Gareth Adams
Shankar P. Gopinath
Claudia S. Robertson
Definition of Intracranial Pressure
Intracranial pressure (ICP) is the pressure of the compartment inside the skull. Since the brain is almost completely incompressible, expansion of the volume in any component within the skull causes an increase in the pressure of the cranial compartment. Increased intracranial pressure can lead to decreased blood flow, resulting in decreased oxygen delivery to the brain and neuronal dysfunction due to global ischemia as well as herniation of the brain with compression of vital structures and death. The principles of increased intracranial pressure are defined by the Monro-Kellie doctrine.
Causes of Increased Intracranial Pressure
Intracranial hypertension complicates many neurologic disorders. Head trauma can result in diffuse brain edema, increased blood flow, and mass lesions such as subdural, epidural, or intraparenchymal hemorrhage. Brain edema following head injury is primarily cellular edema, although vasogenic edema can occur transiently (1). A ruptured aneurysm can cause subarachnoid hemorrhage resulting in vasospasm and hydrocephalus. Ischemic or hemorrhagic strokes can result in a mass lesion and cellular edema. Obstructive or communicating hydrocephalus from multiple causes can result in increased cerebrospinal fluid volume. Fulminant hepatic failure can cause significant cerebral edema. Brain tumors or infection can occupy volume within the intracranial space. In this chapter the focus will be on increased ICP from head injury.
Physiology of the Intracranial Space
Monro-Kellie Doctrine
The major components contained within the intracranial space are the brain, blood, and cerebrospinal fluid (CSF). A mass lesion such as a hematoma or a tumor can also contribute to the volume of the intracranial space. As defined by the Monro-Kellie doctrine, all these components are contained within a fixed volume confined by the skull. The brain itself is not compressible. Thus an increase in the volume of one of the components contained within the intracranial space can occur only at the expense of the other components. As the volume of edema in the brain or an expanding mass lesion increases, initially the CSF volume decreases, then the blood volume decreases. Continued expansion of intracranial volume after these compensatory mechanisms are exhausted causes a rapid increase in ICP and herniation of the brain.
The intracranial compartment is partially divided into two compartments by the tentorium cerebelli. This shelf of dura divides the skull into the supratentorial compartment, containing the cerebral hemispheres, and the infratentorial compartment, containing the brainstem and cerebellum. Increased ICP in the supratentorial compartment can cause uncal herniation, with the temporal lobe herniating through the incisura of the tentorium and compressing the midbrain. Continued increase in the ICP in the superior fossa or increased ICP in the posterior fossa can cause tonsillar herniation with the tonsils of the cerebellum herniating through the foramen magnum with compression of the medulla.
Clinical Pearl
The volume of the intracranial space is fixed by the skull. An increase in volume of any component within the skull causes an increase in the intracranial pressure. Initial increases in volume can be compensated for by displacement of CSF. Once this compensatory reserve is expended, pressure increases quickly.
Circulation of Cerebrospinal Fluid
The CSF contained within the ventricles of the brain provides a buffer to changes in other volumes within the intracranial space. The choroid plexus, which is located within the lateral and fourth ventricles, secretes CSF. CSF drains from the lateral ventricles through the two foramina of Monro into the third ventricle. From the third ventricle it drains via the cerebral aqueduct into the fourth ventricle. From the fourth ventricle it drains into the subarachnoid space through the two lateral foramina of Luschka and the median foramen of Magendie and into the central canal of the spinal cord. In the subarachnoid space the CSF is resorbed at the arachnoid villi. The normal total CSF volume in an adult is 150 mL. In a day, 450 mL of CSF is secreted; thus the CSF volume is recirculated three times each day. Increased secretion or decreased absorption can cause communicating hydrocephalus. Blockage of the circulation of CSF can cause obstructive hydrocephalus. Absorption of CSF is pressure dependent, whereas secretion is not.
Blood Supply of the Brain
The primary arteries supplying blood to the brain are the paired internal carotid and vertebral arteries. Branches of the internal carotid arteries supply the frontal, parietal, and temporal lobes and the basal ganglia. The vertebral arteries merge to form the basilar artery. Branches from the vertebral arteries and the basilar arteries are the primary blood source for the brainstem, cerebellum, and occipital lobes. Some redundancy in blood supply is provided by the communicating arteries forming the circle of Willis. The two internal carotid arteries are connected by the anterior communicating artery. The posterior communicating arteries connect the internal carotid arteries to the basilar artery. There is significant anatomic variation in the circle of Willis. Venous drainage of blood is through the deep and cortical veins to the sinuses and then out of the intracranial vault through the internal jugular veins.
ICP Pressure–Volume Curve
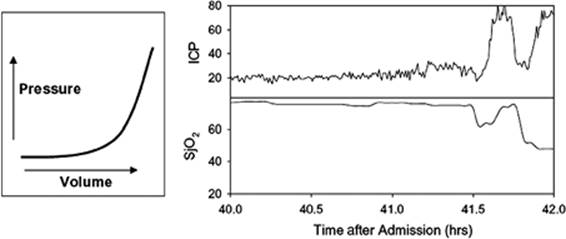
The intracranial pressure as a function of increasing volume in the intracranial space is a nonlinear function (Fig. 146.1). Early increases in volume of a mass lesion result in displacement of CSF from the cranial compartment into the spinal compartment and cause little increase in ICP. Once the maximal amount of CSF has been displaced from the intracranial compartment, the ICP increases rapidly. As ICP increases, cerebral perfusion pressure decreases and blood flow to the brain decreases.
Cerebral Perfusion Pressure
Cerebral perfusion pressure (CPP) is defined as the mean arterial pressure (MAP) minus the ICP and is the driving pressure for cerebral blood flow (CBF). Pressure autoregulation is the intrinsic ability of the cerebral vasculature to maintain flow constant over a wide range of CPP values. Normally, the brain is able to autoregulate and to maintain an adequate CBF at CPP values ranging from 50 to 140 mm Hg. However, dynamic pressure autoregulation is commonly dysfunctional in the injured brain (2). When pressure autoregulation is impaired, the lower limit of autoregulation can be shifted upward from 50 to 60 to 90 mm Hg. When pressure autoregulation is entirely absent, perfusion passively follows CPP. This loss of normal autoregulation requires careful maintenance of a sufficient MAP and adequate control of ICP to avoid hypoperfusion of the brain. Extremely elevated MAP should also be avoided as it can cause increased ICP.
|
|
|
Figure 146.1. The left graph illustrates the nonlinear nature of the relationship between the volume of intracranial contents and intracranial pressure. The right graph shows the actual intracranial pressure (ICP) and jugular venous oxygen saturation (SjO2) tracings in a patient who developed a delayed intracranial hematoma. Initially the rise in ICP was minimal as the brain was able to compensate for the increasing volume from the hematoma. Later when these compensatory mechanisms were exhausted, there was a very rapid increase in ICP associated with compromise of cerebral blood flow and a decrease in SjO2. |
Clinical Pearl
Cerebral perfusion pressure (CPP) is defined as mean arterial pressure (MAP) minus intracranial pressure (ICP). CPP can be increased by lowering ICP or by raising MAP.
Brain Metabolism
Given its relatively small size, the brain consumes a significant fraction of the resources of the body. Fifteen percent of blood flow goes to the brain, providing it with oxygen and glucose. Normal CBF is 50 to 55 mL per 100 g per minute. Each 100 g of brain tissue requires at least 20 mL of blood flow per minute to function normally. Between 20 mL per minute and 8 mL per minute of blood flow, the brain tissue can survive for at least a period of time but does not function normally, and below 8 mL per minute the brain tissue dies. The brain consumes 20% of the oxygen used by the body, using 3.5 mL of oxygen per 100 g per minute. The brain is also dependent on delivery of glucose in the blood as the brain has almost no intrinsic energy stores.
These CBF thresholds were determined in normal brain, but seem to hold true for brain-injured patients, and a low CBF, particularly in the early postinjury period, is highly predictive of a poor outcome. Traumatic brain injury patients with CBF of less than 18 mL/100 g per minute had a significantly worse outcome compared to patients with higher CBF (3). Within 12 hours of injury, every 10 mL/100 g per miute increase in CBF was associated with a threefold increase in the probability of surviving the injury (4).
Brain oxygenation as a measure of oxygen delivery can be monitored with global and local techniques. A snapshot of regional oxygenation can be acquired noninvasively using positron emission tomography (PET). Similarly, cerebral perfusion an be assessed with xenon computed tomography (CT) or perfusion CT imaging. Invasive techniques can be used to continuously monitor brain oxygenation. Combined monitoring of arterial oxygen saturation and the oxygen saturation in the internal jugular vein allows the calculation of the arteriojugular oxygen content difference (AJDO2), which is dependent on the amount of oxygen consumed by the brain (CMRO2) and on CBF by the formula CMRO2/CBF. An increased AJDO2 indicates a deficiency of flow relative to the metabolic needs of the brain. Brain oxygenation can also be monitored continuously in a local region of the brain by inserting an invasive probe to measure the brain tissue oxygen tension (PbO2). Continuous global monitoring provides an overall assessment of brain oxygenation but can miss local changes (5). Continuous local monitoring with an invasive probe provides information on a specific portion of the brain but is dependent on probe location and may not reflect overall brain perfusion (6).
Clinical Pearl
Maintenance of oxygenation of brain tissue is of paramount importance in the injured brain. Cerebral blood flow (CBF) must be maintained at levels high enough to deliver sufficient oxygen. Below a CBF of 20 mL/100 g of brain per minute, the brain does not function normally. Below a CBF of 8 mL/100 g of brain per minute, the neurons die.
Commonly Monitored Parameters in Patients with Elevated ICP
Intracranial Pressure
Elevated ICP can be both measured and treated. Normal ICP in the adult is less than 10 mm Hg. An ICP value of 20 to 30 mm Hg is mild intracranial hypertension, and values above 40 mm Hg are severe, life-threatening intracranial hypertension. The goal of treatment should be to maintain ICP below 20 to 25 mm Hg. Studies of traumatic brain injury patients have demonstrated that patients with elevated ICP have a worse outcome than those with ICP below 20 (7,8,9,10).
The gold standard for monitoring ICP is a ventriculostomy catheter inserted through a burr hole into one of the lateral ventricles. The ventriculostomy catheter is connected to a drainage system and can be used to monitor the ICP through a fluid-coupled external pressure transducer. This system provides the most accurate measurement of ICP and is stable over time (11). A ventriculostomy catheter will also allow drainage of CSF for control of ICP. Problems associated with ventriculostomy catheters include blockage of the catheter, displacement of the catheter from the ventricle, and infection. Antibiotic-impregnated ventriculostomy catheters reduce the risk of infection from 9.4% to 1.3% (12).
Other invasive monitors for ICP include intraparenchymal, subdural, and epidural monitors. These probes use either a strain gauge or a fiberoptic probe. These probes require zeroing prior to insertion and are subject to drift over time. Of these probes, intraparenchymal probes are the most accurate with the least amount of drift. The advantage of these probes is that they do not have to be inserted into the ventricle, which may be difficult to locate if it is collapsed or if there is significant midline shift.
Clinical Pearl
A ventriculostomy catheter placed into one of the lateral ventricles is the gold standard for ICP monitoring. ICP should be maintained at less than 20 to 25 mm Hg.
Cerebral Perfusion Pressure
Cerebral perfusion pressure (CPP) is defined as the mean arterial pressure (MAP) minus the ICP. It can be measured using a combination of an ICP monitor and either an arterial line or a noninvasive blood pressure monitor. CPP is a second major parameter that can be both monitored and treated in the patient with elevated intracranial pressure. CPP can be supported by maintaining MAP with intravenous fluids and vasopressors and by lowering ICP. CPP should be maintained at a level that will allow adequate perfusion of the brain. For most patients, a CPP of at least 60 mm Hg is adequate (13).
Brain Oxygenation
Brain tissue oxygenation can be measured either using PET imaging techniques to obtain a single time point sample of oxygenation across the entire brain or by using probes to continuously monitor brain tissue pO2 (PbO2) and/or jugular venous oxygen saturation (SjO2). PET imaging is not widely available for ICU patients, but has provided important insights into the pathophysiology of brain injury. PbO2 and SjO2 monitoring can be used in any ICU setting.
Brain Tissue Oxygen Tension
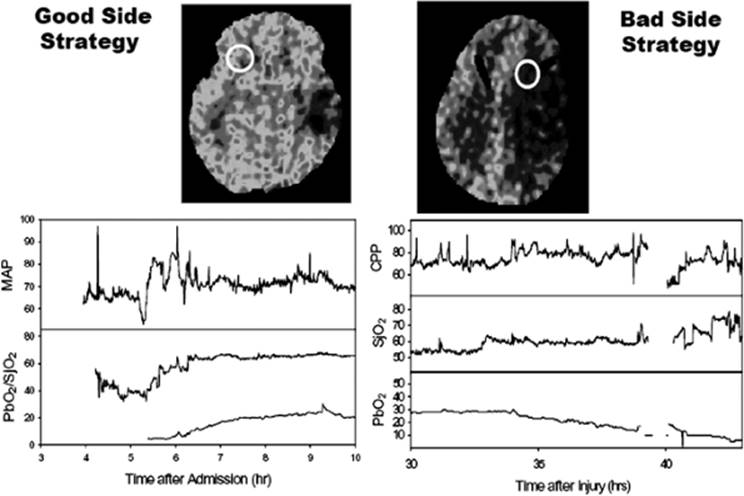
Insertion of a PbO2 probe allows continuous monitoring of oxygenation in a local region of the brain. The location of the probe is critical and determines the nature of the pO2 information that will be obtained (Fig. 146.2). If the probe is inserted near a focal lesion, oxygenation can be monitored in the tissue at greatest risk should the injury expand. Insertion of the probe in uninjured brain allows monitoring of a local area that should be representative of the overall less-injured oxygenation status of the brain. This pO2 value provides information that is similar to SjO2 monitoring (6). Both monitoring strategies have been successfully used.
Normal values and critical threshold values for PbO2 are somewhat less accepted. In anesthetized subjects, PbO2 in normal brain ranges from 20 to 40 mm Hg. Recent studies comparing PbO2 values to PET measurements of oxygen extraction fraction (OEF) found that the PbO2 value associated with an OEF of 40% (the mean value for OEF in normal subjects) was 14 mm Hg (14). Values of PbO2 that indicate tissue hypoxia/ischemia are probably considerably less than 14 mm Hg. Serial measurements of both SjO2 and PbO2 suggest that a PbO2 of 8.5 mm Hg indicates a similar level of oxygenation as a SjO2 of 50% (15).
Prospective studies have demonstrated that PbO2 less than 15 mm Hg is associated with poor outcome (16). Some studies have suggested that a treatment protocol aimed at keeping brain pO2 higher than 25 mm Hg may reduce mortality when compared to patients treated similarly with no brain pO2 probe (17).
Jugular Venous Oxygen Saturation
Oxygen saturation in the jugular bulb can be measured by inserting a catheter into the internal jugular vein and advancing it to the skull base. This allows measurement of the oxygen saturation of the blood exiting the brain, which provides information on the adequacy of cerebral blood flow and oxygen delivery to the brain (5). Episodes of jugular venous oxygen desaturation are associated with worse neurologic outcome (18). Increased SjO2 may indicate decreased oxygen uptake in the brain (19). The major limitation of SjO2monitoring is that it cannot detect local ischemia within the brain.
|
|
|
Figure 146.2. Location of the brain tissue oxygen tension (PbO2) catheter relative to the injured brain determines the nature of the pO2 information that will be obtained. On the left is a patient where the PbO2 catheter was placed in relatively normal brain opposite a temporal contusion. The PbO2 reflected the global oxygenation of the brain measured with jugular venous oxygen saturation (SjO2). On the right is a patient where the PbO2 catheter was placed near a contusion. As this contusion evolved, the PbO2 decreased even though the global measures (SjO2 and cerebral perfusion pressure [CPP]) remained unchanged. MAP, mean arterial pressure. |
Normal values for SjO2 are better established than for PbO2. Gibbs et al. (20) studied 50 normal young males and observed that their SjO2 ranged from 55% to 71% (mean of 61.8%). Some studies suggest that normal SjO2 values may be as low as 45% (21). Normal SjO2 is lower than normal mixed venous oxygen saturation, indicating that the brain normally extracts oxygen more completely from arterial blood than do many other organs.
Clinical Pearl
Brain oxygenation can be measured globally at a single time point using PET imaging. Global brain oxygenation can be monitored continuously using a catheter inserted into the internal jugular vein to measure the oxygen saturation of venous blood from the brain. Local brain oxygen tension can be measured continuously by inserting a pO2 probe directly into the brain parenchyma.
What is the Most Important Physiologic End Point (ICP, CPP, or Brain Oxygenation)?
As it has become possible to measure additional brain-specific physiologic parameters in the ICU, different management strategies have evolved that place special emphasis on parameters other than ICP. For example, some have advocated that ICP is not important as long as CPP is maintained. This philosophy led to the use of CPP-directed therapy where induced hypertension was used to drive CPP to high levels even though ICP was also increased by the therapy (22). However, all of these physiologic parameters are related to outcome, and there is no clear evidence that one parameter is more important than the others. Table 146.1 presents normal values and treatment thresholds for these parameters.
The best circumstance occurs when ICP, CPP, and brain oxygenation are all maintained in normal ranges, and this should probably be the goal of management. When this is not possible, it is important to understand the limitations of each of the monitors when making therapeutic decisions. Additionally, clinical studies are needed to demonstrate what management strategies may best improve neurologic outcome.
|
Table 146.1 Normal values and treatment thresholds for physiologic parameters |
||||||||||||||||||
|
||||||||||||||||||
Immediate Concerns for Treatment
Identification of Patients with Increased ICP
A patient with mildly increased ICP can present with complaints of headache and blurred vision. Further increases in ICP are associated with decreased level of consciousness and symptoms of herniation. Herniation of the temporal lobe over the edge of the tentorium can compress cranial nerve III causing ipsilateral pupillary dilation and decreased reaction to light. Direct compression of the brainstem can cause contralateral posturing or hemiparesis, although if the brainstem is displaced and compressed against the opposite side of the tentorium, there may be ipsilateral symptoms, called Kernohan notch phenomenon. Further symptoms of herniation are hypertension, bradycardia, and widening pulse pressure, which make up the Cushing triad. Respiratory abnormalities may be present, including Cheyne-Stokes respiration, hypoventilation, and central neurogenic hyperventilation. Any patient in whom elevated ICP is suspected should undergo noncontrast CT scan of the brain to evaluate for mass lesions, hydrocephalus, subarachnoid hemorrhage, or other treatable causes.
Clinical Pearl
Symptoms concerning for elevated ICP include decreased level of consciousness, a fixed and dilated pupil, decorticate or decerebrate posturing, or hemiparesis.
Initial Stabilization and Management of Patients with Elevated ICP
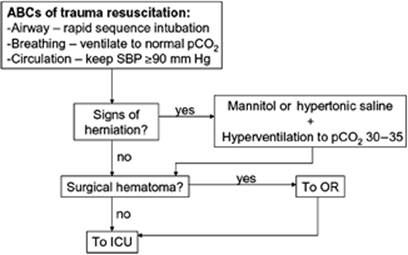
The initial steps in managing any patient with elevated ICP (Fig. 146.3) are defined by the ABCs of trauma resuscitation. Episodes of hypoxemia (arterial pO2 <60 mm Hg) or hypotension (systolic blood pressure <90 mm Hg) have been associated with significantly worse outcome in patients with traumatic brain injury (10,23,24,25,26).
|
|
|
Figure 146.3. Algorithm for initial management of patient with elevated intracranial pressure. ICU, intensive care unit; OR, operating room; SBP, systolic blood pressure. |
Airway protection is essential (27). All patients with a Glasgow coma score (GCS) of 8 or less should be intubated to protect the airway. Patients with GCS above 8 may also need intubation if they cannot adequately protect their airway (28). Supplemental oxygen and mechanical ventilation may be necessary to avoid hypoxemia.
Ideally, airway protection should start in the field. However, some studies have suggested that intubation of brain-injured patients in the field results in higher mortality rates (29). Paramedics must be adequately trained in the technique of rapid-sequence intubation, excessive hyperventilation should be avoided following intubation, and this procedure should not significantly delay transport of the patient (30).
Blood pressure should be supported with fluid resuscitation and vasopressors as necessary. Hypotension should not be attributed to the brain injury unless all other possible causes have been excluded. Patients with elevated ICP should be cared for in a dedicated neurologic intensive care unit with a neurosurgeon included in the care team (31,32).
Immediate aggressive management of elevated ICP should be initiated in patients who demonstrate signs and symptoms of herniation. Mild hyperventilation, to a paCO2 of 30 to 35 mm Hg, can reduce intracranial pressure by constricting cerebral blood vessels. Once fluid resuscitation has been completed, osmotic therapy with either mannitol or hypertonic saline should be initiated. The standard dose for mannitol is an intravenous bolus of 1 g/kg. In patients with a subdural hematoma and signs of herniation, early preoperative administration of high-dose mannitol (1.4 g/kg) significantly improved outcome in one study (33). Mannitol should only be given once fluid resuscitation has been completed as it does result in an osmotic diuresis and can cause hypotension in the incompletely resuscitated patient. An alternative osmotic agent is hypertonic saline, which may have an advantage in the hypotensive patient since it does not induce diuresis. Resuscitation with hypertonic saline has been demonstrated to result in less hypotension when compared with mannitol but has not clearly been shown to result in an improved outcome.
If a surgical lesion is identified, the patient should be immediately taken to the operating room for evacuation of the lesion. Invasive monitoring devices can be inserted either at the bedside or in the operating room if surgery is required. At a minimum, an ICP monitor, preferably a ventriculostomy drain, should be inserted. Other monitoring devices that can be used if available include a brain PbO2 monitor and a SjO2 catheter. An arterial line and a Foley catheter should also be placed. A central venous catheter may be needed, especially if hypotension is present or large doses of mannitol are needed.
|
|
|
Figure 146.4. Algorithm for treatment of elevated intracranial pressure (ICP). CT, computed tomography; CSF, cerebrospinal fluid. |
Clinical Pearl
Initial management of the patient with elevated ICP should focus on the ABCs of the Advanced Trauma Life Support (ATLS) system. When signs of herniation are present, mannitol or hypertonic saline should be given. Early administration of high-dose mannitol (1.4 g/kg) may improve outcome for patients with subdural hematoma and signs of herniation. Surgical mass lesions should be identified and promptly evacuated.
Treatment of Elevated Intracranial Pressure— Principles of CNS Resuscitation
Principles of management of intracranial hypertension (Fig. 146.4) include general measures that are used in all patients to minimize factors that exacerbate ICP, first-line therapies that are applied to patients who subsequently have elevated ICP, and additional treatments that can be used if elevated ICP is refractory to these first-line measures.
General Measures
Prevention or treatment of factors that may aggravate or precipitate intracranial hypertension is the cornerstone of central nervous system (CNS) resuscitation. Specific factors that may aggravate intracranial hypertension include obstruction of venous return (head position, agitation), respiratory problems (airway obstruction, hypoxia, hypercapnia), fever, severe hypertension, hyponatremia, anemia, and seizures. Routine critical care management of the patient at risk for intracranial hypertension should include measures to prevent these factors. Also important are general strategies for maintaining normal brain oxygenation and cerebral perfusion pressure.
Minimize Obstruction to Venous Return
Elevation of the head of the bed and keeping the head in a neutral position to minimize compression of venous return from the brain has been standard neurosurgery practice for management of ICP. Elevation of the head to 30 degrees results in a reduction in ICP without a reduction in either CPP or CBF in most patients (34,35). If head elevation is used, it is important to remember that both the ICP and blood pressure transducers should be zeroed at the same level, i.e., at the level of the foramen of Monro (36).
In multiple-trauma patients, where abdominal injury may also be present, an increased intra-abdominal pressure may also impede venous return from the brain, decrease blood pressure (BP), and increase ICP. Abdominal decompression can improve control of ICP when abdominal compartment syndrome is present (37).
Prevent Fever
Fever is common during the recovery from a head injury. In experimental studies, postinjury fever worsens the outcome from fluid percussion injury (38). Fever is a potent cerebral vasodilator and can raise ICP. In addition, fever can raise cerebral metabolic requirements. Efficient external cooling systems as well as intravascular cooling devices are available and can maintain normal body temperature. When fever occurs, infectious causes should be investigated with appropriate cultures and treated with antibiotics.
Prevent Seizures
Seizures occur in approximately 15% of patients with head injury (39). The risk of seizures is related to the severity of injury.
In a study of 4,541 patients with head injury, the standardized incidence ratio for developing seizures was 1.5 after mild injuries but with no increase over the expected number after 5 years, 2.9 after moderate injuries, and 17.0 after severe injuries (40).
Since patients are often sedated and pharmacologically paralyzed to treat intracranial hypertension, clinical monitoring is often not helpful. Continuous electroencephalogram (EEG) monitoring may be useful for this high-risk group of patients (41).
The use of anticonvulsants to prevent seizures is controversial. Although seizures can dramatically increase cerebral metabolic rate, there is not a clear relationship between the occurrence of early seizures and a worse neurologic outcome (42). Young et al. (43) found no difference in the incidence of seizures with prophylactically administered phenytoin. Temkin et al. (44) recently reported results from a double-blind study in which 404 severely head-injured patients randomly received phenytoin or placebo for 1 year. Phenytoin reduced the incidence of seizures during the first week but not thereafter.
Maintain Brain Oxygenation
Oxygen delivery to the brain is dependent on oxygen content of the blood and cerebral blood flow. Oxygen content of the blood can be increased by ensuring an adequate hemoglobin concentration and by increasing arterial pO2. The optimal hemoglobin concentration for tissue oxygenation is approximately 10 g/dL (45). A lower hemoglobin concentration reduces the oxygen-carrying capacity of the blood more than it improves viscosity. A higher hemoglobin concentration increases viscosity and reduces CBF even though it increases oxygen-carrying capacity.
Increasing arterial pO2 after hemoglobin is nearly 100% saturated increases arterial oxygen content only by a small amount, i.e., by the amount of oxygen dissolved in the blood. However, if tissues are ischemic, even small increases in oxygen content can be important. In addition, studies using PET imaging have suggested that there may be impaired diffusion of oxygen in injured brain tissue (46). Other studies demonstrate mitochondrial dysfunction in injured brain (47). Hyperoxia may improve tissue oxygenation under these conditions. Menzel et al. (48) have observed an increase in PbO2 and a decrease in extracellular lactate concentration in the brain, measured by microdialysis, when patients with very low baseline PbO2 were placed on 100% oxygen. The reduction in lactate accumulation with this therapy suggests that the increase in PbO2 altered ischemic cerebral metabolism favorably.
Maintain Cerebral Perfusion Pressure
Initial volume resuscitation should be aimed at achieving euvolemia. Once an ICP monitor is in place, further blood pressure management should be aimed at maintaining a CPP of at least 60 mm Hg (13). Boluses of intravenous fluid can be used as initial treatment of low CPP. All intravenous fluids given should be isotonic to avoid worsening cerebral edema. If fluid resuscitation is not sufficient to maintain CPP, treatment with pressors should be initiated. Some studies have suggested that treatment with norepinephrine results in a more predictable blood pressure response than treatment with dopamine (49). Treating to a CPP higher than 60 mm Hg has not been shown to improve outcome but does increase the risk of ARDS (50).
First-line Therapies of Intracranial Hypertension
When ICP becomes elevated despite measures to remove exacerbating factors, the first-line therapies include sedation and neuromuscular blockade, osmotic therapy, CSF drainage, and mild hyperventilation.
Sedation and Paralysis
Sedative/analgesic drugs blunt the effect that the routine nursing care of patients has on intracranial pressure. Routine paralysis of all patients with severe head injury increases the risk of pulmonary complications and prolongs the ICU stay (51). However, ICP raised by agitation, posturing, or coughing should be prevented by narcotics and nondepolarizing muscle relaxants that do not alter cerebrovascular resistance. A reasonable regimen is morphine and lorazepam for analgesia/sedation and cisatracurium or vecuronium as a muscle relaxant, with the dose titrated by twitch response to stimulation (52). Although the neurologic examination cannot be closely monitored while the patient is paralyzed, muscle relaxants can be withheld once a day, usually before morning rounds, to obtain a brief neurologic examination.
Osmotic Therapy to Reduce Intracranial Pressure
One of the mainstays of treatment of elevated ICP is osmotic therapy with either mannitol or hypertonic saline. This treatment can be initiated prior to insertion of an ICP monitor if signs and symptoms of herniation are present. The initial dose of mannitol is 1g/kg. Once an ICP monitor has been inserted, further bolus doses can be given to maintain ICP <20 to 25 mm Hg. Bolus dosing of 0.25 mg to 0.5 mg per kg every 2 to 6 hours can be given if the serum osmolality is less than 320. Dosing to a serum osmolality of greater than 320 has not been demonstrated to improve outcome and increases the risk of acute renal failure.
Mannitol is thought to reduce ICP through two mechanisms (53). The first mechanism of action is an immediate expansion of the intravascular volume, resulting in reduced hematocrit and reduced blood viscosity that causes decreased ICP and increased cerebral blood flow and oxygen delivery. The second mechanism of action involves the establishment of osmotic gradients between the plasma and the cells, resulting in decreased cellular volume. Long-term treatment with mannitol can result in build-up of mannitol within the cells. This effect is more marked with continuous infusion of mannitol.
Although mannitol has been more widely studied, some studies have suggested that hypertonic saline is more effective at lowering intracranial pressure. These studies have been small and have not demonstrated a statistically significant difference in outcome. The usual dose form is boluses of 7.5% hypertonic saline (54). Other agents that are under investigation include hypertonic saline hetastarch, with studies comparing bolus doses of 7.2% hypertonic saline hetastarch 200/0.5 with 15% mannitol showing some improved reduction in ICP with the hypertonic saline hetastarch but no significant difference in outcome (55). Another study comparing 7.5% hypertonic saline/6% dextran solution to bolus dosing of 20% mannitol showed improved reduction of ICP but no difference in outcome (56). Further large randomized controlled studies are necessary to define the role of hypertonic saline in the treatment of increased ICP.
Clinical Pearl
Osmotic therapy is one of the mainstays of treatment for elevated ICP. Mannitol can be dosed with an initial bolus of 1 g/kg. Further bolus doses of 0.25 to 0.5 g/kg can be given as necessary. Hypertonic saline is another agent that can be used.
Drainage of Cerebrospinal Fluid
Placement of a ventriculostomy catheter allows drainage of CSF during episodes of increased ICP. Drainage of 3 to 5 mL of CSF can reduce ICP. CSF drainage is a mainstay of therapy for hydrocephalus but may be useful in more diffuse processes where the ventricles are effaced by brain swelling. If CSF is to be drained continuously, the drainage system should not be set lower than 10 cm above the level of foramen of Monro, which is approximated by the external auditory canal.
Hyperventilation
Hyperventilation, to a paCO2 of 30 to 35 mm Hg, can reduce intracranial pressure by constricting cerebral blood vessels and reducing cerebral blood volume. The effect of changes in paCO2 on cerebral vessels is mediated by the change in pH induced in the extracellular fluid (57). The effects of hyperventilation on ICP are immediate, but the duration of the effect is brief because the pH of the brain, at least in normal individuals, soon equilibrates to the lower pCO2 level.
Long-term hyperventilation (to paCO2 of 25–30 mm Hg) has been demonstrated to result in worse outcomes after traumatic brain injury, possibly secondary to reduction in CBF (58). The authors of this study recommended using hyperventilation only in patients with intracranial hypertension rather than as a routine in all head-injured patients.
In patients who have been chronically hyperventilated, abruptly returning the pCO2 to normal can result in a dramatic increase in ICP. In experimental studies, this phenomenon occurs after 24 hours of hyperventilation and is associated with vasodilation of cerebral vessels as the CSF pH equilibrates at the new lower pCO2 level (59). Hyperventilation should be withdrawn over several days to avoid this increase in ICP.
Additional Treatments for Refractory Intracranial Hypertension
All of the treatments outlined below have been shown in clinical studies to significantly reduce ICP, even when the ICP is refractory to initial treatments. However, none of these treatments have been demonstrated to improve neurologic outcome. There are also no data to suggest which of these treatments is most effective or has the least morbidity. For these reasons, such additional therapies are usually applied selectively to patients who are judged to have some potential for neurologic recovery if their ICP can be controlled.
When refractory intracranial hypertension occurs, it is also important to consider whether a delayed intracranial hematoma may have developed. A follow-up CT scan may be indicated before advancing to these additional treatments of elevated ICP. Any surgical intracranial hematoma should be evacuated.
Pentobarbital Coma
If CSF drainage and mannitol fail to control elevated ICP, other techniques need to be considered. One intervention that can lower ICP is administration of pentobarbital to achieve burst suppression on EEG. However, routine use of barbiturates in unselected patients has not been consistently effective in reducing morbidity or mortality after severe head injury (60,61). A randomized multicenter trial demonstrated that instituting barbiturate coma in patients with refractory intracranial hypertension resulted in a twofold greater chance of controlling the ICP (62).
A simple dosing scheme from the randomized clinical trial is a loading dose of 10 mg/kg intravenously (IV) over 30 minutes followed by 5 mg/kg per hour for three doses followed by a maintenance dose of 1 mg/kg per hour (62). The maintenance dose rate should be titrated to achieve a level of 3.5 to 5.0 mg% (35–50 ug/mL). If available, continuous EEG may be used to monitor for burst suppression to ensure maximal therapeutic effect. Significant hypotension may develop during administration of the loading dose (61). Consideration should be given to placement of a Swan-Ganz catheter for continuous cardiac monitoring during loading. The initial loading dose should be given by slow intravenous push with close monitoring of the blood pressure. Hypotension during induction of barbiturate coma usually responds to fluid bolus. Higher doses of pentobarbital can cause myocardial depression and require inotropic or even vasopressor support. Laboratory studies suggest that for the treatment of hypotension associated with barbiturate coma, volume resuscitation may be better than dopamine (63). In these studies, dopamine infusion increased cerebral metabolic requirements and partially offset the beneficial effects of barbiturates on metabolism.
The goal of treatment is to reduce ICP to <20 to 25 mm Hg. After ICP has been controlled for 24 to 48 hours, pentobarbital can be weaned and stopped. If ICP increases during reduction of pentobarbital, stop titration and increase the dose until ICP is controlled. Pentobarbital is thought to work through multiple mechanisms, including alterations in vascular tone, decreased cerebral metabolism, and decreased free radical production (64,65).
Pentobarbital coma is associated with significant morbidity. Hypotension is a common complication (61) and may require concomitant administration of vasopressors, which should be available when loading. Pentobarbital coma increases the risk of pneumonia, pressure ulcers, and paralytic ileus.
Several studies have tried to predict which patient would be most likely to respond to pentobarbital coma. Characteristics that may indicate a favorable ICP response to barbiturates include younger age, diffuse rather than focal brain injury, a relatively high level of brain electrical activity, and a high cerebral metabolic rate prior to treatment (66,67). Preservation of CO2 reactivity predicted a good ICP response to barbiturates in one study (68).
Clinical Pearl
Routine use of barbiturate coma does not improve neurologic outcome. If CSF drainage and osmotic therapy are not sufficient to control elevated ICP, barbiturate coma has been shown to double the chances of controlling ICP. Hypotension is the most common complication of barbiturate coma.
Decompressive Craniectomy
Initially described by Kocher in 1901, craniectomy for control of elevated intracranial pressure has a long history. Studies in the 1970s initially showed improved outcome (69) resulting in increased usage of the procedure, but follow-up studies did not demonstrate the same improved outcome (70) and craniectomy became less popular. More recent studies have demonstrated benefit (71) and craniectomy is being used more often for control of elevated ICP. Craniectomy can be considered in patients with elevated intracranial pressure uncontrolled by CSF drainage and osmotic therapy. If the injury primarily involves a single hemisphere of the brain, a hemicraniectomy can provide adequate decompression. A large bone flap should be removed with particular focus on decompressing the anterior temporal lobe and visualization of the floor of the middle fossa. Diffuse injury involving both hemispheres may necessitate bifrontal craniectomy, with removal of a large bone flap and decompression of both temporal lobes and both frontal lobes.
Craniectomy has been demonstrated to result in a significant decrease in ICP and in retrospective review has been suggested to improve outcome (72), although further prospective trials are needed. Two randomized clinical trials of decompressive craniectomy, Rescue ICP (73) and DECRAN, for traumatic brain injury are currently underway. Craniectomy can also be useful in treatment of elevated ICP secondary to malignant middle cerebral artery stroke, with significant reduction in mortality and improved outcome if performed prior to the development of symptoms of herniation (74,75,76).
Hypothermia
Hypothermia has several potentially neuroprotective effects, including reducing cerebral metabolism and decreasing ICP. Although a randomized, controlled trial in humans did not show a significant improvement in neurologic outcome, ICP was better controlled during hypothermia (77). Routine induction of hypothermia is not indicated at present, but hypothermia may be an effective adjunctive treatment for increased ICP refractory to other medical management.
Steroids
Multiple randomized, controlled studies have demonstrated no benefit in treating patients with traumatic brain injury with steroids (78). The recently completed Corticosteroid Randomization After Significant Head injury (CRASH) trial observed an increased risk of death in patients receiving methylprednisolone for 48 hours after injury (79). As a result, steroids are not recommended for treatment of head injury. Steroids are also not recommended for treatment of the cellular edema accompanying stroke.
Steroids, however, can be useful in treating vasogenic edema associated with brain tumors or selected infections such as neurocysticercosis (80). Dosing schemes are relatively arbitrary with a typical dosing scheme for a patient with significant symptomatic vasogenic edema from a tumor being dexamethasone, 4 mg IV every 6 hours.
Completion of Treatment
Treatment should be titrated off once ICP has been controlled below 20 mm Hg for 24 to 48 hours. ICP monitors can be removed if ICP is stable below 20 mm Hg for 48 hours off any intervention. It is important to remember that patients may develop delayed increase in ICP secondary to blossoming of contusions, evolution of a stroke, or development of new mass lesions.
Outcome with Intracranial Hypertension
Patients with traumatic brain injury can be classified based on initial GCS. Patients with mild head injury, with a GCS of 13 to 15, have a minimal chance (3%) of deteriorating into a coma and developing elevated ICP. Patients with moderate head injury, with a GCS from 9 to 12, have a moderate chance (10%–20%) of deteriorating into a coma and developing elevated ICP. Patients with a GCS of 8 or less, defined as a severe head injury, have the highest chance of developing elevated ICP and also have the worst outcome. Patients classified as having a severe head injury are more likely to develop increased ICP if they have an abnormal head CT or if they meet two of the three criteria of age older than 40 years, unilateral or bilateral motor posturing, or SBP <90 mm Hg (81).
No study clearly demonstrates that monitoring and treating ICP improves neurologic outcome, but there are strong associations between intracranial hypertension and a poor outcome, and the development of refractory intracranial hypertension has a very high mortality rate (7,8,9,10,60). In the randomized trial of pentobarbital coma for refractory intracranial hypertension, control of the elevated ICP determined the outcome, with 92% of patients where ICP was controlled surviving, and 83% of patients where ICP was not controlled dying (62).
Historically, institution of treatment protocols aimed at controlling elevated intracranial pressure has significantly improved outcome in traumatic brain injury. Jennett et al. (82) in 1977 reported a mortality rate of 50% in comatose patients (GCS <8) in a cohort treated without ICP monitoring. Becker et al. (83) reported significantly lower mortality of 30% in a similar patient cohort using ICP monitoring. Mortality rates at 30 days for patients with severe brain injury have been slowly declining with mortality of patients in the Traumatic Coma Data Bank reduced from 39% in 1984 to 27% in 1996 (84).
The availability of multiple monitoring modalities has provided more information on which patients have poor outcomes and has provided treatment goals. Treatment strategies are now aimed at maintaining ICP less than 20–25 mm Hg, maintaining CPP greater than 60 mm Hg, and maintaining brain tissue oxygenation at greater than 10 mm Hg. Even with these advances in treatment, traumatic brain injury is still associated with significant morbidity and mortality in both the short and long term.
Summary
Elevated intracranial pressure can be caused by multiple injuries, including stroke, subarachnoid hemorrhage, mass lesion, hydrocephalus, and traumatic brain injury. Initial management is focused on maintaining oxygenation and perfusion using the ABCs of resuscitation. If signs of herniation are present, an initial bolus of mannitol should be given. Surgical mass lesions should be identified and evacuated. A ventriculostomy catheter should be inserted to allow monitoring of ICP. Treatment should be initiated with the goal of maintaining ICP below 20 to 25 mm Hg. Cerebral perfusion pressure should be maintained at 60 mm Hg. Initial treatments of elevated ICP include sedation and paralysis, drainage of CSF, mild hyperventilation, and bolus administration of osmotic agents such as mannitol. If ICP is not controlled with these measures, additional treatments including pentobarbital coma, hypothermia, or decompressive craniectomy can be considered. Other parameters that can be monitored and treated include jugular venous oxygen saturation and brain tissue oxygenation. Therapy should be continued until ICP is controlled at less than 20 mm Hg for 24 to 48 hours. Mortality rates for severe head injury have shown a steady decrease but are still 27%.
References
1. Marmarou A, Signoretti S, Fatouros PP, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104:720.
2. Hlatky R, Furuya Y, Valadka AB, et al. Dynamic autoregulatory response after severe head injury. J Neurosurg. 2002;97:1054.
3. Bouma GJ, Muizelaar JP, Choi SC, et al. Cerebral blood flow and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg. 1991;75:685.
4. Hlatky R, Contant CF, Diaz Marchan P, et al. Significance of a reduced CBF during the first 12 hours following traumatic brain injury. Neurocrit Care. 2004;1:69.
5. Robertson CS, Gopinatb SP, Goodman JC, et al. SjvO2 monitoring in head-injured patients. J Neurotrauma. 1995;12:891.
6. Gopinath SP, Valadka A, Uzura M, et al. Comparison of brain tissue pO2 and jugular venous oxygen saturation as monitors of cerebral oxygenation. Crit Care Med. 1999;27:2337.
7. Marshall LF, Smith RW, Shapiro HM. The outcome with aggressive treatment in severe head injuries, I: significance of intracranial pressure monitoring. J Neurosurg. 1979;50:20.
8. Miller JD, Butterworth J, Gudeman SK, et al. Further experience in the management of severe head injury. J Neurosurg. 1981;54:289.
9. Saul TG, Ducker TB. Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. J Neurosurg. 1982;56:498.
10. Marmarou A, Anderson RL, Ward JD, et al. Impact of ICP instability and hypotension on outcome in patients with severe head injury. J Neurosurg. 1991;75:S59.
11. Bullock RM, Chesnut R, Clifton GL, et al. Management and prognosis of severe traumatic brain injury, 1: guidelines for the management of severe traumatic brain injury: recommendations for intracranial pressure monitoring technology. J Neurotrauma. 2000;17:497.
12. Zabramski JM, Whiting D, Darouiche RO, et al. Efficacy of antimicrobial-impregnated external ventricular drain catheters: a prospective, randomized, controlled trial. J Neurosurg. 2003;98:725.
13. Robertson CS, Valadka AB, Hannay HJ, et al. Prevention of secondary insults after severe head injury. Crit Care Med. 1999;27:2086.
14. Johnston AJ, Steiner LA, Coles JP, et al. Effect of cerebral perfusion pressure augmentation on regional oxygenation and metabolism after head injury. Crit Care Med. 2005;33:189.
15. Kiening KL, Unterberg AW, Bardt TF, et al. Monitoring of cerebral oxygenation in patients with severe head injuries: brain tissue PO2 versus jugular vein oxygen saturation. J Neurosurg. 1996;85:751.
16. Valadka A, Gopinath SP, Contant CF, et al. Critical values for brain tissue PO2 to outcome after severe head injury. Crit Care Med. 1998;26:1576.
17. Stiefel MF, Spiotta A, Gracias VH, et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J Neurosurg. 2005;103:805.
18. Gopinath SP, Robertson CS, Contant CF, et al. Jugular venous desaturation and outcome after head injury. J Neurol Neurosurg Psych. 1994;57:717.
19. Cormio M, Valadka AB, Robertson CS. Elevated jugular bulb oxygen saturation after severe head injury. J Neurosurg. 1999;90:9.
20. Gibbs EL, Lennox WG, Nims LF, et al. Arterial and cerebral venous blood. Arterial-venous differences in man. J Biol Chem. 1942;144:325.
21. Chieregato A, Calzolari F, Trasforini G, et al. Normal jugular bulb oxygen saturation. J Neurol Neurosurg Psychiatry 2003;74:784.
22. Rosner MJ, Daughton S. Cerebral perfusion pressure management in head injury. J Trauma. 1993;30:933.
23. Fearnside MR, Cook RJ, McDougall P, et al. The Westmead Head Injury Project outcome in severe head injury. A comparative analysis of pre-hospital, clinical and CT variables. Br J Neurosurg. 1993;7:267.
24. Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216.
25. Miller JD. Head injury and brain ischaemia—implications for therapy. Br J Anaesth. 1985;57:120.
26. Pigula FA, Wald SL, Shackford SR, et al. The effect of hypotension and hypoxia on children with severe head injuries. J Pediatr Surg. 1993;28:310.
27. Winchell RJ, Hoyt DB. Endotracheal intubation in the field improves survival in patients with severe head injury. Trauma Research and Education Foundation of San Diego. Arch Surg. 1997;132:592.
28. Hsiao AK, Michelson SP, Hedges JR. Emergent intubation and CT scan pathology of blunt trauma patients with Glasgow Coma Scale scores of 3-13. Prehosp Disaster Med. 1993;8:229.
29. Davis DP, Hoyt DB, Ochs M, et al. The effect of paramedic rapid sequence intubation on outcome in patients with severe traumatic brain injury. J Trauma. 2003;54:444.
30. Davis DP, Fakhry SM, Wang HE, et al. Paramedic rapid sequence intubation for severe traumatic brain injury: perspectives from an expert panel. Prehosp Emerg Care. 2007;11:1.
31. American College of Surgeons Committee on Trauma. Resources for the Optimal Care of the Injured Patient. Chicago: American College of Surgeons. 2006.
32. Bullock RM, Chesnut R, Clifton GL, et al. Management and prognosis of severe traumatic brain injury, 1: guidelines for the management of severe traumatic brain injury: trauma systems. J Neurotrauma. 2000;17:457.
33. Cruz J, Minoja G, Okuchi K. Improving clinical outcomes from acute subdural hematomas with the emergency preoperative administration of high doses of mannitol: a randomized trial. Neurosurgery. 2001;49:864.
34. Feldman Z, Kanter MJ, Robertson CS, et al. Effect of head elevation on intracranial pressure, cerebral perfusion pressure, and cerebral blood flow in head-injured patients. J Neurosurg. 1992;76:207.
35. Meixensberger J, Baunach S, Amschler J, et al. Influence of body position on tissue-pO2, cerebral perfusion pressure and intracranial pressure in patients with acute brain injury. Neurol Res. 1997;19:249.
36. Rosner MJ, Coley IB. Cerebral perfusion pressure, intracranial pressure, and head elevation. J Neurosurg. 1986;65:636.
37. Bloomfield GL, Dalton JM, Sugerman HJ, et al. Treatment of increasing intracranial pressure secondary to the acute abdominal compartment syndrome in a patient with combined abdominal and head trauma. J Trauma. 1995;39:1168.
38. Dietrich WD, Alonso O, Halley M, et al. Delayed posttraumatic brain hyperthermia worsens outcome after fluid percussion brain injury: a light and electron microscopic study in rats. Neurosurgery. 1996;38:533.
39. Jennett B. Epilepsy after Nonmissile Head Injuries. 2nd ed. London, England: Heineman; 1976.
40. Annegers JF, Hauser WA, Coan SP, et al. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20.
41. Vespa PM, Nenov V, Nuwer MR. Continuous EEG monitoring in the intensive care unit: early findings and clinical efficacy. J Clin Neurophysiol. 1999;16:1.
42. Lee ST, Lui TN, Wong CW, et al. Early seizures after severe closed head injury. Can J Neurol Sci. 1997;24:40.
43. Young B, Rapp RP, Norton NA, et al. Failure of prophylactically administered phenytoin to prevent early posttraumatic seizures. J Neurosurg. 1983;58:231.
44. Temkin NR, Dikmen SS, Wilensky AJ, et al. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497.
45. Kee DB, Wood JH. Rheology of the cerebral circulation. Neurosurgery. 1984;15:125.
46. Menon DK, Coles JP, Gupta AK, et al. Diffusion limited oxygen delivery following head injury. Crit Care Med. 2004;32:1384.
47. Daugherty WP, Levasseur JE, Sun D, et al. Effects of hyperbaric oxygen therapy on cerebral oxygenation and mitochondrial function following moderate lateral fluid-percussion injury in rats. J Neurosurg. 2004;101:499.
48. Menzel M, Doppenberg EMR, Zauner A, et al. Increased inspired oxygen concentration as a factor in improved brain tissue oxygenation and tissue lactate levels after severe human head injury. J Neurosurg. 1999;91:1.
49. Steiner LA, Johnston AJ, Czosnyka M, et al. Direct comparison of cerebrovascular effects of norepinephrine and dopamine in head-injured patients. Crit Care Med. 2004;32:1049.
50. Contant CF, Valadka AB, Hannay HJ, et al. ARDS as a complication of induced hypertension after severe head injury. J Neurosurg. 2003;95:560.
51. Hsiang J, Chesnut RM, Crisp CB, et al. Early, routine paralysis for ICP control in severe head injury: Is it necessary? Crit Care Med. 1994;22:1471.
52. Schramm WM, Jesenko R, Bartunek A, et al. Effects of cisatracurium on cerebral and cardiovascular hemodynamics in patients with severe brain injury. Acta Anaesthesiol Scand. 1997;41:1319.
53. Muizelaar JP, Lutz HA, Becker DP. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984;61:700.
54. Vialet R, Albanese J, Thomachot L, et al. Isovolume hypertonic solutes (sodium chloride or mannitol) in the treatment of refractory posttraumatic intracranial hypertension: 2 mL/kg 7.5% saline is more effective than 2 mL/kg 20% mannitol. Crit Care Med. 2003;31:1683.
55. Harutjunyan L, Holz C, Rieger A, et al. Efficiency of 7.2% hypertonic saline hydroxyethyl starch 200/0.5 versus mannitol 15% in the treatment of increased intracranial pressure in neurosurgical patients—a randomized clinical trial [ISRCTN62699180]. Crit Care. 2005;9:R530.
56. Battison C, Andrews PJ, Graham C, et al. Randomized, controlled trial on the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury. Crit Care Med. 2005;33:196.
57. Kontos HA, Wei EP, Raper AJ, et al. Local mechanism of CO2 action on cat pial arterioles. Stroke. 1977;8:226.
58. Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731.
59. Muizelaar JP, van der Poel HG, Li ZC, et al. Pial artery diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. 1988;69:923.
60. Schwartz ML, Tator CH, Rowed DW, et al. The University of Toronto Head Injury Treatment Study: A prospective, randomized comparison of pentobarbital and mannitol. Can J Neurol Sci. 1984;11:434.
61. Ward JD, Becker DP, Miller JD, et al. Failure of prophylactic barbiturate coma in the treatment of severe head injury. J Neurosurg. 1985;62:383.
62. Eisenberg HM, Frankowski RF, Contant CF, et al. High-dose barbiturate control of elevated intracranial pressure in patients with severe head injury. J Neurosurg. 1988;69:15.
63. Sato M, Niiyama K, Kuroda R, et al. Influence of dopamine on cerebral blood flow, and metabolism for oxygen and glucose under barbiturate administration in cats. Acta Neurochir (Wien). 1991;110:174.
64. Goodman JC, Valadka AB, Gopinath SP, et al. Lactate and excitatory amino acids measured by microdialysis are decreased by pentobarbital coma in head-injured patients. J Neurotrauma. 1996;13:549.
65. Kassell NF, Hitchon PW, Gerk MK, et al. Alterations in cerebral blood flow, oxygen metabolism, and electrical activity produced by high dose sodium thiopental. Neurosurgery. 1980;7:598.
66. Miller JD, Piper IR, Dearden NM. Management of intracranial hypertension in head injury: matching treatment with cause. Acta Neurochir Suppl (Wien). 1993;57:152.
67. Cormio M, Gopinath SP, Valadka AB, et al. Cerebral hemodynamic effects of pentobarbital coma in head injured patients. J Neurotrauma. 1999;16:927.
68. Messeter K, Nordstrom CH, Sundbarg G, et al. Cerebral hemodynamics in patients with acute severe head trauma. J Neurosurg. 1986;64:231.
69. Ransohoff J, Benjamin V. Hemicraniectomy in the treatment of acute subdural haematoma. J Neurol Neurosurg Psychiatry. 1971;34:106.
70. Cooper PR, Rovit RL, Ransohoff J. Hemicraniectomy in the treatment of acute subdural hematoma: a re-appraisal. Surg Neurol. 1976;5:25.
71. Aarabi B, Hesdorffer DC, Ahn ES, et al. Outcome following decompressive craniectomy for malignant swelling due to severe head injury. J Neurosurg. 2006;104:469.
72. Sahuquillo J, Arikan F. Decompressive craniectomy for the treatment of refractory high intracranial pressure in traumatic brain injury. Cochrane Database Syst Rev. 2006 Jan 25;(1):CD003983.
73. Hutchinson PJ, Corteen E, Czosnyka M, et al. Decompressive craniectomy in traumatic brain injury: the randomized multicenter RESCUEicp study (www.RESCUEicp.com). Acta Neurochir Suppl. 2006;96:17.
74. Mori K, Nakao Y, Yamamoto T, et al. Early external decompressive craniectomy with duroplasty improves functional recovery in patients with massive hemispheric embolic infarction: timing and indication of decompressive surgery for malignant cerebral infarction. Surg Neurol. 2004;62:420.
75. Cho DY, Chen TC, Lee HC. Ultra-early decompressive craniectomy for malignant middle cerebral artery infarction. Surg Neurol. 2003;60:227.
76. Schwab S, Steiner T, Aschoff A, et al. Early hemicraniectomy in patients with complete middle cerebral artery infarction. Stroke. 1998;29:1888.
77. Clifton GL, Miller ER, Choi SC, et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;344:556.
78. Bullock RM, Chesnut R, Clifton GL, et al. Management and prognosis of severe traumatic brain injury, 1: guidelines for the management of severe traumatic brain injury: role of steroids. J Neurotrauma. 2000;17:531.
79. Edwards P, Arango M, Balica L, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005;365:1957.
80. French LA, Galicich JH. The use of steroids for control of cerebral edema. Clin Neurosurg. 1964;10:212.
81. Narayan RK, Kishore PRS, Becker DP, et al. Intracranial pressure: to monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg. 1982;56:650.
82. Jennett B, Teasdale G, Galbraith S, et al. Severe head injuries in three countries. J Neurol Neurosurg Psychiatry. 1977;40:291.
83. Becker DP, Miller JD, Ward JD, et al. The outcome from severe head injury with early diagnosis and intensive management. J Neurosurg. 1977;47:491.
84. Lu J, Marmarou A, Choi S, et al. Mortality from traumatic brain injury. Acta Neurochir Suppl. 2005;95:281.