Carl W. Peters
Mihae Yu
Robert N. Sladen
Andrea Gabrielli
A. Joseph Layon
Immediate Concerns
Major Problems
The acute respiratory distress syndrome (ARDS) is characterized by nonhydrostatic pulmonary edema and hypoxemia associated with a variety of etiologies that cause both direct and indirect insults to the lungs. The process develops acutely (usually within 72 hours of the precipitating event), requires immediate recognition, and often leads to death despite maximal medical support. The therapeutic goals in the setting of ARDS are to provide appropriate resuscitation measures and to quickly identify, address, and eliminate the precipitating event if possible. Adequate tissue perfusion and oxygenation must be maintained to support vital organs. Prevention of complications and the prompt recognition of their presence are critical to prevent late deaths (1).
Stress Points
1. ARDS is commonly seen with the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS).
2. Risk factors for developing ARDS include SIRS, sepsis, pulmonary contusion, aspiration, inhalation of toxic substances, near-drowning, long bone fractures, pancreatitis, diffuse pneumonia, and multiple blood transfusions.
3. Most patients with ARDS demonstrate similar clinical and pathologic features, irrespective of the cause of the acute lung injury (ALI).
4. The lung's response to injury can be divided into an exudative phase, a proliferative phase, and a fibrotic phase.
5. A variety of inflammatory mediators have been implicated in the pathogenesis of ALI.
6. The neutrophil plays a central role in ALI.
7. The severe hypoxemia associated with this syndrome is caused by intrapulmonary shunting that occurs with interstitial edema, and alveolar flooding and collapse.
8. A reduction in functional residual capacity (FRC) and lung compliance are the hallmarks of ARDS.
9. Radiographic changes seen in patients with ARDS are characteristic but nonspecific, and rarely reveal the etiology of the syndrome.
Essential Diagnostic Tests and Procedures
1. History and physical examination
2. Chest radiograph
3. Arterial blood gas measurements
4. Further diagnostic tests based on the clinical circumstances
Initial Therapy
1. Most patients require early endotracheal intubation and positive pressure ventilation.
2. The goal of mechanical ventilation is to provide adequate oxygenation and carbon dioxide elimination while keeping complications, such as oxygen toxicity, ventilator-associated lung injury, and hemodynamic compromise, to a minimum.
Overview
ARDS is a devastating injury to the lungs, characterized by diffuse pulmonary inflammation, hypoxemia, and respiratory distress. ARDS was described by Ashbaugh et al. in 1967 (2), although the syndrome of acute pulmonary failure was recognized by military physicians during World War I (3). Initially described as acute respiratory failure, the constellation of the signs and symptoms were first termed “adult” respiratory distress syndrome (4). In 1994, the American-European Consensus Committee on ARDS changed the word “adult” back to “acute” because development of ARDS was not restricted to adults (5). The consensus meeting further defined diagnostic criteria to include (a) acute onset; (b) bilateral radiographic infiltrates; (c) pulmonary artery occlusion pressure (PAOP) ≤18 mm Hg, or no evidence of left atrial hypertension; and (d) PaO2/FiO2 ratio of ≤300 mm Hg for ALI and ≤200 mm Hg for ARDS.
This definition is far from perfect. Respiratory distress, characterized by tachypnea, dyspnea, and acute respiratory alkalosis not relieved by correcting hypoxemia, is common to many pulmonary processes. Bilateral radiographic infiltrates may be seen with cardiogenic edema, pneumonitis, and several other entities. The PaO2/FiO2 ratio may be influenced by therapy, especially positive end-expiratory pressure (PEEP) and the FiO2 itself. It seems specious to separate “acute lung injury” from ARDS when the two terms reflect only somewhat different severity of the same processes. Heart failure may be present at a PAOP <18 mm Hg and may coexist with ARDS, but heart failure may not be present with a PAOP of 18 mm Hg or higher. Nonetheless, although presently undergoing revision, this definition has stood the test of time and forms the basis for all investigation done on ARDS in the past decade.
|
Table 136.1 Synonyms of ARDS |
||
|
A long list of synonyms of this syndrome exists in the literature, usually describing either the clinical situation or radiologic and pathologic changes (Table 136.1) (6). To better define the range of lung injury, Murray et al. in 1988 described the Lung Injury Score (LIS) based on chest radiographic findings, degree of hypoxemia (using PaO2/FiO2 values), compliance of the pulmonary system (if ventilated), and PEEP levels (7) (Table 136.2). A patient was considered to have ARDS if the score was >2.5. Whether this scoring system contributes additional descriptive value is debatable, since the mortality rate was impacted more by the comorbidities, such as sepsis or cirrhosis (8,9), than by the LIS value, and the LIS did not add accuracy to the definitions of the consensus statement (10).
Multiple risk factors for ALI/ARDS have been identified, with sepsis syndrome having the highest prevalence (30%–50%) (10,11,12,13,14,15,16). The pathogenesis for pulmonary and extrapulmonary causes for ALI may be different (17); the Consensus Committee categorized ARDS into direct versus indirect causes (Tables 136.3A and 136.3B) (5). Secondary predisposing factors described in the literature are alcohol abuse, chronic lung disease, and a low systemic pH (14).
While ARDS is usually considered a homogeneous entity, it should be considered the final common pathway of a very heterogeneous group of insults. Although the pulmonary injury is widespread, it does not uniformly affect lung tissue; this nonuniformity has important therapeutic consequences. There are also two broad etiologies of ARDS (17): In pulmonary ARDS (generally corresponding to “direct” disease), there is primary lung injury (e.g., pneumonia) that involves the alveolar epithelium, and may be confined to single organ failure.
In extrapulmonary ARDS (generally corresponding to “indirect” disease), the inflammatory effect of a remote insult—usually sepsis—reaches the capillary endothelium via a SIRS phenomenon, and lung failure becomes one more component of MODS. Although there are important differences in pathophysiology, the outcome between ARDS of pulmonary and extrapulmonary origin does not appear to differ greatly. While the vast majority of studies reviewed here consider ARDS to be a single entity, questions remain as to whether this is true.
|
Table 136.2 Lung Injury Score |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Table 136.3A Major Categories of ARDS Risk |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Table 136.3B Conditions Associated with ARDS |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The reported incidence of ARDS is variable. In 1972, the National Heart and Lung Institute Task Force on Respiratory Disease estimated the incidence to be 150,000 cases per year, or 71 patients per 100,000 people. Although the “true” incidence of ARDS as defined by the LIS may be lower—1.5 to 8 cases per 100,000 people—the incidence of ALI was found to be 89 cases per 100,000, which approximates the previous value (15,18). A more recent study reported the incidence of ALI to be 78.8 cases per 100,000 and for ARDS to be 58.7 cases per 100,000 (16).
Outcome
The cause of death in ARDS patients is more often associated with MODS than deficient oxygenation. The overall mortality rate has declined from 68% in the 1980s to 36% in 1993 (18), and presently ranges widely from 30% to 58% (4,13,16,18,19,20,21,22,23,24), depending on the specific patient group—based on age and etiology of lung injury—being studied. ARDS patients who leave the hospital seem to have no increased risk of subsequent death when matched for comorbidities (25).
Families and intensive care unit (ICU) patients frequently ask about the long-term outcomes and quality of life after ARDS. As with all heterogeneous diseases, outcome varies. Lung mechanics in ALI/ARDS survivors may return to normal in the year after hospital discharge, but pulmonary gas exchange abnormalities may persist (26). Spirometry is likely to be normal at 6 months, but the Short Form General Health Survey (SF-36) score was low in one study (27). Mild to moderate deterioration in health-related quality of life (QOL), as measured by the Sickness Impact Profile, has been reported (28). Thus, ARDS appears to add a functional burden of reduced QOL to survivors compared to non-ARDS patients who survived a major illness (29,30); nonetheless, as many as 78% of patients return to work (27). Determining whether the quality of life is “good” after a devastating illness is likely a personal decision.
Patients with ARDS who die within the first several days do so because of the underlying condition and respiratory failure. Many of those who survive the original insult succumb to sepsis or MODS. Of those who survive ARDS, most return to their premorbid state of respiratory function by about 6 months after extubation (28).
Pathophysiology
The inciting process in ALI is the pathologic loss of integrity of the alveolar–capillary membrane complex associated with exuberant inflammation, with increased endothelial and epithelial permeability and leakage of proteinaceous edema and cellular components into the interstitial and alveolar spaces. This occurs in response to some provocative stimulus, which may arise from various disease processes or physical or chemical insults, including primary pulmonary or extrapulmonary events (Tables 136.3A and 136.3B). While the details of lung injury may differ between primary and secondary causes (31), the differences in overt clinical consequences are difficult to identify when comparing patients from either general category.
The initial acute event that induces disruption of the alveolar epithelial or capillary endothelial cells in the exudative phase of ARDS yields denuded alveolar basement membrane and dysfunctional or destroyed surfactant and type 1 and 2 pneumocytes (Table 136.4). Demarginated “activated” neutrophils within the pulmonary circulation release inflammatory mediators, degrading the integrity of capillary endothelial cell junctions and allowing the influx of proteinaceous plasma fluid, erythrocytes, and inflammatory cells into the interstitium (32,33). Interstitial fluid volume eventually exceeds lymphatic clearance capabilities, flooding the alveoli with hemorrhagic plasma. Thickened interstitium is “stiffer” and worsens pulmonary compliance, yielding a scenario of restrictive physiology. Loss and dysfunction of surfactant (34) increases alveolar surface tension, thus producing alveolar collapse. Ongoing inflammation initiates the coagulation cascade within the microcapillaries, with platelet deposition (35) obliterating the capillary luminal cross-sectional area, disrupting blood flow, and raising pulmonary artery pressure. Further recruitment of activated neutrophils into the interstitium (32,33) augments the inflammatory cycle of capillary permeability, interstitial edema, and continuous alveolar macrophage activation (36) (Fig. 136.1).
|
Table 136.4 Histopathologic Changes in ARDS |
||||||||||||||||
|
Accumulation of proinflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-8 in the alveolar fluid of ARDS patients (37) portends the amplified production of cytokine and toxic reactive oxygen and nitrogen radical species (38,39) (Table 136.5). Activated complement components accumulate with fibrin and immunoglobulins to form alveolar hyaline membranes, further worsening compliance. Fibroproliferation and accelerated collagen deposition may begin early in the inflammatory sequence and continue into the proliferative phase (7–21 days) (40,41), with thickening of the alveolar walls already denuded of type 1 pneumocytes (36,42).
|
|
||||||||||||||||||||||||||||||||||
|
Figure 136.1. Pictorial detail of the pathogenesis of acute respiratory distress syndrome. (With permission from Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202[2]:145–156.) |
||||||||||||||||||||||||||||||||||
|
Table 136.5 Inflammatory mediators in ARDS |
||||||||||||||||||||||||||||||||||
|
While the original inciting event may resolve, judicious correction of persistent metabolic and infectious issues, and meticulous attention to appropriate ventilatory techniques, must continue in order to minimize iatrogenic contributions to self-sustaining inflammation (see Ventilator Associated Lung Injury below). Evolution into the fibrotic phase occurs, generally, after 3 to 4 weeks. Variable degrees of fibrosis and parenchymal tissue loss (40) yield “diffuse alveolar damage,” the histologic correlate of advanced ARDS, characterized by widespread and severe damage to the alveolar–capillary unit (40). Micro- and macrocystic areas abut dilated ectatic bronchi, with fibrotic noncompliant septa and collapsed alveoli—no longer tethered open by healthy surrounding tissue—and interwoven with thrombosed capillaries that provide no capacity for gas exchange (i.e., dead space ventilation) (40). Hypoxemia from tenaciously collapsed, fibrotic, shunt-producing alveoli accompanies the hypercarbia and respiratory acidosis of large dead space fractions from non gas exchanging overdistended alveoli, dilated cystic areas, and thrombosed non–CO2-excreting pulmonary capillaries.
Clinical Presentation
Physical Examination
After the inciting event, several hours to a day may pass before clinically apparent respiratory failure ensues. Based on work by Gomez (43), the clinical findings in ARDS may be roughly grouped into four phases (Table 136.6). Tachypnea and tachycardia usually develop during the first 12 to 24 hours. The skin may appear moist and cyanotic. Intercostal and accessory respiratory muscles become actively involved in supporting ventilation. A dramatic increase in work of breathing can be appreciated at a glance from the bedside. High-pitched end-expiratory crackles are heard throughout all lung fields. Increasing agitation, lethargy, and obtundation may occur as the syndrome progresses. Because these clinical findings may become apparent long after hypoxemia develops, careful attention to blood gas analysis is warranted in patients at risk for ARDS.
|
Table 136.6 Progression of Clinical Findings in ARDS |
|||||||||
|
|||||||||
|
|
|||||||||
|
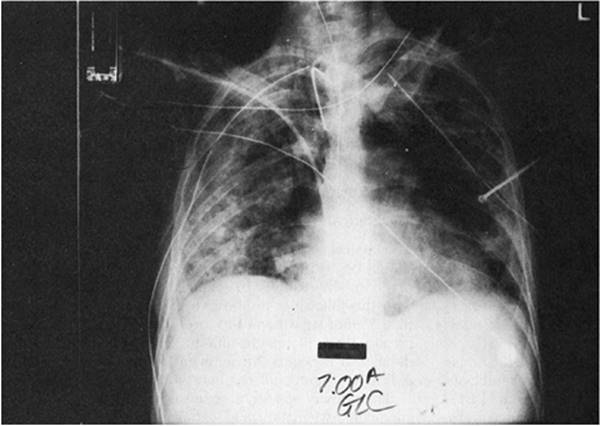
Figure 136.2. Diffuse interstitial and panacinar infiltrates are seen in a 36-year-old patient with acute respiratory distress syndrome. Also notice one of the complications of the respiratory support—a right mainstem intubation. |
Lung Imaging
The changes seen on the chest radiograph in ARDS are characteristic but nonspecific, rarely revealing the etiology of the syndrome. Acutely, pulmonary edema is seen. Interstitial infiltrates progress to a diffuse, fluffy, panacinar pattern (Fig. 136.2). Although it may be difficult to differentiate from cardiogenic pulmonary edema, there is generally an absence of pulmonary vascular redistribution, pleural effusion, or cardiomegaly. The panacinar infiltrates may consolidate and, with time, take on a patchy or nodular pattern. If the patient improves, radiographic results may revert to normal. If the disorder progresses, a pattern of diffuse interstitial fibrosis may ensue (Fig. 136.3).
Therapeutic interventions may alter the radiographic findings. Pulmonary infiltrates may increase with injudicious fluid administration. Positive pressure ventilation and PEEP may lead to hyperinflation, and subcutaneous, mediastinal, retroperitoneal, and intraperitoneal emphysema, or pneumothorax. Mainstem bronchus intubation may lead to ipsilateral pneumothorax or contralateral lung collapse (Fig. 136.4).
|
|
|
Figure 136.3. A pattern of diffuse interstitial fibrosis has developed in this 52-year-old patient with acute respiratory distress syndrome. |
|
|
|
Figure 136.4. This 70-year-old patient with acute respiratory distress syndrome has a right tension pneumothorax and right mainstem intubation. |
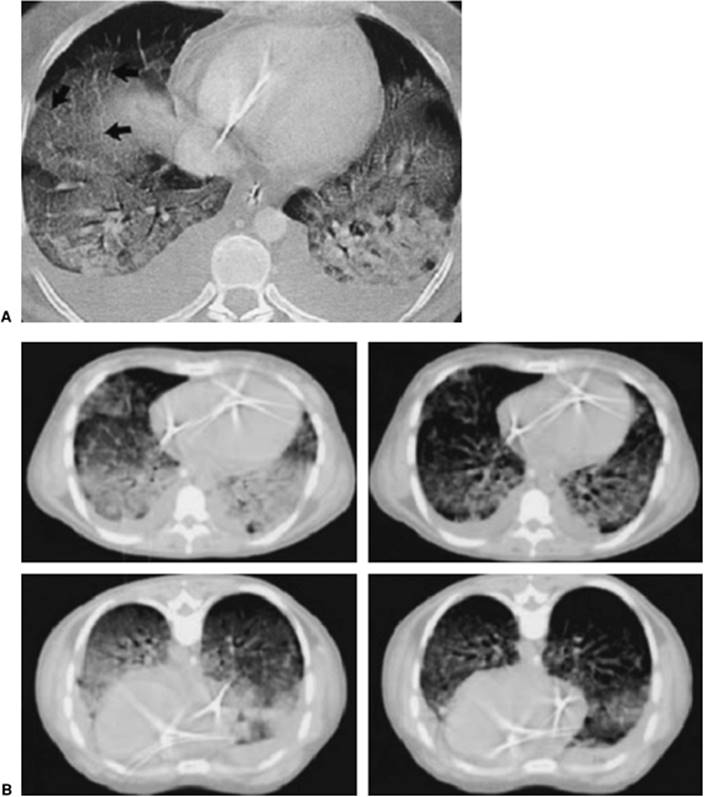
Whereas a two-dimensional chest radiograph may suggest diffuse homogeneous infiltrates, the chest computed tomography (CT) scan usually demonstrates remarkably inhomogeneous lung involvement. Dependent regions of the lung appear to be much more involved than nondependent regions. Although chest CT scanning is not always practical in the day-to-day management of patients with ARDS, in investigational trials, it has provided a vivid image of dramatically reduced lung volumes. The chest CT also may be useful in demonstrating the presence and magnitude of pneumothoraces and pleural effusions not well visualized on the standard chest radiograph. It is also useful for the positioning of thoracostomy tubes in patients with loculated pneumothoraces.
Treatment
General Therapeutic Measures
Nutritional Support
The gut serves a critical function beyond the absorption and transport of nutrients. Enteral nutrition seems to have an advantage over parenteral nutrition in preventing gastrointestinal atrophy, maintaining normal gut flora, and preserving immune function in surgical patients (44). Chapters 64 and 65 detail the importance of nutrition in the critically ill.
Fluid Management
Fluid management in ARDS has been controversial. As the permeability of the alveolar–capillary membrane increases, pulmonary edema develops at lower pulmonary capillary pressures. The Starling equation predicts mathematically what is seen clinically. When a strategy of fluid restriction and diuresis is undertaken, extravascular lung water (EVLW) is decreased, as is the duration of mechanical ventilation; mortality in ARDS seems to be associated with net fluid gain. Adequate intravascular volume must be maintained to avoid tissue hypoperfusion, although we recommend that the minimal amount of fluid be given, and that judicious attempts at diuresis be undertaken in the hemodynamically stable patient (45). A large study conducted by the National Heart, Lung, and Blood Institute (NHLBI) Acute Respiratory Distress Syndrome Clinical Trials Network (46) found no difference in 60-day mortality when comparing liberal and conservative fluid management strategies. Although the time allowed to enrollment was long (48 hours) and may not have captured the initial resuscitation, in light of the shorter ventilator and ICU days with conservative fluid management and associated improvement in pulmonary function when compared to liberal use of fluid, our routine practice is a conservative fluid strategy.
Bronchodilators
Multiple factors may lead to airflow obstruction in patients with ARDS, including mucosal and interstitial edema, airway secretions, and atelectasis. Airway hyperreactivity also contributes to increased airflow resistance in many patients with ARDS, in both the acute and chronic phases. Aerosolized β-agonists can decrease airway resistance, even in patients without underlying chronic obstructive pulmonary disease or asthma. By reducing airway resistance, the work of breathing can be decreased. We recommend a therapeutic trial of inhaled bronchodilators in patients with wheezing, in those with increased resistance as measured directly, or in patients with high peak airway pressures (47).
Steroids
The use of corticosteroids in the treatment of the various phases of ARDS is a basis of controversy and ongoing investigation.
The cytokine-mediated inflammatory response to an inciting event in ARDS intuitively suggests that suppression of that response would be therapeutic, but studies are equivocal in reporting benefit. Steroid use in different phases of ARDS has been meticulously investigated, but the dynamic nature of the inflammatory process has made the findings in individual studies difficult to extrapolate to varying illnesses at varying times. Furthermore, infectious risks of corticosteroid use aside, their prolonged use risks profoundly detrimental neuromuscular effects, even further compounded when employed with nondepolarizing neuromuscular blocking agents—often utilized in ARDS patients to facilitate efficient mechanical ventilation (48). Thus, routine use of corticosteroids is not advocated, especially in the acute phase of ARDS. During the late phase, fibroproliferation often occurs in response to tissue injury and is associated with persistent inflammation. In this setting, fever and SIRS are present in the absence of infection. A small uncontrolled trial suggested that improvement in “late” ARDS patients—those mechanically ventilated for ~15 days—with progressive fibroproliferation may be seen when corticosteroid treatment begins during that period (49). Proponents of this therapy recommend that a trial of corticosteroids be instituted in such patients after infection has been excluded. More recently, the NHLBI ARDS Clinical Trials Network (50) conducted a randomized multicenter controlled trial of steroid use in 180 patients with ARDS of at least 7 days' duration. While there was no difference overall in mortality at 60 and 180 days, steroids imparted a higher number of ventilator-free days and earlier departure from the ICU in the first 28 days. Those given methylprednisolone after day 13 of ARDS, however, had a higher mortality than controls. Meduri et al. (51) recently found reductions in length of mechanical ventilation and ICU stay and in mortality in early septic/ARDS patients receiving “low-dose” methylprednisolone infusions. While there have been several recent reviews and meta-analyses addressing corticosteroid use in ARDS (52,53,54,55,56), varying population groups and treatment regimens and differing end points and definitions of “success” in the studies make broadly inclusive recommendations difficult to formulate. Even the impact of steroids on mortality varies positively or negatively with different groups of patients. In general, corticosteroids are not effective in ameliorating cytokine-induced inflammation in ARDS in a clinically significant way, and routine use of corticosteroids is, therefore, not advocated, especially in the acute phase. There are, however, some subgroups of patients upon which corticosteroids may have a positive effect. One example may be the late phase, during which fibroproliferation often occurs in response to tissue injury. This response is damaging to the lung and is associated with persistent cytokine-mediated inflammation (57). Lung injury is characterized by endothelial and epithelial damage, as well as augmented fibroblast proliferation, which may be lessened by steroid treatment. Proponents of this therapy recommend that a trial of corticosteroids be instituted in patients with severe ARDS after infection has been excluded (49).
Monitoring
Monitoring the patient with ARDS is similar to that performed on other critically ill patients (Table 136.7). Chapters 16 through 22 detail descriptions of monitoring techniques that are essential to reduce or prevent the occurrence of significant complications. Careful titration of therapy is best guided by monitoring clinical, laboratory, and cardiorespiratory variables. Our standard practice is to always use a minimum of an arterial line, pulse oximetry, and capnography in patients with ALI/ARDS. More invasive devices—central venous pressure (CVP), pulmonary artery catheter—may be required based on the clinical situation.
|
Table 136.7 Monitoring the Patient with ARDSa |
||||||||||||||||||||
|
Standard Management
Progress has been made in the management of ARDS, as suggested by the number of large studies and meta-analyses published in the last decade. Over this time, data have been gathered addressing modes of therapy and ancillary support techniques previously initiated and practiced empirically.
Despite considerable progress, however, many questions still await definitive resolution, as will become apparent in the discussion that follows. Due to the complex metabolic and pulmonary aberrations that characterize ARDS/ALI, treatment strategies can be divided into those directed toward respiratory support and all other therapeutic measures.
Respiratory Support
Mechanical ventilatory support, most often via an endotracheal tube, is fundamental to the management of ARDS, as perturbations of gas exchange and respiratory mechanics associated with this syndrome exceed the limits of compensation that most individuals are able to muster without mechanical assistance; it is as fundamental and integral to the management of the patient with ALI as is exogenous insulin to the diabetic or antibiotics to the treatment of infections. The indications for respiratory support are well defined (58), and include hemodynamic instability, protection and maintenance of the airway, inability to maintain PaO2 above 55 mm Hg on an FiO2 = 60%, need for positive airway pressure, and progressive ventilatory insufficiency with rising respiratory rate and hypercarbia. The presence of several or all of these features in most individuals with ALI mandates endotracheal intubation and mechanical ventilation to optimize gas exchange and minimize work of breathing. Noninvasive positive pressure ventilation (NIPPV) has been employed in some instances for those with less severe pulmonary impairment and preserved mental status (59), although studies are few with fairly high rates of eventual endotracheal intubation (60,61). Broad recommendations regarding the use of NIPPV in ARDS are difficult to make in the absence of large prospective studies due to the heterogeneity of patient populations, comorbidities, and diversity of the inciting pathophysiology (61).
Lung CT studies have demonstrated the distribution of areas of alveolar collapse and distention characteristic of ARDS to be regional rather than diffuse. Alveolar collapse predominates in dependent areas, producing venous admixture and hypoxemia, while nondependent areas manifest airway destruction with hyperinflation, often to the point of exclusion of pulmonary capillary blood flow (dead space) (62,63). These alveolar morphologies, however, are not strictly related to dependency within the chest cavity, as is clearly visible in Figure 136.5. Areas of atelectasis, producing shunt (solid arrows) and airway/alveolar destruction, and areas of overdistention, producing dead space (dashed arrows), may be randomly distributed and interspersed with areas of spared pulmonary tissue, thereby generating profound ventilation/perfusion mismatch.
The use of mechanical ventilation in ALI/ARDS has evolved dramatically over the last 30 years. Techniques of mechanical ventilation (MV) commonly employed through the decade of the 1980s led to use of what would now be described by most practitioners and investigators as “high” tidal volumes, with FiO2 supplemented well above ambient. Subsequently, the observation was made (64,65) that ventilation of healthy laboratory animals with high tidal volumes induced profound clinical and histologic deterioration that was difficult to distinguish from those of ARDS. In 1990, Hickling et al. noted improved mortality in ARDS patients with lower than “traditional” tidal volumes (66). Subsequent investigations yielding conflicting results mandated the ARMA (Acute Respiratory Distress Syndrome Network Low Tidal Volume) trial of mechanical ventilation, with limited tidal volume and plateau pressures compared to the higher values in common use at the time (67). The result was a reduction in mortality from 40% to 31% with the experimental protocol parameters. While the ARMA study has been criticized from a number of standpoints (68), none is sufficiently compelling to negate the persuasiveness of its results. In our practice, low tidal volume ventilation (Vt = 6–8 mL/kg ideal body weight) is considered standard, maintaining a plateau pressure <30 cm H2O. Tidal volumes exceeding these parameters have been implicated in generating lung injury caused by mechanical ventilation itself. This phenomenon, termed ventilator-associated lung injury (VALI), is a by-product of the interaction of mechanical ventilation and the cytokine proliferation that is a fundamental pathophysiologic feature of ARDS (69,70). As described below, components of VALI include (a) barotrauma, the appearance of air outside the airways and alveoli, attributed to airway pressures that exceed certain thresholds (71,72); (b) volutrauma, increased alveolar and capillary permeability due to alveolar overdistention and leading to pulmonary edema (73); and (c) atelectrauma, the destructive repetitive opening and closing of stiff, collapsed, surfactant-depleted, fibrotic alveoli with thickened interstitium that are associated with cyclic positive pressure ventilation (74). Excessive alveolar stretch is associated with inflammatory cytokine proliferation, in particular during excursions into ranges of tidal volume that induce VALI (74,75) (Fig. 136.6). Of note, this cytokine proliferation can be limited by using a lung-protective ventilation strategy (using a pressure–volume curve to determine tidal volume and PEEP) (75).
|
|
|
Figure 136.5. Computed tomography scan of the chest of a patient with acute respiratory distress syndrome. Solid arrows show dense parenchymal opacification resulting in shunt. The broken lines show relatively “normal”-appearing lung but that can suffer from overdistention, resulting in dead space. (With permission from Desai SR. Acute respiratory distress syndrome: imaging of the injured lung. Clin Radiol. 2002;57[1]:8–17.) |
The importance of limitation of tidal volume as a guide to appropriate MV in the acutely injured lung may be more easily understood when viewed within a conceptional framework of patchy, unevenly distributed alveolar injury. When such an injured lung receives a positive pressure breath, the gas distribution is impacted by the variability of compliance and resistance in the injured and healthy areas. Flow preferentially enters unaffected (i.e., low resistance, relatively high compliance) pulmonary tissue, risking unintentional overdistention and injury of these normal areas (76) despite inflation with an “appropriate” tidal volume based on body weight. This is often termed the baby lung phenomenon (77), since the volume of unaffected lung parenchyma within the ARDS patient's thorax more closely approximates that of a child than an adult. Delivered tidal volumes, therefore, must more closely approximate those appropriate for a smaller lung, usually on the order of 6 to 8 mL/kg; exceeding these volumes risks iatrogenic perpetuation of lung injury, since a positive pressure breath inflates a smaller volume of lung tissue than would be predicted by ideal body weight.
The importance of PEEP and recruitment maneuvers in providing efficient ventilator management of ARDS patients warrants further discussion. While the traditional approach of oxygen supplementation may improve the PaO2 within the limits of a marginal FRC, such supplementation should be looked upon only as a temporizing measure. Prolonged high FiO2 use risks toxicity and absorption atelectasis, while leaving the underlying cause of hypoxemia neither identified nor corrected. Recovery of FRC by reinflation of atelectatic areas using recruitment maneuvers and PEEP will restore gas flow to previously nonaerated areas of lung (78,79,80,81,82). These modalities of treatment are commonly utilized in the modern strategy of ARDS treatment (83,84). Areas of particularly tenacious atelectasis will often require an inspiratory time (Ti) equal to several inspiratory time-constants (one “time-constant” equals product of compliance and resistance, both easily measurable by current ventilators) to achieve inflation. Insufficient Ti may leave such areas persistently collapsed, worsening shunt fraction and compromising FRC and oxygenation. Despite the most heroic efforts, a substantial percentage of ARDS patients harbor lung tissue that is only variably “PEEP recruitable” (81). The benefit of recruitment maneuvers and PEEP can be understood within the context of the Law of LaPlace (actually the Young-LaPlace equation), which states that the pressure difference across a fluid interface is equal to the surface tension times the mean curvature of the surface. In pulmonary physiology and ARDS, this means that the pressure difference between alveolar gas and alveolar epithelium contracts the alveolus inward unless counteracted by surfactant. Furthermore, the relationship between surface tension and alveolar radius is inverse. Thus, the smaller the alveolar radius, the greater the force contracting it even further inward (i.e., toward collapse). Since surfactant decreases surface tension, the inward force within a collapsed alveolus is greater than that within its surfactant-replete, healthy, “noncollapsed” neighbor with lower surface tension, resulting in a temporary high-pressure requirement to open a collapsed alveolus. The alveolus may then be maintained open, with PEEP exceeding the alveolar closing pressure. Because low tidal volume (6 mL/kg) followed by PEEP in itself is generally ineffective in expanding collapsed alveoli, a “recruitment maneuver,” the temporary application of airway pressure far above any possible alveolar retractive force, may be warranted to open and stabilize collapsed alveoli (85,86,87), preventing exposure to repetitive cyclic collapse and associated destructive shear forces by maintaining an “open lung” (Fig. 136.7).
|
|
|
Figure 136.6. The effect of ventilation strategy on inflammatory mediator concentration. High tidal volume strategy resulted in higher levels. C, control: Vt = 7 mL/kg, PEEP = 3 cm H2O; MVHP, moderate-volume, high PEEP: Vt = 15 mL/kg, PEEP = 10 cm H2O; HVZP, high-volume, zero PEEP: Vt = 40 mL/kg; MVZP, moderate-volume, zero PEEP: Vt = 15 mL/kg. TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IL-6, = interleukin-6; MIP-2, macrophage inflammatory protein 2; IFNγ, interferon-γ. (With permission from Tremblay L, Valenza F, Ribeiro SP, et al. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997;99[5]:944–952.) |
The benefit of PEEP was recently addressed and revealed, surprisingly, that no difference was achieved in discharge or survival between ICU patients receiving high- or low-PEEP regimens (83). More recently, no significant improvement in all-cause mortality rates was noted in two studies of ARDS patients managed with an “open-lung” approach, involving the addition of modestly higher levels of PEEP and recruitment maneuvers compared to a regimen utilizing a tidal volume of 6 mL/kg and approximately 10 cm H2O PEEP (84). Nonetheless, most clinicians feel that utilizing a level of PEEP above the lower inflection point (LIP) on a pressure–volume curve (see below) improves FRC and oxygenation by inflating recruitable alveoli, and thus decreasing venous admixture.
|
|
|
Figure 136.7. Atelectrauma and the interdependence of lung units. (With permission from Moloney ED, Griffiths MJ. Protective ventilation of patients with acute respiratory distress syndrome. Br J Anaesth. 2004;92[2]:261–270.) |
The selection of tidal volume is intimately linked to the pressure–volume curve. Optimal gas exchange with minimal alveolar injury is achieved when the lung is positioned on the vertical portion of the pressure–volume curve (Fig. 136.8). This minimizes collapse in areas of high time-constants and overdistention in normal areas. Once alveolar re-expansion is optimized, which may take several hours of vigilance to titrate tidal volume and mean airway pressure, optimal inflation is maintained with PEEP as ventilation is then conducted along the expiratory limb of the curve, lowering mean pressures overall (Fig. 136.8). There is wide acceptance of the use of low tidal volume/limited plateau pressure ventilation techniques, directed toward gas exchange along the expiratory curve once inflation has been achieved, with the goal of preserving the integrity of pulmonary parenchyma not yet affected by inflammation and to allow healing of diseased areas. Since compliance varies between individual alveoli, a given inspiratory pressure may hold some in overdistention while others are minimally opened; the curve depicted in Figure 136.8 actually represents an averaged compliance. A not uncommon observation when monitoring gas exchange in ARDS is hypercarbia with mild acidemia, often more uncomfortable for the clinician to observe than the patient to experience. However, “permissive hypercapnia” is safe and acceptable (88) when not contraindicated by underlying medical condition (e.g., elevated intracranial pressure), though it often warrants protocol-delivered sedation. This may be understood by visualizing a variety of pressure–volume curves depicting compliance curves for variously distensible alveoli. The pressure to aerate sufficient numbers of tenaciously collapsed alveoli may overdistend more compliant areas such that the increased dead space precludes adequate ventilation.
The mode of mechanical ventilation used in ALI/ARDS is likely more dependent on the comfort level of the practitioner than on “best evidence.” Pressure control ventilation (PCV) offers the theoretical advantages of limiting peak airway pressure, a component that may be associated with ventilator-induced lung injury (70). PCV may decrease work of breathing, possibly due to the variable flow rate (89). PCV is a ventilatory mode that is time initiated, pressure limited, and time cycled. PCV delivers a square pressure wave that provides tight control of the inflation pressure equal to the applied pressure plus PEEP. This mode also allows precise adjustment of inspiratory time at the expense of expiratory time—that is, increased inspiratory to expiratory (I:E) ratio, or “inverse ratio ventilation” (IRV). Mean airway pressure is substantially increased without an increase in peak airway pressure, promoting alveolar recruitment while—again, theoretically—attenuating barotrauma and volutrauma. With PC-IRV, mean airway pressures are typically increased from less than 10 to between 20 and 30 cm H2O; inspiratory time—if the ventilator is set, for example, at 10 breaths per minute— between 3 and 5 seconds; and I:E ratio between 1:1 and 3:1. Indeed, IRV may be considered an alternative (or adjunct) to PEEP in providing airway pressure therapy during inspiration instead of expiration, and with limited peak airway pressure.
|
|
|
Figure 136.8. Pressure–volume curve of an idealized lung, showing both the inspiratory and expiratory limbs as well as the upper and lower inflection points. |
To date, the hypothesis that PC-IRV results in a better outcome than standard volume-limited ventilation has not bee rigorously tested (90,91). Moreover, IRV may result in inadequate exhalation time, air trapping, and the generation of intrinsic PEEP (“auto-PEEP”), leading to barotrauma and CO2 retention. Paradoxically, hypercarbia occurring during PC-IRV may be improved by decreasing the ventilator rate to allow additional time for CO2 elimination. Nonetheless, high-time-constant, low-compliance lung segments may benefit from the ability to control the inspiratory time and the prolonged, but controlled, plateau pressures that PCV allows (92).
Airway pressure release ventilation (APRV), also known as invasive bilevel ventilation, combines the advantages of improved alveolar recruitment, lung protection, and spontaneous ventilation. In this mode, a sustained 3- to 4-second high airway pressure—the upper PEEP level—of 20 to 30 cm H2O is intermittently released for about a second to the lower level of PEEP (5–10 cm H2O), while allowing spontaneous breathing to occur throughout the cycle (93). This technique optimizes alveolar recruitment by increasing mean airway pressure while restricting the peak airway pressure to the upper PEEP level, and can maintain oxygenation and ventilation at lower airway pressures than conventional ventilation (93). This mode is useful in the transition from PC-IRV to ventilatory weaning with IMV or pressure support, but it has not been subjected to randomized outcome trials (94,95).
Advantageous aspects of both volume- and pressure-control ventilation can be combined in advanced circuitry ventilators in a mode termed pressure-regulated volume control (VC+). This mode allows the practitioner to select the mechanical rate, tidal volume, inspiratory time, pressure support level (if desired), FiO2, PEEP level, and maximal values for peak inspiratory pressure and tidal volume. When VC+ is selected, the ventilator adjusts the pressure to deliver the desired tidal volume, changing the pressure by about 3 cm H2O every third breath or so. As compliance worsens, tidal volume is maintained up to the maximal set peak inspiratory pressure, which will not be exceeded. When compliance improves, the ventilator automatically decreases the inspiratory pressure to keep the tidal volume within the set range. A single tidal volume delivered above the set maximal tidal volume generates a ventilator alarm. Thus, the potential problems one might see with standard volume ventilation (excessive peak pressure to deliver the target tidal volume) or PC ventilation (improving compliance, producing a dangerously high tidal volume) are obviated with this mode of ventilation.
Prone Positioning
The typical pattern of distribution of inflammatory edema and alveolar inflation in ARDS is noted in Figure 136.9A. A progressive decrease in transpulmonary pressure—the force distending the alveoli, defined as the difference between alveolar pressure (PA) and pleural pressure (Ppl)—with dependency manifests itself as airway collapse in the dependent portions of the inflamed lung. When proceeding from ventral to dorsal areas in the supine position, transpulmonary pressure—the outward traction force keeping the airways “tethered” open—no longer exceeds alveolar surface tension, and collapse occurs in dependent areas. In the absence of adequate PEEP, inflation of dependent alveoli, once achieved, cannot be maintained, and inspiratory volume is preferentially directed into nondependent areas of the lung (96). Preferential distribution of perfusion to dependent areas with collapsed alveoli contributes to ventilation/perfusion mismatch and intrapulmonary shunt (97). A logical step to realign distribution of inflated alveoli with pulmonary perfusion is to turn the patient prone, alleviating many factors contributing to airway collapse. These factors include the position of cardiac mass that no longer impinges on the retrocardiac lung parenchyma, patterns of diaphragm movement, gravitational redistribution of perfusion, and chest wall mechanics (Fig. 136.9B) (96).
Several large studies have documented improvements in oxygenation without significant improvement in mortality. Considerable skill and experience are needed to pronate and support a critical patient bearing invasive monitoring and therapeutic devices. The risks to potential pressure-bearing ventral body structures—face, eyes, chest, and knees—and of accidental device removal must be weighed against the benefits (98,99,100). Proning beds are available commercially, but may have certain restrictions such as cervical spine clearance and weight limits.
High-frequency Ventilation
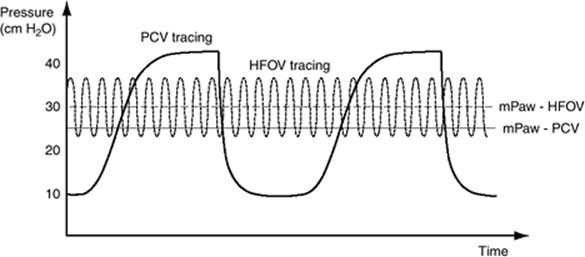
High-frequency ventilation (HFV) is a technique that minimizes the risk of VALI and atelectrauma by avoiding both excessive inspiratory volumes and repetitive airway collapse produced by conventional cyclic ventilation in the noncompliant ARDS lung, while maintaining higher mean airway pressures (Fig. 136.10).
Subcategories of HFV include high-frequency positive pressure ventilation, high-frequency oscillatory ventilation, and high-frequency jet ventilation. Respiratory rates range from 50 to 2,400 breaths/minute, the latter rate produced in oscillatory ventilation. Gas transport and exchange occur through several mechanisms, some occurring simultaneously in a given patient. These include bulk convection, to a lesser degree than in conventional ventilation; asymmetric velocity profiles, causing a simultaneous, opposite directional flow of oxygen and carbon dioxide in different regions of the airways; Pendelluft, the asynchronous filling and emptying of alveoli with nonhomogeneous time constants; Taylor dispersion, wherein shear forces augment forward gas diffusion; and mixing due to cardiogenic oscillations and molecular diffusion (101,102). Effective gas exchange may take place despite the use of tidal volumes well below dead space volume (103).
High-frequency oscillation (HFO) potentially provides lung protection in ARDS by avoiding alveolar distention and collapse (104). Oscillation is provided at rates of 180 to 900 cycles per minute—or 3 to 15 Hz, where 1 Hz = 60 cycles per minute or 1 cycle per second—with below dead space tidal volumes (0.1–0.3 mL/kg), high gas flow, and an active expiratory phase. High levels of PEEP are necessary to support the mean airway pressure and maintain alveolar recruitment. In the HFO ventilator, an adjustable power control determines the amplitude of piston displacement and peak and trough pressure excursions (ΔP) above and below the mean airway pressure. The oscillation frequency (Hz) determines the time for piston displacement, and thus a lower Hz will lead to larger bulk tidal volumes. Oxygenation is determined by the FiO2 and by mean airway pressure, whereas ventilation and CO2 elimination are determined by ΔP and oscillation frequency (Hz). Occasionally, it may be necessary to create a small endotracheal tube cuff leak to facilitate CO2 washout.
HFO provides a number of management challenges, including the necessity for a firm bed surface, with increased risk of pressure injury, and difficulty with humidification of inspired gas. Nonetheless, HFO has established itself as a ventilatory mode in pediatric ICUs and trauma units, where it facilitates ventilation in the presence of abdominal compartment syndrome and constrained lung volume (105). Thus far, only one large randomized trial has compared HFO with conventional ventilation in adults. After 2 to 4 days of conventional ventilation, 150 patients were randomized to HFO or PC-IRV (tidal volume 6–10 mL/kg) (106). Patients who received HFO had improved PaO2/FiO2 ratios at 24 hours, but there was no statistical difference in mortality—37% versus 52% (p = 0.1). Clearly, there is a need for a large randomized trial where HFO is instituted at an early stage of ARDS.
|
|
|
Figure 136.9. A: Consolidated lung in a patient with acute respiratory distress syndrome. Note the air bronchograms and dependent consolidation. (With permission from Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342[18]:1334–1349.) B: Improvement in dependent consolidation once proning has occurred. (With permission from Pelosi P, Brazzi L, Gattinoni L. Prone position in acute respiratory distress syndrome. Eur Respir J. 2002;20[4]:1017–1028.) |
Extracorporeal Life Support
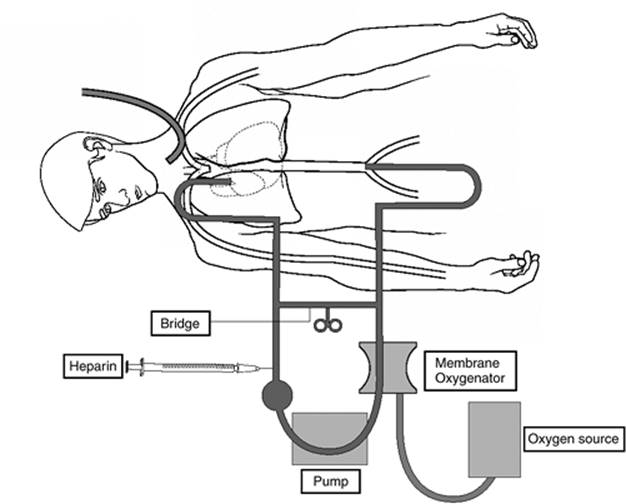
ARDS-related respiratory failure particularly refractory to the most aggressive support measures may warrant the temporary use of mechanical gas exchange devices for pulmonary support while native lung tissue recovers. The process, known as extracorporeal life support (ECLS) or ECMO (extracorporeal membrane oxygenation), employs a membrane oxygenator, blood warmer, and pump systems in parallel with components of the central circulation (Fig. 136.11), depending on the intensity of support required. Blood is withdrawn from the venous system—typically via the internal jugular vein—anticoagulated, oxygenated, decarbonated, adjusted to appropriate temperature, and then returned to the patient. Blood is returned via the femoral vein (venovenous ECLS) or the right carotid or femoral artery (venoarterial ECLS), depending on the physiologic system(s) requiring support (Fig. 136.11). Either function can be supported alone or together. ECLS is supportive only, bridging the patient's vital cardiopulmonary functions until definitive therapy is instituted. ECLS should not be used as a salvage procedure once irreversible loss of organ function is thought to have occurred (107). During the actual functioning of the bypass circuit, ventilator settings are turned to minimal, thereby avoiding the additional pulmonary insult that VALI would impart.
|
|
|
Figure 136.10. Depiction of airway pressures—peak and mean—in high-frequency oscillatory ventilation (HFOV) as compared to pressure control ventilation (PCV). (With permission from Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest. 2007;131[6]:1907–1916.) |
Initial studies, such as the U.S. ECMO trial (1974–1977), used ECMO with complete lung collapse; the unfortunate result was dismal survival (9%). Over the next 10 years, Gattinoni et al. demonstrated the effectiveness of maintaining low levels of lung ventilation (pressure limit 35 cmH2O, rate 3–5 breaths/minute) by utilizing low flow venovenous ECMO for CO2 removal (108). In Gattinoni's hands, this approach, termed low-frequency positive pressure ventilation with extracorporeal CO2 removal (LFPPV-ECCO2R), was associated with a 49% survival in very severe ARDS patients (108). In survivors, lung function improved within 48 hours. In a subsequent randomized study carried out in the United States, Morris et al. compared LFPPV-ECCO2R with PC-IRV using computerized protocols in 40 patients (109). There was no statistical significance in 30-day survival—33% versus 42% (p = 0.8)— but the study size was small.
|
|
|
Figure 136.11. Depiction of extracorporeal membrane oxygenation. (With permission from Brown JK, Haft JW, Bartlett RH, et al. Acute lung injury and acute respiratory distress syndrome: extracorporeal life support and liquid ventilation for severe acute respiratory distress syndrome in adults. Semin Respir Crit Care Med. 2006;27[4]:416–425.) |
At present, ECLS is well established in neonatology and pediatrics (110,111), but use in the adult population is less widespread. In the most experienced center, the University of Michigan at Ann Arbor (http//www.med.umich.edu/ecmo/intro.htm), consideration is given to use of ECLS when all maximally supportive measures yield an arterial-alveolar DO2 >600 with hypercarbia and persistently reduced compliance (107). Survival may exceed 50%, despite several potential complications, including coagulopathy with bleeding, stroke, pulmonary thromboembolism, ischemic bowel, sepsis, and MODS. Considerable experience and expertise in this complex, expensive, and resource-intensive procedure is required to maximize outcome. Venovenous ECMO may be a life-saving intervention in selected patients with primary ARDS, especially ischemic-perfusion injury after double lung transplantation. A salutary outcome is predicated on good cardiovascular function, the absence of MODS, and relatively rapid (<72-hour) improvement in lung function.
Inhaled Vasodilators
Fundamental to the pathophysiology of ALI/ARDS is the phenomenon of ventilation/perfusion mismatch–induced shunt-related hypoxemia. The phenomenon of hypoxic pulmonary vasoconstriction (HPV), which may be viewed evolutionarily as a mechanism to “isolate and exclude” the pathologic hypoxic collapsed alveoli, carries a price of right heart pressure elevation. The influence of HPV extends beyond the collapsed areas; well-aerated alveoli may abut remotely constricted vessels, increasing dead space ventilation and further straining the right ventricle. When airway inflation and stabilization via optimization of mechanical ventilation do not suffice to alleviate collapse, vasodilators may be employed. Intravenous agents, such as sodium nitroprusside, affect all vessels, frequently worsening hypoxemia by dilating and perfusing collapsed areas. Aerosolized vasoactive medications, such as nitric oxide or prostaglandin-I, diffuse from ventilated alveoli and result in relaxation of endothelial smooth muscle within remotely constricted vascular beds, thus improving ventilation/perfusion matching while vessels adjacent to collapsed alveoli remain unaffected. Selective vasodilation in ventilated areas decreases shunt fraction and contributes to alleviation of pulmonary hypertension (112,113) (Fig. 136.12).
|
|
|
Figure 136.12. Intravenous versus inhaled vasodilator effects on pulmonary circulation. A: Shows two idealized alveoli, one occluded (left) and the other normal; both have hypoxic pulmonary vasoconstriction (HPV)-induced decreased pulmonary blood flow. B: Shows the result of using an intravenous vasodilator: HPV is removed to both the occluded and nonoccluded alveoli, resulting in significant shunt. C: Shows the result of utilization of inhaled nitric oxide (NO): HPV is reversed in the area of the ventilated alveolus, but not the obstructed one. NTP, nitroprusside. (With permission from Lunn RJ. Inhaled nitric oxide therapy. Mayo Clin Proc. 1995;70[3]:247–255.) |
Nitric oxide (NO) was discovered to be an endogenous compound with vasoactive properties in the late 1980s (114,115). The mechanism of action is through the generation of cyclic guanosine monophosphate (cGMP) (115). Its rapid absorption and inactivation by hemoglobin restrict its effects to the pulmonary circulation (116). While clinical trials have repeatedly documented improvement in pulmonary artery pressures and oxygenation with NO in ALI/ARDS, there is no evidence that overall mortality is reduced (117,118). Haphazard use of NO is inadvisable in that substantial potential toxicities exist, including free radical formation, production of nitrogen dioxide (NO2) (119), and generation of methemoglobin. The rate of formation of NO2 from oxygen and NO depends on the concentration of oxygen and the square of the NO concentrations. The Occupational Safety and Health Administration has set safety limits of 5 ppm for NO2, as it can cause pathologic changes to the lungs at doses of 25 ppm. At extremely high doses, pulmonary edema, hemorrhage, and death have been seen in animal models. NO2 levels should be monitored as closely as possible to the endotracheal tube (120). In clinical trials using NO at 5 to 40 ppm, NO2 has not been a significant problem.
Methemoglobinemia is another potential but rare complication of NO administration. About 80% to 90% of inhaled NO is absorbed within the bloodstream, where it reacts with hemoglobin within the red blood cell to form nitrosyl-hemoglobin and methemoglobin. The primary factor determining the development of methemoglobinemia is the dose of NO, although the hemoglobin level, oxygen saturation, and methemoglobin reductase also play a role. In the United States, Native Americans more frequently have methemoglobin reductase deficiency—either partial or complete—and therefore are more susceptible to methemoglobinemia. Closer monitoring of such patients is warranted. In clinical trials using NO at 5 to 40 ppm, methemoglobinemia has not been a significant problem. Finally, the cost of inhaled NO is surprisingly steep (121), warranting the closest scrutiny of its use in “marginal” situations.
Liquid Ventilation
Pulmonary gas transport using complex low-surface tension, high vapor pressure fluorocarbon molecules in the liquid state has been investigated for use in patients with severe ALI. Respiratory gases are carried in solution, and either partial- or full-liquid ventilation is conducted with a conventional or “liquid ventilator,” respectively. Perfluorocarbons possess anti-inflammatory properties and, by virtue of their liquid state, localize particularly in dependent areas where airway collapse is most prevalent and thus most in need of PEEP in the ARDS lung (122). These features of perfluorocarbons tend to stabilize collapsed dependent alveoli without the risks of high pressures associated with conventional ventilation. Animals have survived the imposition of liquid ventilation in experimental circumstances (123). Studies on adult humans, however, have not revealed a benefit to this form of treatment, with no overall improvement in outcome (124,125) (Fig. 136.13).
|
|
|
Figure 136.13. All-cause mortality for all patients enrolled into the three studied groups over the first 28 days of the study. Treatment groups: solid line—low-dose perfluorocarbon; dotted line—high-dose perfluorocarbon; dashed line—conventional ventilation. (With permission from Kacmarek RM, Wiedemann HP, Lavin PT, et al. Partial liquid ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;173[8]:882–889.) |
Surfactant Replacement
Surfactant is a phospholipid, a protein material produced by type II epithelial pneumocytes, secreted along the alveolar surface and acting to decrease surface tension to prevent alveolar collapse. Hydrophilic surfactant proteins A and D contribute to the immune response (126), while hydrophobic types B and C facilitate monolayer formation within the alveolus (127). Pulmonary epithelial injury and surfactant dysfunction from direct or indirect injury destabilize alveoli, leading to the collapse, venous admixture, hypoxemia, and a decreased lung compliance characteristic of ARDS.
Surfactant replacement therapy in adults has been, thus far, unsuccessful. After an encouraging phase I/II trial of recombinant surfactant protein C–based surfactant supplement (128), a phase III trial of the same material improved oxygenation without lowering mortality or the number of ventilator days in adult patients with ARDS (129). Further investigation of this promising modality in adults is warranted based on its clear success in neonatology.
Complications
Significant morbidity or mortality may occur during supportive therapy for ALI/ARDS. Most aspects of supportive care transcend the specifics of ALI/ARDS, and the clinician should be aware of these potential complications, many of which are outlined by Pingleton (130) (Table 136.8). Attention to detail decreases complications and may improve outcome in ARDS. As suggested earlier, the ARDS patient is so exquisitely sensitive to the smallest subtleties of mechanical ventilation that discussion of the main potential sequela of suboptimal mechanical ventilation, namely VALI, is provided in detail below.
Ventilator-associated Lung Injury
To date, the only mode of management with which outcome in ARDS patients can predictably be improved is the optimal use of mechanical ventilation, using low tidal volumes with PEEP (131). As in all things medical, specific techniques of ventilator management carry risks and benefits. While mechanical assistance with gas exchange may be life saving, mechanical ventilation may contribute simultaneously to worsening the overall inflammatory process and the eventual triumph of MODS leading to death. In fact, the very process of mechanical ventilatory support can, if conducted suboptimally, provoke edematous morphologic and microscopic pulmonary changes indistinguishable from those associated with commonly recognized ARDS-inducing processes (71). Again, in ARDS, nonhomogeneous distribution of areas of consolidated, noncompliant lung, featuring thickened interstitium and fluid- and debris-filled alveoli residing adjacent to unaffected areas, causes maldistribution of positive pressure ventilation, exceeding injurious pressure and volume thresholds in healthy areas, while not affecting inflamed tissues. The additional inflammatory insult induced by suboptimal mechanical ventilation technique is termed ventilator-associated lung injury. The ARMA study (67) revealed the importance of low-volume ventilation in ARDS, with substantial improvement in several indices using 6 mL/kg rather than 12 mL/kg tidal volumes. While criticized, the findings document the importance of avoiding several putative mechanisms of pathologic effect:
|
Table 136.8 Complications Associated with ARDS |
||||||||||||
|
1. Excess alveolar hyperinflation with associated increased permeability and cytokine release
2. Escape of alveolar air outside the confines of alveoli
3. Destructive sheer-stress influence of repetitive inflation/ collapse of unstable alveoli
Each of these phenomena contributes to pulmonary dysfunction and perpetuation of the inflammatory response in the ARDS patient, and thus each has been classified. Barotrauma refers to the presence of air outside the alveoli when receiving positive pressure ventilation. Air leaks track along the perivascular sheath to the mediastinum and pleural cavities, or along fascial planes to extrathoracic areas (132). It seems intuitive that such occurrences are related to pressures exceeding the limits of tissue structural integrity, but the issue is clearly more complicated, since musicians are repeatedly able to generate 150 cm H2O airway pressure with no sequelae (133). It is speculated that barotrauma represents regional overinflation in areas of diseased lung, such areas thereby being particularly at risk for structural failure and air leak (133). Volutrauma occurs when excessive inspiratory volumes induce microvascular edema (134); the offending agent appears to be excessive volume, rather than the excessive pressure required to supply that volume (71). Resultant mechanical stretch triggers changes in the alveolar–capillary barrier (73) and in proliferation of inflammatory cytokines (75), resulting in interstitial and alveolar proteinaceous edema, decreased compliance, and hyaline membrane formation. Compromised surfactant production and function leads to increased surface tension, provoking alveolar collapse with increased venous admixture and subjecting alveolar epithelium to the tissue-destructive shear stresses of recruitment/derecruitment in the process known as atelectrauma. While the use of PEEP to maintain diseased distal alveoli splinted “open” has not definitively been demonstrated to improve outcome (75, and earlier discussion, above), improvements in oxygenation and pulmonary compliance with PEEP mandate its routine use in ARDS. Of note is the complex relationship between mechanical ventilation and patchy maldistribution throughout the lung in areas of varying ratios of ventilation and perfusion, with atelectatic areas abutting hyperinflated bullous and cystic areas. While collapsed noncompliant airways require high initial opening pressures consistent with the Law of Laplace (within the context of pulmonary physiology, the following formula is appropriate: P = 2t/R, where P = pressure, t = tension, and R = radius), such high pressure is transmitted throughout the lung, overdistending more compliant airways both through direct influence and by an unequally distributed traction force upon the adjacent airways, as depicted in Figure 136.7. Implicit in such heterogeneous patterns of gas distribution is the initiation of the destructive sequelae associated with inflation of each subregion of lung, as delineated above.
Furthermore, over the last few years, there has been recognition of the inflammatory cytokine release, as well as alveolar and interstitial neutrophil infiltration associated with ventilator-related pulmonary disruption, leading to MODS (69,135). While it is indisputable that “excessive” tidal volume ventilation augments systemic cytokine levels (75), the specific causal relationship with worse outcome has yet to be validated. The ALVEOLI study (83), which examined the variation of short-term indices—28-day mortality and number of ventilator-free days—with modulation of PEEP, was discontinued early based on lack of improvement in outcome with high PEEP levels. Thus, the optimal inspiratory pressure, or level of PEEP for a given ARDS patient's pressure–volume curve, can be exceedingly difficult to identify despite the potentially severe consequences of failure to do so. While dependent areas of tenaciously collapsed, high time-constant alveoli may require the equivalent of repeated and prolonged high-pressure recruitment maneuvers to achieve inflation and avoid atelectrauma, simultaneous transmission of such pressure to compliant alveoli incurs the risk of inducing volutrauma and inciting inflammation. Clearly, in those with advanced lung injury, the “optimal” inspiratory pressure, in reality, reflects a statistical bell curve of widely variable individual alveolar compliances. The clinician must vary inspiratory time, plateau pressure, PEEP, and tidal volume to inflate stiff alveoli while not persistently overdistending the normal ones. Such important actions are required because of the dynamic and changing compliance profiles of the inflamed lung.
References
1. Kollef MH, Schuster D. The acute respiratory distress syndrome. N Engl J Med. 1995;332:27.
2. Ashbaugh DG, Bigelow DB, Petty TL, et al. Acute respiratory distress in adults. Lancet. 1967;ii:319.
3. Simeone FA. Pulmonary complications of nonthoracic wounds: a historical perspective. J Trauma. 1968;8:625.
4. Petty TL, Ashbaugh DG. The adult respiratory distress syndrome: clinical features, factors influencing prognosis and principles of management. Chest. 1971;60:233.
5. Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Am J Respir Crit Care Med. 1994;149:818.
6. Balk R, Bone RC. The adult respiratory distress syndrome. Med Clin North Am. 1983;67:685.
7. Murray JF, Matthay MA, Luce JM, et al. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:720.
8. Matthay MA. Conference summary—acute lung injury. Chest. 1999;116: 119S–126S.
9. Zilberberg MD, Epstein SK. Acute lung injury in the medical ICU: comorbid conditions, age, etiology, and hospital outcome. Am J Respir Crit Care Med. 1998;157(Pt 1):1159–1164.
10. Moss M, Goodman PL, Heinig M, et al. Establishing the relative accuracy of three new definitions of the adult respiratory distress syndrome. Crit Care Med. 1995;23:1629.
11. Fowler AA, Hamman RF, Good JT, et al. Adult respiratory distress syndrome. Risk with common predisposition. Ann Intern Med. 1983;98:593.
12. Pepe PE, Potkin RT, Reus DH, et al. Clinical predictors of adult respiratory distress syndrome. Am J Surg. 1982;144:124.
13. Hudson LD, Milberg JA, Anardi D, et al. Clinical risks of the development of the acute respiratory distress syndrome. Am J Respir Care. 1995;151:293.
14. Hudson LD, Steinberg KP. Epidemiology of acute lung injury and ARDS. Chest. 1999;116:74S.
15. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334.
16. Rubenfield GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685.
17. Rocco PRM, Zin WA. Pulmonary and extrapulmonary acute respiratory distress syndrome: are they different? Curr Opin Crit Care. 2005;11:10.
18. Milberg JA, Davis DR, Steinberg KP, et al. Improved survival of patients with acute respiratory distress syndrome (ARDS): 1983–1993. JAMA. 1995;273(4):306–309.
19. Abel SJC, Finney SJ, Brett SJ, et al. Reduced mortality in association with acute respiratory distress syndrome (ARDS). Thorax. 1998;53:292.
20. Petty TL. Adult respiratory distress syndrome: definition and historical perspective. Clin Chest Med. 1982;3:3.
21. Taylor RW, Duncan CA. The adult respiratory distress syndrome. Res Med. 1983;1:17.
22. Brun-Buisson C, Minelli C, Bertolini G, et al. Epidemiology and outcome of acute lung injury in European intensive care units: results from the ALIVE study. Intensive Care Med. 2004;30:51–61.
23. Luhr OR, Antonsen K, Karlsson M, et al. Incidence and mortality after acute respiratory failure and acute respiratory distress syndrome in Sweden, Denmark, and Iceland: the ARF Study Group. Am J Respir Crit Care Med. 1999;159:1849–1861.
24. Bersten AD, Edibam C, Hunt T, et al. Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian states. Am J Respir Crit Care Med. 2002;165:443–448.
25. Davidson TA, Rubenfeld GD, Caldwell ES, et al. The effect of acute respiratory distress syndrome on long-term survival. Am J Respir Crit Care Med. 1999;160:1838.
26. Luce JM. Acute lung injury and the acute respiratory distress syndrome. Crit Care Med. 1998;26(2):369–376.
27. Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348:683.
28. McHugh LG, Milberg JA, Whitecomb ME, et al. Recovery of function in survivors of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150:90.
29. Davidson TA, Caldwell ES, Curtis JR, et al. Reduced quality of life in survivors of acute respiratory distress syndrome compared with critically ill control patients. JAMA. 1999;281:354.
30. Weinert CR, Gross CR, Kangas JR, et al. Health-related quality of life after acute lung injury. Am J Respir Crit Care Med. 1997;156:1120.
31. Pelosi P, D'Onofrio D, Chiumello D, et al. Pulmonary and extrapulmonary acute respiratory distress syndrome are different. Eur Respir J (Suppl). 2003;42:48s–56s.
32. Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7(1):1–7.
33. Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4 Suppl):S195–199.
34. Frerking I, Günther A, Seeger W, et al. Pulmonary surfactant: functions, abnormalities and therapeutic options. Intensive Care Med. 2001;27(11): 1699–1717.
35. Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. 2003;31(4 Suppl):S213–220.
36. Pittet JF, Mackersie RC, Martin TR, et al. Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med. 1997;155(4):1187–1205.
37. Park WY, Goodman RB, Steinberg KP, et al. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1896–1903.
38. Fink MP. Role of reactive oxygen and nitrogen species in acute respiratory distress syndrome. Curr Opin Crit Care. 2002;8(1):6–11.
39. Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202(2):145–156.
40. Tomashefski JF Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 2000;21(3):435–466.
41. Bellingan GJ. The pulmonary physician in critical care: the pathogenesis of ALI/ARDS. Thorax. 2002;57(6):540–546.
42. Marshall RP, Bellingan G, Webb S, et al. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med. 2000;162(5):1783–1788.
43. Gomez AC: Pulmonary insufficiency in non-thoracic trauma [discussion]. J Trauma. 1968;8:666.
44. Moore FA, Feliciano DV, Andrassy RJ, et al. Early enteral feeding, compared with parenteral, reduces postoperative septic complications. Ann Surg. 1992;216:172.
45. Schuster D. Fluid management in ARDS: “keep them dry” or does it matter? Intensive Care Med. 1995;21:101.
46. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354(24): 2564–2575.
47. Wright P, Carmichael L, Bernard G. Effect of bronchodilators on lung mechanics in the acute respiratory distress syndrome (ARDS). Chest. 1994;106:157.
48. Stoelting RK. Pharmacology and Physiology in Anesthetic Practice. 3rd ed. Philadelphia: Lippincott–Raven; 1999:196.
49. Meduri GU, Chinn AJ, Leeper KV, et al. Corticosteroid rescue treatment of progressive fibroproliferation in late ARDS. Patterns of response and predictors of outcome. Chest. 1994;105(5):1516–1527.
50. Steinberg KP, Hudson LD, Goodman RB, et al., and the National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354(16):1671–1684.
51. Meduri GU, Golden E, Freire AX, et al. Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest. 2007; 131(4):954–963.
52. Meduri GU, Marik PE, Chrousos GP, et al. Steroid treatment in ARDS: a critical appraisal of the ARDS network trial and the recent literature. Intensive Care Med. 2008;34(1):61–69.
53. Peter JV, John P, Graham PL, et al. Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: meta-analysis. BMJ. 2008;336(7651):1006–1009.
54. Calfee CS, Matthay MA. Nonventilatory treatments for acute lung injury and ARDS. Chest. 2007;131(3):913–920.
55. Hudson LD, Hough CL. Therapy for late-phase acute respiratory distress syndrome. Clin Chest Med. 2006;27(4):671–677.
56. Bream-Rouwenhorst HR, Beltz EA, Ross MB, et al. Recent developments in the management of acute respiratory distress syndrome in adults. Am J Health Syst Pharm. 2008;65(1):29–36.
57. Meduri GU, Headley S, Tolley E, et al. Plasma and BAL cytokine response to corticosteroid rescue treatment in late ARDS. Chest. 1995;103:1315.
58. Tobin MJ. Principles and Practice of Mechanical Ventilation. 2nd ed. New York: McGraw Hill; 2006:38, 129–154, 782–784.
59. Antonelli M, Conti G, Esquinas A, et al. A multiple-center survey on the use in clinical practice of noninvasive ventilation as a first-line intervention for acute respiratory distress syndrome. Crit Care Med. 2007;35(1):18–25.
60. Ferrer M, Esquinas A, Leon M, et al. Noninvasive ventilation in severe hypoxemic respiratory failure: a randomized clinical trial. Am J Respir Crit Care Med. 2003;168(12):1438–1444.
61. Keenan SP, Sinuff T, Cook DJ, et al. Does noninvasive positive pressure ventilation improve outcome in acute hypoxemic respiratory failure? A systematic review. Crit Care Med. 2004;32(12):2516–2523.
62. Rouby JJ, Puybasset L, Cluzel P, et al. Regional distribution of gas and tissue in acute respiratory distress syndrome. II. Physiological correlations and definition of an ARDS Severity Score. CT Scan ARDS Study Group. Intensive Care Med. 2000;26(8):1046–1056.
63. Puybasset L, Gusman P, Muller JC, et al. Regional distribution of gas and tissue in acute respiratory distress syndrome. III. Consequences for the effects of positive end-expiratory pressure. CT Scan ARDS Study Group. Adult Respiratory Distress Syndrome. Intensive Care Med. 2000;26(9): 1215–1227.
64. Kolobow T, Moretti MP, Fumagalli R, et al. Severe impairment in lung function induced by high peak airway pressure during mechanical ventilation. An experimental study. Am Rev Respir Dis. 1987;135(2):312– 315.
65. Tsuno K, Prato P, Kolobow T. Acute lung injury from mechanical ventilation at moderately high airway pressures. J Appl Physiol. 1990;69(3):956–961.
66. Hickling KG, Henderson SJ, Jackson R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med. 1990;16(6):372–377.
67. Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342(18):1301–1308.
68. Tremblay LN, Slutsky AS. Ventilator-induced lung injury: from the bench to the bedside. Intensive Care Med. 2006;32(1):24–33.
69. Tremblay L, Valenza F, Ribeiro SP, et al. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997;99(5):944–952.
70. Slutsky AS. Lung injury caused by mechanical ventilation. Chest. 1999;116 (1 Suppl):9S–15S.
71. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110(5):556–565.
72. Petersen GW, Baier H. Incidence of pulmonary barotrauma in a medical ICU. Crit Care Med. 1983;11(2):67–69.
73. Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157(1):294–323.
74. Belperio JA, Keane MP, Lynch JP 3rd, et al. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med. 2006;27(4):350–364.
75. Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282(1):54– 61.
76. Terragni PP, Rosboch G, Tealdi A, et al. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007;175(2):160–166.
77. Gattinoni L, Pesenti A. The concept of “baby lung.” Intensive Care Med. 2005;31(6):776–784.
78. Barbas CS, de Matos GF, Pincelli MP, et al. Mechanical ventilation in acute respiratory failure: recruitment and high positive end-expiratory pressure are necessary. Curr Opin Crit Care. 2005;11(1):18–28.
79. Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338(6):347–354.
80. Lachmann B. Open up the lung and keep the lung open. Intensive Care Med. 1992;18(6):319–321.
81. Gattinoni L, Caironi P, Cressoni M, et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006;354(17):1775–1786.
82. Dueck R. Alveolar recruitment versus hyperinflation: a balancing act. Curr Opin Anaesthesiol. 2006;19(6):650–654.
83. Brower RG, Lanken PN, MacIntyre N, et al., National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351(4):327–336.
84. Meade MO, Cook DJ, Guyatt GH, et al., Lung Open Ventilation Study Investigators. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2008;299(6):637–645.
85. Borges JB, Okamoto VN, Matos GF, et al. Reversibility of lung collapse and hypoxemia in early acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;174(3):268–278.
86. Lapinsky SE, Mehta S. Bench-to-bedside review: recruitment and recruiting maneuvers. Crit Care. 2005;9(1):60–65.
87. Medoff BD, Harris RS, Kesselman H, et al. Use of recruitment maneuvers and high-positive end-expiratory pressure in a patient with acute respiratory distress syndrome. Crit Care Med. 2000;28(4):1210–1216.
88. Laffey JG, O'Croinin D, McLoughlin P, et al. Permissive hypercapnia—role in protective lung ventilatory strategies. Intensive Care Med. 2004;30(3):347–356.
89. Kallet RH, Campbell AR, Alonso JA, et al. The effects of pressure control versus volume control assisted ventilation on patient work of breathing in acute lung injury and acute respiratory distress syndrome. Respir Care. 2000;45(9):1085–1096.
90. Shanholtz C, Brower R. Should inverse ratio ventilation be used in adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;149(5):1354–1358.
91. Mercat A, Graïni L, Teboul JL, et al. Cardiorespiratory effects of pressure-controlled ventilation with and without inverse ratio in the adult respiratory distress syndrome. Chest. 1993;104(3):871–875.
92. Esteban A, Alía I, Gordo F, et al. Prospective randomized trial comparing pressure-controlled ventilation and volume-controlled ventilation in ARDS. For the Spanish Lung Failure Collaborative Group. Chest. 2000;117(6):1690–1696.
93. Habashi NM. Other approaches to open-lung ventilation: airway pressure release ventilation. Crit Care Med. 2005;33(3 Suppl):S228–240.
94. Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care. 2001;5(4):221–226.
95. Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med. 2001;164(1):43–49.
96. Pelosi P, Brazzi L, Gattinoni L. Prone position in acute respiratory distress syndrome. Eur Respir J. 2002;20(4):1017–1028.
97. West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung; relation to vascular and alveolar pressures. J Appl Physiol. 1964;19: 713–724.
98. Gattinoni L, Tognoni G, Pesenti A, et al., Prone-Supine Study Group. Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med. 2001;345(8):568–573.
99. Guerin C, Gaillard S, Lemasson S, et al. Effects of systematic prone positioning in hypoxemic acute respiratory failure: a randomized controlled trial. JAMA. 2004;292(19):2379–2387.
100. Mancebo J, Fernández R, Blanch L, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;173(11):1233–1239.
101. Pillow JJ. High-frequency oscillatory ventilation: mechanisms of gas exchange and lung mechanics. Crit Care Med. 2005;33(3 Suppl):S135–141.
102. Chang HK. Mechanisms of gas transport during ventilation by high-frequency oscillation. J Appl Physiol. 1984;56(3):553–563.
103. Slutsky AS, Drazen FM, Ingram RH Jr, et al. Effective pulmonary ventilation with small-volume oscillations at high frequency. Science. 1980;209 (4456):609–671.
104. Derdak S. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adult patients. Crit Care Med. 2003;31(4 Suppl):S317–323.
105. Wunsch H, Mapstone J, Takala J. High-frequency ventilation versus conventional ventilation for the treatment of acute lung injury and acute respiratory distress syndrome: a systematic review and Cochrane analysis. Anesth Analg. 2005;100(6):1765–1772.
106. Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med. 2002;166(6):801–808.
107. Brown JK, Haft JW, Bartlett RH, et al. Acute lung injury and acute respiratory distress syndrome: extracorporeal life support and liquid ventilation for severe acute respiratory distress syndrome in adults. Semin Respir Crit Care Med. 2006;27(4):416–425.
108. Gattinoni L, Pesenti A, Mascheroni D, et al. Low-frequency positive-pressure ventilation with extracorporeal CO2 removal in severe acute respiratory failure. JAMA. 1986;256:881–886.
109. Morris AH, Wallace CJ, Menlove RL, et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;149:295–305.
110. Lequier L. Extracorporeal life support in pediatric and neonatal critical care: a review. J Intensive Care Med. 2004;19(5):243–258.
111. Skinner SC, Hirschl RB, Bartlett RH. Extracorporeal life support. Semin Pediatr Surg. 2006;15(4):242–250.
112. Pison U, López FA, Heidelmeyer CF, et al. Inhaled nitric oxide reverses hypoxic pulmonary vasoconstriction without impairing gas exchange. J Appl Physiol. 1993;74(3):1287–1292.
113. Rossaint R, Falke KJ, López F, et al. Inhaled nitric oxide for the adult respiratory distress syndrome. N Engl J Med. 1993;328(6):399–405.
114. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526.
115. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–142.
116. Cooper CE. Nitric oxide and iron proteins. Biochim Biophys Acta. 1999;1411(2–3):290–309.
117. Taylor RW, Zimmerman JL, Dellinger RP, et al., Inhaled Nitric Oxide in ARDS Study Group. Low-dose inhaled nitric oxide in patients with acute lung injury: a randomized controlled trial. JAMA. 2004;291(13):1603–1609.
118. Adhikari NK, Burns KE, Friedrich JO, et al. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. BMJ. 2007;334(7597):779.
119. Lowson SM. Inhaled alternatives to nitric oxide. Crit Care Med. 2005;33(3 Suppl):S188–195.
120. Puybasset L, Rouby JJ, Mourgeon E, et al. Factors influencing cardiopulmonary effects of inhaled nitric oxide in acute respiratory failure. Am J Respir Crit Care Med. 1995;152(1):318–328.
121. Pierce CM, Peters MJ, Cohen G, et al. Cost of nitric oxide is exorbitant [Letter to the Editor]. BMJ. 2002;325(7359):336.
122. Wiedemann HP. Partial liquid ventilation for acute respiratory distress syndrome. Clin Chest Med. 2000;21(3):543–554.
123. Clark LC Jr, Gollan F. Survival of mammals breathing organic liquids equilibrated with oxygen at atmospheric pressure. Science. 1966;152(730):1755–1756.
124. Kacmarek RM, Wiedemann HP, Lavin PT, et al. Partial liquid ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006;173(8):882–889.
125. Hirschl RB, Croce M, Gore D, et al. Prospective, randomized, controlled pilot study of partial liquid ventilation in adult acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;165(6):781– 787.
126. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58–68.
127. Stevens TP, Sinkin RA. Surfactant replacement therapy. Chest. 2007; 131(5):1577–1582.
128. Spragg RG, Lewis JF, Wurst W, et al. Treatment of acute respiratory distress syndrome with recombinant surfactant protein C surfactant. Am J Respir Crit Care Med. 2003;167(11):1562–1566.
129. Spragg RG, Lewis JF, Walmrath HD, et al. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med. 2004;351(9):884–892.
130. Pingleton SK. Complications associated with the adult respiratory distress syndrome. Clin Chest Med. 1982;5:143.
131. Villar J, Kacmarek RM, Pérez-Méndez L, et al. A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: a randomized, controlled trial. Crit Care Med. 2006;34(5):1311–1318.
132. Gammon RB, Shin MS, Buchalter SE. Pulmonary barotrauma in mechanical ventilation. Patterns and risk factors. Chest. 1992;102(2):568– 572.
133. Bouhuys A. Physiology and musical instruments. Nature. 1969;221(5187): 1199–2004.
134. Dreyfuss D, Soler P, Saumon G. Mechanical ventilation-induced pulmonary edema. Interaction with previous lung alterations. Am J Respir Crit Care Med. 1995;151(5):1568–1575.
135. Kawano T, Mori S, Cybulsky M, et al. Effect of granulocyte depletion in a ventilated surfactant-depleted lung. J Appl Physiol. 1987;62(1):27–33.