Howard M. Saal
Cleft lip (CL) with or without cleft palate (CL/P) and isolated cleft palate (CP), collectively termed oral clefts (OCs), are among the most common birth defects, with a prevalence that ranges between 1 case in every 500 to 1000 newborns. The prevalence of CL/P varies according to ethnicity, gender, and socioeconomic factors (Croen et al., 1998; Bender, 2000). Native Americans have the highest reported prevalence at birth, with 3.6 CL/P cases per 1000 births. Asians have the second highest prevalence at birth for CL/P, with 2.1 cases per 1000 Japanese and 1.7 cases per 1000 Chinese live births (Croen et al., 1998). Gender seems to play a role in the etiology of CL/P since males predominate by a 2:1 ratio (Tolarova, 1987). Also, population studies have shown that individuals born in more rural, lower socioeconomic conditions have a higher risk for CL/P compared to ethnically similar groups with a higher socioeconomic status (Chung et al., 1987; Cembrano et al., 1995).
The incidence of CP is much less dependent on racial and ethnic factors, being approximately 1 in 2000 live births (Vanderas, 1987; Gorlin et al., 1990). In contrast to CLP, however, there is a female predominance of CP, the ratio being approximately 3:2 females to males (Cohen, 2000).
There are many genetic and developmental conditions that can cause CL/P or CP. The Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/Omim/) lists over 174 Mendelian disorders associated with CL/P and 312 associated with CP. The London Dysmorphology Database, which also lists many teratogenic and other nongenetic disorders, identifies 205 disorders associated with CL/P and 441 with CP (Winter and Baraitser, 1996). These resources do not include the substantial number of chromosomal disorders that can be associated with OCs. However, despite these figures, approximately 75% of CL/P and 50% of CP cases will be isolated, nonsyndromic OCs (Table 5.1) (Milerad et al., 1997; Saal, 1998, 2000; Tolarova and Cervenka, 1998; Jones, 2000; Stoll et al., 2000).
It is important to distinguish between syndromic and nonsyndromic CL/P and CP. This distinction has significant implications for determining management and recurrence risks for patients and families. In addition, the success of genetic studies, which search for genes that cause OCs, depends on accurate clinical diagnosis. What differentiates syndromic from nonsyndromic CL/P and CP is often subjective, differing in basic definition from one center to the next. There are no specific guidelines which define nonsyndromic OCs, although one author has specified nonsyndromic OCs as those associated with no or one major anomaly or two or fewer minor anomalies (Jones, 1988). Another study precluded consideration of a nonsyndromic cleft if there was any other major anomaly or three or more minor anomalies in addition to the OC (Tolarova and Cervenka, 1998). Major anomalies are those of functional or cosmetic significance requiring some degree of medical intervention (Spranger et al., 1982). Minor anomalies are those of minimal or no cosmetic or functional significance, which occur in less than 5% of the population (Jones, 1997; Spranger et al., 1982). If one is to equate nonsyndromic OCs with isolated OCs, it seems reasonable to include only those cases where no additional major anomaly exists. The distinctions between syndromic and nonsyndromic OCs become less clear in the presence of minor anomalies. The most helpful studies have shown that the presence of three or more minor anomalies is strongly associated with the presence of a major anomaly (Marden et al., 1964; Leppig et al., 1987, 1988). Therefore, for the purpose of definition, it is reasonable to limit nonsyndromic OC to those associated with no additional malformations and two or fewer minor anomalies. However, some syndromic OCs may be associated with two or fewer minor anomalies. For example, Van der Woude's syndrome is characterized by positive family history for OC and the presence of lower lip pits. Similarly the Robin sequence with CP and mild ear dysplasia or preauricular sinuses are found in individuals with a mild form of the branchio-oto-renal syndrome. In Stickler's syndrome, the only presenting sign may be the Robin sequence and myopia with a positive family history for CP. The diagnosis is even more challenging in the nonocular forms of Stickler's syndrome or if an affected infant has a new mutation with no family history of CP.
|
TABLE 5.1. Studies of Syndromic vs. Nonsyndromic Oral Clefts |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
Oral clefts can be classified on the basis of etiology and/or pathogenesis. There have been multiple classifications of OC based on anatomic and embryological considerations (Fogh-Andersen, 1942, 1971; Millard, 1976). Fogh-Anderson (1942) divided OCs into three main groups: (1) CL extending to the the incisive foramen and including clefts of the alveolus (primary palate); (2) CL and CP (CLP), including unilateral and bilateral CLP; and (3) CP identified as being median and not extending beyond the incisive foramen. Another comprehensive approach was taken by the International Confederation for Plastic and Reconstructive Surgery, which at their 1967 congress established a classification of OC based on the embryology of the developing structures. They identified three major groups of OC (Millard, 1976).
Group 1. Clefts of the primary palate
a. Lip
b. Alveolus
Group 2. Clefts of the primary and secondary palate
a. Lip
b. Alveolus
c. Hard palate (secondary palate)
Group 3. Clefts of the secondary palate
a. Hard palate
b. Soft palate
An anatomic diagnosis is clearly advantageous to the surgeon, who must decide on the best approach and timing for the surgical treatment. However, some problems with diagnosis based solely on anatomy and embryology are that it does not explain the cause of the cleft, does not allow for a comprehensive management plan of the patient for medical conditions that are not directly related to the cleft, and does not give sufficient information regarding possible recurrence risks. Classification of OCs based solely on etiology or pathogenesis supplies insufficient information regarding the severity of the cleft and the types of direct medical and surgical management required. Therefore, it is necessary to combine the two types of classification in order to optimally diagnose and treat any patient with an OC.
Cleft Lip with or without Cleft Palate
Nonsyndromic CL/P accounts for between 70% and 80% of all cases (Milerad et al., 1997; Saal, 1998, 2000; Tolarova and Cervenka, 1998; Jones, 2000; Stoll et al., 2000). Affected individuals may have either CL or CLP. In nonsyndromic CL/P, the cleft is not in the midline and there is laterality to the CL. Midline CL is indicative of another underlying disorder, such as one of the oral-facial-digital syndromes or the holoprosencephaly sequence. The London Dysmorphology Database identifies at least 40 different conditions with midline clefts (Winter and Baraitser, 1996).
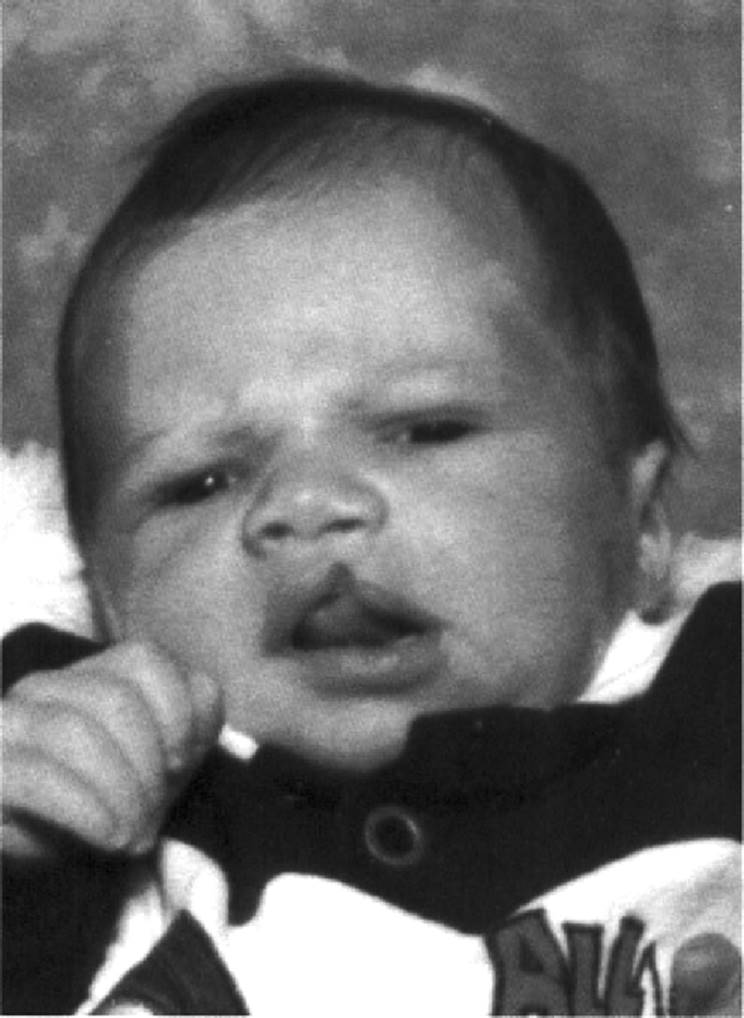
In nonsyndromic CL, unilateral clefts are more common than bilateral involvement (Fig. 5.1). In unilateral CL, the prevalence of left-sided CL is greater than that of right-sided CL. The left-sided:right-sided:bilateral CL/P ratio is 6:3:1 (Lettieri, 1993).
Anatomically, the CL may be complete or incomplete. Complete CL refers to the cleft involving the entire upper lip and extending into the naris. In incomplete CL, there is a variable amount of tissue that bridges the upper lip. The connecting tissue may comprise only a narrow band, called Simonart band (Millard, 1976).
|
|
|
FIG. 5.1. Infant boy with isolated left-sided cleft lip. |
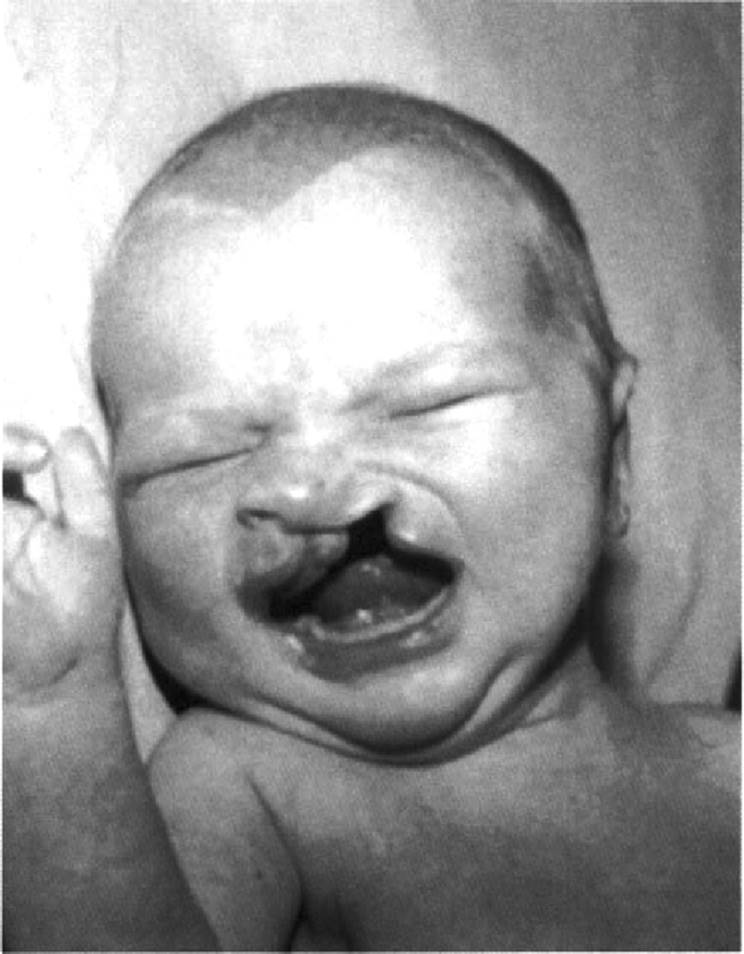
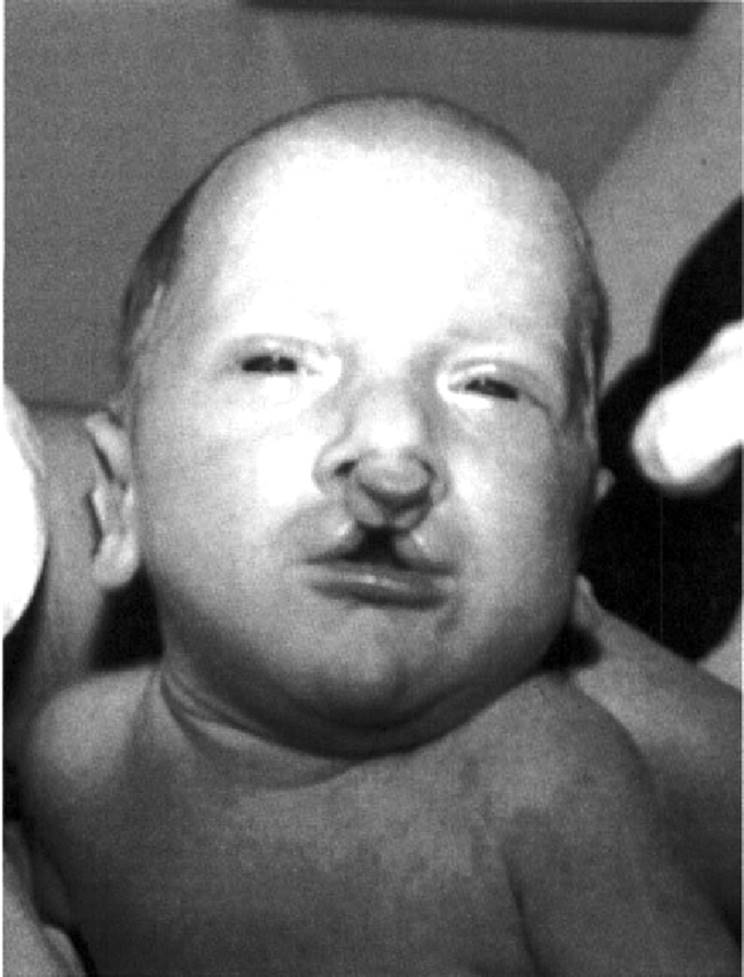
Often, CL and CP occur concurrently. In most cases of CLP, there is a cleft of both the primary and secondary palates. Again, when there is a unilateral CLP, the cleft of the primary palate occurs on the side of the CL (Fig. 5.2). There are uncommon cases of CL with intact primary palate but clefting of the secondary palate. Such cases are often syndromic, and one must exclude associations, sequences (especially Robin sequence), and other associated anomalies. Bilateral CLP is the most severe presentation (Fig. 5.3). In this classification, there is involvement of the primary palate on the left and the right with, in most cases, clefting of the secondary palate. Although possible in patients with CLP, there is no specific pattern of expected minor anomalies. Probably, the most common minor anomaly in patients with nonsyndromic CL/P is hypertelorism. This also may be a sign of an underlying genetic condition, such as the Opitz G/BBB syndrome (Saal, 2000).
Recurrence risk for nonsyndromic CLP depends on several factors, including severity of the cleft, number of affected relatives, gender of the affected individual, and degree of genetic relationship to the affected individual (e.g., first-degree relatives have higher risk than second- and third-degree relatives). As noted, more severe clefts are associated with a higher recurrence risk. For example, for a child with a unilateral incomplete CL and a negative family history for clefts, the recurrence risk would be 3%. However, if the child were born with a bilateral complete CLP, the recurrence risk would be 5% (Gorlin et al., 1990; Harper, 1993; Bender, 2000).
Cleft Palate
Nonsyndromic CP refers to abnormal development of the secondary palate. The prevalence at birth of CP is estimated to be 1 in 2000 live births (Murray, 1995; Wyszynski et al., 1996). In contrast to CL/P, where there are obvious racial and ethnic genetic factors that contribute to the prevalence in certain populations, CP has no such population propensity. Instead, the prevalence of CP is similar in all populations studied.
|
|
|
FIG. 5.2. Infant with unilateral complete cleft lip and cleft palate. |
|
|
|
FIG. 5.3. Infant with complete bilateral cleft lip and cleft palate. |
CP is genetically distinct from CL/P. In families and individuals with nonsyndromic CL/P, the increased recurrence risk is for CL/P; however, the risk for having a child with CP remains at the general population level (1 in 2000). The same is true for families and individuals with nonsyndromic CP. Moreover, when individuals in the same family are born with CL/P and CP, one must look for an underlying genetic etiology, such as Van der Woude's syndrome, one of a very few syndromes where one can see both CP and CL/P (Murray, 1995).
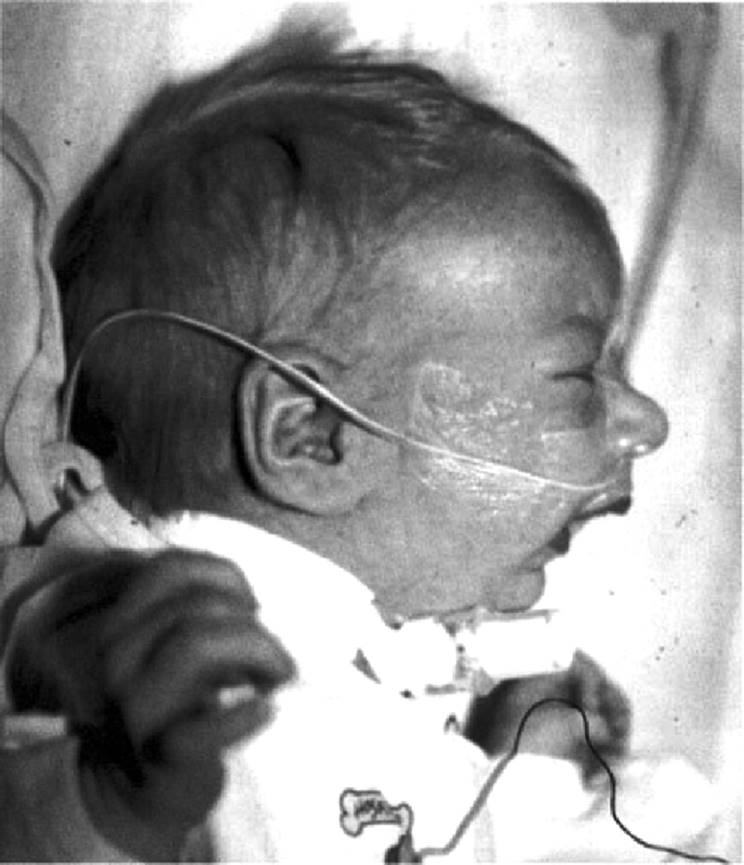
Nonsyndromic CP is pathogenetically unrelated to the CP in patients with CLP. In CLP, the CP is a secondary phenomenon related to failure of the lip to close (Trasler and Fraser, 1977). It may be a primary malformation, reflecting abnormal palate development in one of the many critical steps including elevation, merging, and fusion of the palatal shelves. It may also be a consequence of other events, which do not primarily involve palate development, such as the Robin sequence. This condition is related to developmental factors that cause either retrognathia or micrognathia, with secondary posterior positioning of the tongue. This, in turn, may cause interference with elevation and merging of the lateral palatal shelves (Fig. 5.4) (Cohen, 1999). In the classic presentation, the CP is U-shaped, reflecting fusion of the palatal shelves around the tongue (Fig. 5.5). Another complication of the Robin sequence is respiratory distress from pharyngeal airway obstruction from the posteriorly placed tongue (glossoptosis). Many cases of Robin sequence will present with a V-shaped CP, making it difficult to distinguish between CP as a malformation and as a component of the Robin sequence. Children with sequence and V-shaped CP remain at high risk for glossoptosis. Therefore, it is often not possible to clinically differentiate CP as a malformation from secondary component of a sequence of events related to micrognathia or retrognathia (Cohen, 1999).
|
|
|
FIG. 5.4. Infant with micrognathia and Pierre Robin sequence. |
|
|
|
FIG. 5.5. Cleft palate secondary to Pierre Robin sequence. Note Ushaped cleft of the secondary palate. |
An additional difficulty in categorizing CP is classifying submucous CP and bifid uvula. Submucous CP appears to represent a microform or forme fruste of CP. As in CP, approximately 50% of cases of submucous CP are associated with underlying syndromes (Saal, 2000).
In contrast to CLP, CP is much more likely to be syndromic or associated with additional malformations (Jones, 1988,2000; Tolarova and Cervenka, 1998; Saal, 2000; Stoll et al., 2000). In several studies, the incidence of nonsyndromic CP has ranged from 41% to to 55% (Jones, 2000; Saal, 2000; Stoll et al., 2000). The most common diagnosis is Stickler's syndrome (Saal, 2000). Stickler's syndrome has also been identified as the most common syndrome causing Robin sequence (Hanson and Smith, 1975; Cohen, 1979; Gorlin et al., 1990). It is genetically heterogeneous, with at least three genes identified (Brunner et al., 1994; Annunen et al., 1999; Martin et al., 1999; Snead and Yates, 1999). With both ocular and nonocular forms, the diagnosis may be difficult to make in infants and young children with what appear to be nonsyndromic CP. This is especially true in nonocular Stickler's syndrome, where the patient may have a mildly affected parent or a spontaneous gene mutation and absence of myopia in infancy or early childhood as a diagnostic sign.
Therefore, the diagnosis of nonsyndromic CP is challenging. In many cases, the final diagnosis of nonsyndromic CP cannot be made in infancy and must be delayed until later childhood, emphasizing the need for clinical genetics follow-up. This is especially true when there are associated developmental disabilities, including mental retardation, since these clinical signs may not become evident for several years.
Conclusions
Oral clefts are among the most common birth defects. The causes of CL/P and CP are heterogeneous and include genetic and environmental etiologies. To provide optimal long-term management and adequate genetic counseling and recurrence risks, identification of the etiology of the OC is essential. A large percentage of OC are syndromic, approximately 25% of CL/P and 50% of CP cases. Therefore, the diagnosis of isolated, nonsyndromic OC is one of exclusion of other diagnoses. This has significant implications for physicians, patients, and families since management and genetic counseling issues are often closely tied to specific diagnosis. This also has implications for those who do research in the area of OCs since much of the genetic research depends on an accurate diagnosis.
References
Annunen, S, Korkko, J, Czarny, M, et al. (1999). Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet 65: 974–983.
Bender, PL (2000). Genetics of cleft lip and palate. J Pediatr Nurs 15: 242–249.
Brunner, HG, van Beersum, SE, Warman, ML, et al. (1994). A Stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum Mol Genet 3: 1561–1564.
Cembrano, JRJ, Vera, JS, Joaquino, JB, et al. (1995). Familial risk of recurrence of cleft lip and palate. Philippine J Surg Spec 50: 37–40.
Chung, CS, Mi, MP, Beechert, AM (1987). Genetic epidemiology of cleft lip with or without cleft palate in the population of Hawaii. Genet Epidemiol 4: 415–423.
Cohen, MM Jr (1979). Syndromology's message for craniofacial biology. J Maxillofac Surg 7: 89–109.
Cohen, MM Jr (1999). Robin sequences and complexes: causal heterogeneity and pathogenetic/phenotypic variability [editorial]. Am J Med Genet 84: 311–315.
Cohen, MM Jr (2000). Etiology and pathogenesis of orofacial clefting. Oral Maxillofac Clin North Am 12: 379–397.
Croen, LA, Shaw, GM, Wasserman, CR, Tolarova, MM (1998). Racial and ethnic variations in the prevalence of orofacial clefts in California, 1983–1992. Am J Med Genet 79: 2–7.
Fogh-Andersen, P (1942). Inheritance of Harelip and Cleft Palate. Copenhagen: Munksgaard.
Fogh-Andersen, P (1971). Epidemiology and etiology of clefts. In Birth Defects: Original Article Series (Volume VII, Part 7), edited by D. Bergsma. Baltimore: Williams and Wilkins Company, pp. 50–53.
Gorlin, R, Cohen, MJ, Levin, L (1990). Syndromes of the Head and Neck, 3rd ed. New York: Oxford University Press.
Hanson, JW, Smith, DW (1975). U-shaped palatal defect in the Robin anomalad: developmental and clinical relevance. J Pediatr 87: 30–33.
Harper, PS (1993). Practical Genetic Counseling, 4th ed. Oxford: Butterworth-Heinemann.
Jones, K (1997). Smith's Recognizable Patterns of Human Malformation, 5th ed. Philadelphia: Saunders.
Jones, MC (1988). Etiology of facial clefts: prospective evaluation of 428 patients. Cleft Palate J 25: 16–20.
Jones, MC (2000). Cleft lip with or without cleft palate and cleft palate alone: a clinic based population revisited to determine the frequency of multiple malformation syndromes within the population and to define subgroups among individuals with isolated clefts. Presented at the David W. Smith Workshop on Morphogenesis and Malformations, San Diego, August 9, 2000.
Leppig, KA, Werler, MM, Cann, CI, et al. (1987). Predictive value of minor anomalies. I. Association with major malformations. J Pediatr 110: 531–537.
Leppig, KA, Werler, MM, Cann, CI, et al. (1988). Minor malformations: significant or insignificant [letter]. Am J Dis Child 242: 1274.
Lettieri, J (1993). Human Malformations and Related Anomalies, edited by RE Stevenson, JG Hall, and RM Goodman. New York: Oxford University Press, pp. 367–381.
Marden, PM, Smith, DW, McDonald, MJ (1964). Congenital anomalies in the newborn infant including minor variations. J Pediatr 64: 357.
Martin, S, Richards, AJ, Yates, JR, et al. (1999). Stickler syndrome: further mutations in COL11A1 and evidence for additional locus heterogeneity. Eur J Hum Genet 7: 807–814.
Milerad, J, Larson, O, et al. (1997). Associated malformations in infants with cleft lip and palate: a prospective, population-based study. Pediatrics 200: 180–186.
Millard, DR Jr (1976). Cleft Craft, 1st ed., vol. 1. Boston: Little, Brown.
Murray, JC (1995). Face facts: genes, environment, and clefts [editorial; comment]. Am J Hum Genet 57: 227–232.
Saal, HM (1998). Syndromes and malformations associated with cleft lip with or without cleft palate. Am J Hum Genet 64: All8.
Saal, HM (2000). A prospective analysis of cleft palate: associated syndromes and malformations. Presented at the XXI David W. Smith Workshop on Morphogenesis and Malformations, San Diego, August 9, 2000.
Snead, MP, Yates, JR (1999). Clinical and molecular genetics of Stickler syndrome. J Med Genet 36: 353–359.
Spranger, J, Benirschke, K, Hall, JG, et al. (1982). Errors of morphogenesis: concepts and terms. Recommendations of an international working group. J Pediatr 200: 160–165.
Stoll, C, Alembik, Y, Dott, B, Roth, MP (2000). Associated malformations in cases with oral clefts. Cleft Palate Craniofac J 37: 41–47.
Tolarova, M (1987). Orofacial clefts in Czechoslovakia. Incidence, genetics and prevention of cleft lip and palate over a 19 year period. Scand J Plast Reconstructive Surg 22: 19–25.
Tolarova, MM, Cervenka, J (1998). Classification and birth prevalence of orofacial clefts. Am J Med Genet 75: 126–137.
Trasler, DG, Fraser, EC (1977). Time-position relationships with particular reference to cleft lip and cleft palate. In: Handbook of Teratology, Vol. 2, edited by JG Wilson and EC Eraser. New York: Plenum Press, 265–281.
Vanderas, AP (1987). Incidence of cleft lip, cleft palate, and cleft lip and palate among races: a review. Cleft Palate J 24: 216–225.
Winter, RM, Baraitser, M (1996). London Dysmorphology Database. London: Oxford University Press.
Wyszynski, DF, Beaty, TH, Maestri, NE (1996). Genetics of nonsyndromic oral clefts revisited. Cleft Palate Craniofac J 33: 16406–16417.