleft lip with or without cleft palate: associations with transforming growth factor alpha and retinoic acid receptor loci. Am J Hum Genet 51: 1377–1385.
Condie, BG, Bain, G, Gottlieb, DI, Capecchi, MR (1997). Cleft palate in mice with a targeted mutation in the gamma-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci USA 94: 11451–11455.
Culiat, CT, Stubbs, LJ, Woychik, RP, et al. (1995). Deficiency of the beta3 subunit of the type A gamma-aminobutyric acid receptor causes cleft palate in mice. Nat Genet 11: 344–346.
Damm, K, Heyman, RA, Umesono, K, Evans, RM (1993). Functional inhibition of retinoic acid response by dominant negative retinoic acid receptor mutants. Proc Natl Acad Sci USA 90: 2989–2993.
Darvasi, A (1998). Experimental strategies for the genetic dissection of complex traits in animal models. Nat Genet 18: 19–24.
Davidson, JG, Eraser, EC, Schlager, G (1969). A maternal effect on the frequency of spontaneous cleft lip in the A-J mouse. Teratology 2: 371–376.
De Felice, M, Ovitt, C, Biffali, E, et al. (1998). A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet 19: 395–398.
Diehl, SR, Erickson, RP (1997). Genome scan for teratogen-induced clefting susceptibility loci in the mouse: evidence of both allelic and locus heterogeneity distinguishing cleft lip and cleft palate. Proc Natl Acad Sci USA 94: 5231–5236.
Diewert, VM (1986). Craniofacial growth during human secondary palate formation and potential relevance of experimental cleft palate observations. J Craniofac Genet Dev Biol (Suppl)2: 267–276.
Diewert, VM, Wang, KY (1992). Recent advances in primary palate and midface morphogenesis research. Crit Rev Oral Biol Med 4: 111–130.
Dreyer, CJ, Preston, CB (1974). Classification of cleft lip and palate in animals. Cleft Palate J 11: 327–332.
Eggenschwiler, J, Ludwig, T, Fisher, P, et al. (1997). Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev 11: 3128–3142.
Falconer, DS (1981). Introduction to Quantitative Genetics, 2nd ed. New York: Longman, pp. 270–280.
Francis, BM (1973). Influence of sex-linked genes on embryonic sensitivity to cortisone in three strains of mice. Teratology 7: 119–126.
Frankel, WN, Taylor, BA, Noebels, JL, Lutz, CM (1994). Genetic epilepsy model derived from common inbred mouse strains. Genetics 138: 481–489.
Fraser, FC (1976). The multifactorial/threshold concept—uses and misuses. Teratology 14: 267–280.
Fraser, FC (1980). The William Allan Memorial Award address. Evolution of a palatable multifactorial threshold model. Am J Hum Genet 32: 796–813.
Gasser, DL, Goldner-Sauve, A, Katsumata, M, Goldman, AS (1991). Restriction fragment length polymorphisms, glucocorticoid receptors, and phenytoin-induced cleft palate in congenic strains of mice with steroid susceptibility differences. J Craniofac Genet Dev Biol 11: 366–371.
Gasser, DL, Mele, L, Lees, DD, Goldman, AS (1981). Genes in mice that affect susceptibility to cortisone-induced cleft palate are closely linked to Ir genes on chromosomes 2 and 17. Proc Natl Acad Sci USA 78: 3147–3150.
Gasser, DL, Yadvish, KN, Trammell, MA, Goldman, AS (1988). Recombinants in the H-2S/H-2D interval of mouse chromosome 17 define the map position of a gene for cleft palate susceptibility. Teratology 38: 571–577.
Goldman, AS, Baker, MK, Gasser, DL (1983a). Susceptibility to phenytoin-induced cleft palate in mice is influenced by genes linked to H-2 and H-3. Immunogenetics 18: 17–22.
Goldman, AS, Fishman, CL, Baker, MK (1983b). Phenytoin teratogenicity in the primary and secondary mouse embryonic palate is influenced by the H-2 histocompatibility locus. Proc Soc Exp Biol Med 173: 82–86.
Gong, SG, White, NJ, Sakasegawa, AY (2000). The Twirler mouse, a model for the study of cleft lip and palate. Arch Oral Biol 45: 87–94.
Graham, B, Battey, J, Jordan, E (2001). Report of second follow-up workshop on priority setting for mouse genomics. Mamm Genome 12: 1–2.
Griffith, AJ, Radice, GL, Burgess, DL, et al. (1996). Location of the 9257 and ataxia mutations on mouse chromosome 18. Mamm Genome 7: 417–419.
Gruneberg, H. (1952). Harelip and cleft palate. In: The Genetics of the Mouse, 2nd ed., edited by H Gruneberg. The Hague: Martinus Nijhoff, pp. 363–370.
Gruneberg, H, Truslove, GM (1960). Two closely linked genes in the mouse. Genet Res 1: 69–90.
Gupta, C, Katsumata, M, Goldman, AS (1984). H-2 influences phenytoin binding and inhibition of prostaglandin synthesis. Immunogenetics 20: 667–676.
Hackman, RM, Brown, KS (1972). Corticosterone-induced isolated cleft palate in A/J mice. Teratology 6: 313–316.
Hovland, DN, Cantor, RM, Lee, GS, et al. (2000) Identification of a murine locus conveying susceptibility to cadmium-induced forelimb malformations. Genomics 63: 193–201.
Huie, ML, Kasper, JS, Arn, PH, et al. (1999). Increased occurrence of cleft lip in glycogen storage disease type II (GSDII): exclusion of a contiguous gene syndrome in two patients by presence of intragenic mutations including a novel nonsense mutation Gln58Stop. Am J Med Genet 85: 5–8.
Hynes, RO (1994). Genetic analyses of cell-matrix interactions in development. Curr Opin Genet Dev 4: 569–574.
Illera, MJ, Cullinan, E, Gui, Y, et al. (2000). Blockade of the alpha(v)beta(3) integrin adversely affects implantation in the mouse. Biol Reprod 62: 1285–1290.
Jacob, HJ, Lindpaintner, K, Lincoln, SE, et al. (1991). Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell 67: 213–224.
Johnson, DR (1969). Brachyphalangy, an allele of extra-toes in the mouse. Genet Res 13: 275–280.
Juriloff, DM (1980). Genetics of clefting in the mouse. Prog Clin Biol Res 46: 39–71.
Juriloff, DM (1982). Differences in frequency of cleft lip among the A strains of mice. Teratology 25: 361–368.
Juriloff, DM (1987). Maternal treatment with cortisone accelerates eyelid closure and other developmental fusion processes in fetal mice. Development 100: 611–618.
Juriloff, DM (1993). Current status of genetic linkage studies of a major gene that causes CL/P in mice: exclusion map. J Craniofac Genet Dev Biol 13: 223–229.
Juriloff, DM (1995). Genetic analysis of the construction of the AEJ.A congenic strain indicates that nonsyndromic CL/P in the mouse is caused by two loci with epistatic interaction. J Craniofac Genet Dev Biol 15: 1–12.
Juriloff, DM, Fraser, FC (1980). Genetic maternal effects on cleft lip frequency in A/J and CL/Fr mice. Teratology 21: 167–175.
Juriloff, DM, Harris, MJ (1983). Abnormal facial development in the mouse mutant first arch. J Craniofac Genet Dev Biol 3: 317–337.
Juriloff, DM, Harris, MJ, Brown, CJ (2001). Unravelling the complex genetics of cleft lip in the mouse model. Mamm Genome 12: 426–435.
Juriloff, DM, Harris, MJ, Mah, DG (1996). The clf1 gene maps to a 2- to 3-cM region of distal mouse chromosome 11. Mamm Genome 7: 789.
Juriloff, DM, Mah, DG (1995). The major locus for multifactorial nonsyndromic cleft lip maps to mouse chromosome 11. Mamm Genome 6: 63–69.
Juriloff, DM, Roberts, CW (1975). Genetics of cleft palate in chickens and the relationship between the occurrence of the trait and maternal riboflavin deficiency. Poult Sci 54: 334–346.
Juriloff, DM, Trasler, DG (1976). Test of the hypothesis that embryonic face shape is a causal factor in genetic predisposition to cleft lip in mice. Teratology 14: 35–41.
Kaartinen, V, Voncken, JW, Shuler, C, et al. (1995). Abnormal lung development and cleft palate in mice lacking TGF-beta3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 11: 415–421.
Kadowaki, S, Sakamoto, M, Kamiishi, H, Tanimura, T (1997). Embryologic features of term fetuses and newborns in CL/Fr mice with special reference to cyanosis. Cleft Palate Craniofac J 34: 211–217.
Kalter, H (1979). The history of the A family of inbred mice and the biology of its congenital malformations. Teratology 20: 213–232.
Karolyi, IJ, Liu, S, Erickson, RP (1987). Susceptibility to phenytoin-induced cleft lip with or without cleft palate: many genes are involved. Genet Res 49: 43–49.
Karolyi, J, Erickson, RP, Lui, S, Killewald, L (1990). Major effects on teratogen-induced facial clefting in mice determined by a single genetic region. Genetics 126: 201–205.
Katsumata, M, Baker, MK, Goldman, AS, Gasser, DL (1981). Influence of H-2-linked genes on glucocorticoid receptors in the fetal mouse palate. Immunogenetics 13: 319–325.
Kraus, P, Lufkin, T (1999). Mammalian Dlx homeobox gene control of craniofacial and inner ear morphogenesis. J Cell Biochem Suppl 32-33: 133–140.
Lander, ES, Botstein, D (1989). Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199.
Lankas, GR, Wise, LD, Cartwright, ME, et al. (1998). Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reprod Toxicol 12: 457–463.
Loevy, H, Feynes, V (1968). Spontaneous cleft palate in a family of Siamese cats. Cleft Palate J 5: 57–60.
Lyon, MF (1958). Twirler: a mutant affecting the inner ear of the house mouse. J Embryol Exp Morphol 6: 105–116.
Lyon, MF, Quinney, R, Glenister, PH, et al. (1996). Doublefoot: a new mouse mutant affecting development of limbs and head. Genet Res 68: 221–231.
MacKenzie, A, Ferguson, MWJ, Sharpe, PT (1992). Expression patterns of the homeobox gene Hox-8 in the mouse embryo suggest a role in specifying tooth initiation and shape. Development 115: 403–420.
Maddox, BK, Garofalo, S, Horton, WA, et al. (1998). Craniofacial and otic capsule abnormalities in a transgenic mouse strain with a Col2al mutation. J Craniofac Genet Dev Biol 18: 195–201.
Manly, KF, Olson, JM (1999). Overview of QTL mapping software and introduction to Map Manager QT. Mamm Genome 10: 327–334.
Matsushima, Y, Ohne, M, Irino, T, Maki, M (1992). Spontaneous cleft palate in CF:l/Ohu mice. Jikken Dobutsu 41: 83–85.
Matzuk, MM, Kumar, TR, Bradley, A (1995a). Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 374: 356–360.
Matzuk, MM, Kumar, TR, Vassalli, A, et al. (1995b). Functional analysis of activins during mammalian development. Nature 374: 354–356.
Matzuk, MM, Lu, N, Vogel, H, et al. (1995c). Multiple defects and perinatal death in mice deficient in follistatin. Nature 374: 360–363.
McNeish, JD, Thayer, J, Walling, K, et al. (1990). Phenotypic characterization of the transgenic mouse insertional mutation, legless. J Exp Zool 253: 151–162.
Meier, P, Koedood, M, Philipp, J, et al. (1995). Alternative mRNAs encode multiple isoforms of transcription factor AP-2 during murine embryogenesis. Dev Biol 169: 1–14.
Miettinen, PJ, Berger, JE, Meneses, J, et al. (1995). Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376: 337–341.
Miettinen, PJ, Chin, JR, Shum, L, et al. (1999). Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat Genet 22: 69–73.
Millicovsky, G, Ambrose, LJ, Johnston, MC (1982). Developmental alterations associated with spontaneous cleft lip and palate in CL/Fr mice. Am J Anat 164: 29–44.
Mitchell, LE, Risch, N (1992). Mode of inheritance of nonsyndromic cleft lip with or without cleft palate: a reanalysis. Am J Hum Genet 51: 323–332.
Mitchell, PJ, Timmons, PM, Hebert, JM, et al. (1991). Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev 5: 105–119.
Moon, RT, Brown, JD, Torres, M (1997). WNTs modulate cell fate and behavior during vertebrate development. Trends Genet 13: 157–162.
Morasso, MI, Grinberg, A, Robinson, G, et al. (1999). Placental failure in mice lacking the homeobox gene Dlx3. Proc Natl Acad Sci USA 96: 162–167.
Nakamura, S, Stock, DW, Wydner, KL, et al. (1996). Genomic analysis of a new mammalian distal-less gene: Dlx7. Genomics 38: 314–324.
Nottoli, T, Hagopian-Donaldson, S, Zhang, J, et al. (1998). AP-2- null cells disrupt morphogenesis of the eye, face, and limbs in chimeric mice. Proc Natl Acad Sci USA 95: 13714–13719.
Parr, BA, Shea, MJ, Vassileva, G, McMahon, AP (1993). Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119: 247–261.
Perou, CM, Perchellet, A, Jago, T, et al. (1997). Comparative mapping in the beige-satin region of mouse chromosome 13. Genomics 39: 136–146.
Peters, H, Neubuser, A, Kratochwil, K, Balling, R (1998). Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 12: 2735–2747.
Pourtois, M (1967). Influence of cleft lip upon palatal closure in A/Jax mice. Cleft Palate J 4: 120–123.
Price, JA, Bowden, DW, Wright, JT, et al. (1998). Identification of a mutation in DLX3 associated with tricho-dento-osseous (TDO) syndrome. Hum Mol Genet 7: 563–569.
Proetzel, G, Pawlowski, SA, Wiles, MV, et al. (1995). Transforming growth factor-beta3 is required for secondary palate fusion. Nat Genet 11: 409–414.
Qiu, M, Bulfone, A, Ghattas, I, et al. (1997). Role of the Dlx homeobox genes in proximodistal patterning of the branchial arches: mutations of Dlx-1, Dlx-2, and Dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Dev Biol 185: 165–184.
Rittenhouse, E, Dunn, LC, Cookingham, J, et al. (1978). Cartilage matrix deficiency (cmd): a new autosomal recessive lethal mutation in the mouse. J Embryol Exp Morphol 43: 71–84.
Roelink, H, Nusse, R (1991). Expression of two members of the Wnt family during mouse development—restricted temporal and spatial patterns in the developing neural tube. Genes Dev 5: 381–388.
Sampson, SB, Higgins, DC, Elliot, RW, et al. (1998). An edited linkage map for the AXB and BXA recombinant inbred mouse strains. Mamm Genome 9: 688–694.
Sanford, LP, Ormsby, I, Gittenberger-de Groot, AC, et al. (1997). TGFbetal knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout pheno-types. Development 224: 2659–2670.
Satokata, I, Ma, L, Ohshima, H, et al. (2000). Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat Genet 24: 391–395.
Satokata, I, Maas, R (1994). Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6: 348–356.
Schorle, H, Meier, P, Buchert, M, et al. (1996). Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 381: 235–238.
Schutte, BC, Murray, JC (1999). The many faces and factors of orofacial clefts. Hum Mol Genet 8: 1853–1859.
Seegmiller, RE, Fraser, FC (1977). Mandibular growth retardation as a cause of cleft palate in mice homozygous for the chondrodysplasia gene. J Embryol Exp Morphol 38: 227–238.
Siebert, JR, Williams, B, Collins, D, et al. (1998). Spontaneous cleft palate in a newborn gorilla (Gorilla gorilla gorilla). Cleft Palate Craniofac J 35: 436–441.
Silver, LM (1995). Classical linkage analysis and mapping panels. In: Mouse Genetics, edited by LM Silver. New York: Oxford University Press, pp. 195–263.
Staats, J (1972). Standardized nomenclature for inbred strains of mice: fifth listing. Cancer Res 32: 1609–1646.
Stills, HF, Jr, Bullock, BC (1981). Congenital defects of squirrel monkeys (Saimiri sciureus). Vet Pathol 18: 29–36.
Sulik, KK, Johnston, MC, Ambrose, LJ, Dorgan, D (1979). Phenytoin (dilantin)-induced cleft lip and palate in A/J mice: a scanning and transmission electron microscopic study. Anat Rec 195: 243–255.
Supp, DM, Brueckner, M, Kuehn, MR, et al. (1999). Targetted deletion of the ATP binding domain of left-right dynein confirms its role in specifying development of left-right asymmetries. Development 126: 5495–5504.
Swartz, HA, Vogt, DW, Kinter, LD (1982). Chromosome evaluation of Angus calves with unilateral congenital cleft lip and jaw (cheilognathoschisis). Am J Vet Res 43: 729–731.
Takagi, T, Moribe, H, Kondoh, H, Higashi, Y (1998). DeltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development 125: 21–31.
Takihara, Y, Tomotsune, D, Shirai, M, et al. (1997). Targeted disruption of the mouse homologue of the Drosophila polyhomeotic gene leads to altered anteroposterior patterning and neural crest defects. Development 124: 3673–3682.
Tanksley, SD (1993). Mapping polygenes. Annu Rev Genet 27: 205–233.
Taya, Y, O'Kane, S, Ferguson, MW (1999). Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 126: 3869–3879.
Trasler, DG (1968). Pathogenesis of cleft lip and its relation to embryonic face shape in A-J and C57BL mice. Teratology 1: 33–49.
Trasler, DG, Fraser, FC (1963). Role of the tongue in producing cleft palate in mice with spontaneous cleft lip. Dev Biol 6: 45–60.
Trasler, DG, Kemp, D, Trasler, TA (1984). Increased susceptibility to 6-aminonicotinamide-induced cleft lip of heterozygote Dancer mice. Teratology 29: 101–104.
Trasler, DG, Leong, S (1982). Mitotic index in mouse embryos with 6-aminonicotinamide-induced and inherited cleft lip. Teratology 25: 259–265.
Trasler, DG, Trasler, TA (1984). Left cleft lip predominance and genetic similarities of L line and CL/Fr strain mice. Teratology 30: 423–427.
Tyan, ML, Tyan, DB (1993). Vitamin A-enhanced cleft palate susceptibility gene maps between C4 and B144 within the H-2 complex. Proc Soc Exp Biol Med 202: 482–486.
van den Boogaard, MH, Dorland, M, Beemer, FA, van Amstel, HKP (2000). MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 24: 342–343.
Vekemans, M, Fraser, FC (1979). Stage of palate closure as one indication of “liability” to cleft palate. Am J Med Genet 4: 95–102.
Vekemans, M, Fraser, FC (1982). Susceptibility to cleft palate and the major histocompatibility complex (H-2) in the mouse. Teratology 25: 267–270.
Vekemans, M, Taylor, BA, Fraser, FC (1981). The susceptibility to cortisone-induced cleft palate of recombinant inbred strains of mice: lack of association with the H-2 haplotype. Genet Res 38: 327–331.
Wang, KY, Juriloff, DM, Diewert, VM (1995). Deficient and delayed primary palatal fusion and mesenchymal bridge formation in cleft lip-liable strains of mice. J Craniofac Genet Dev Biol 15: 99–116.
Wang, W, Chen, X, Xu, H, Lufkin, T (1996). Msx3: a novel murine homologue of the Drosophila msh homeobox gene restricted to the dorsal embryonic central nervous system. Mech Dev 58: 203–215.
Wenngren, BI, Anniko, M (1989). Vestibular hair cell pathology in the dancer mouse mutant. Acta Otolaryngol (Stockh) 107: 182–190.
Winograd, J, Reilly, MP, Roe, R, et al. (1997). Perinatal lethality and multiple craniofacial malformations in MSX2 transgenic mice. Hum Mol Genet 6: 369–379.
Wise, TL, Pravatcheva, DD (1997). Perinatal lethality in H19 enhancers-Igf2 transgenic mice. Mol Reprod Dev 48: 194–207.
Zhang, H, Hu, G, Wang, H, et al. (1997). Heterodimerization of Msx and Dlx homeoproteins results in functional antagonism. Mol Cell Biol 17: 2920–2932.
Zhang, J, Hagopian-Donaldson, S, Serbedzija, G, et al. (1996). Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 381: 238–241.
Zhao, Y, Guo, YJ, Tomac, AC, et al. (1999). Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene Ihx8. Proc Natl Acad Sci USA 96: 15002–15006.