Steven G. Achinger
Juan Carlos Ayus
Fluid and electrolyte disorders are very common in critically ill patients. There are some circumstances in which a patient is placed in intensive care for the management of a specific electrolyte disturbance such as hyponatremia or hyperkalemia. The development of electrolyte disturbances among critically ill patients is also common due to breakdown of homeostatic mechanisms that prevent the development of electrolyte disturbances. These impairments in homeostatic function are numerous such as renal failure, use of diuretics, and nonosmotic release of antidiuretic hormone (ADH) due to nausea, pain, or other stimuli. In this chapter the pathophysiology of electrolyte disturbances will be addressed with a focus on presentations common among intensive care unit (ICU) patients. Disorders of water balance (hyponatremia and hypernatremia) will be addressed in more detail due to the importance of these disorders as a cause of morbidity and mortality and the prevalence of impaired water balance in ICU patients.
General Comments
In an average person without extremes of weight, approximately 60% of total body weight is water. Of this total body water, two thirds (40% of total body weight) is in the intracellular (ICF) space, and one third (20% of total body weight) is outside the cells, i.e., extracellular (ECF) space. Extracellular volume is divided into intravascular space and interstitial fluid (5% and 15% of total body weight, respectively). The intravascular space containing plasma is the most mobile fluid compartment and the first to be released to areas of injury and the first to be repleted through intravenous infusion. In the intravascular space are red cells in addition to plasma. Red cell volume plus plasma volume compose the blood volume (BV), which is estimated to be 7% of the total body weight. In a 70-kg person, total body water is 42 L (60% × 70 kg), circulating BV is 4.9 L (7% × 70 kg), ICF is 28 L (40% × 70 kg), and ECF is 14 L (20% × 70 kg). The percentage of weight used will vary depending on deviation from ideal body weight and sex (see Blood Volume chapter).
Water is freely permeable between body compartments and migrates to areas of higher solutes, but this takes time. Therefore, chronic hypotonic losses with ability to “borrow” from ICF (28 L) is better tolerated than acute isotonic losses, which has only the ECF (14 L) to borrow from. Osmolality (tonicity) defined as number of particles in solution is normally 280 to 300 mOsm/L in the serum.
The ICF concentration of solutes is vastly different from that of the ECF, and approximately 80% of adenosine triphosphate (ATP) generated is used to maintain this gradient. Some of the ECF cations (in mEq/L) are as follows: sodium (Na+), 142; potassium (K+), 4; calcium (Ca), 5; and magnesium (Mg), 3. The ICF cations (in mEq/L) are: K+, 150; Mg, 40; and Na+, 10. Some of the ECF anions (in mEq/L) are chloride (Cl-), 103; and bicarbonate, 27. The major ICF anions (in mEq/L) are phosphates, 107; proteins, 40; and sulfates, 43. This difference in ICF and ECF electrolyte composition explains the clinical observations that large amounts of certain electrolytes are needed to replete small deficiencies in the serum (ECF) if the ICF stores are depleted. One of our limitations is the inability to assess ICF electrolyte composition easily at the bedside.
Due to water being freely permeable, the osmolality of all body compartments should be the same, but in reality, the protein concentration in the plasma to interstitial fluid is 16:1, generating an oncotic pressure difference between the two compartments. Starling forces describe net fluid flux between intravascular space and the interstitium:
Q = Kf{(Pc - Pi) - σ(πc - πi)}
where Q is the net fluid flux (mL/minute), (Pc - Pt) is hydrostatic pressure difference between capillary (c) and interstitium (i), and (πc - πi) is the oncotic pressure difference between the capillary and interstitium. Kf is the filtration coefficient for that membrane (mL/minute per mm Hg), and is the product of capillary surface area and capillary hydraulic conductance, and σ is the permeability factor (i.e. reflection coefficient) with one being impermeable, and zero being completely permeable.
The permeability factor explains why, in times of capillary leak as in shock states, colloids cannot maintain an oncotic pressure difference and tend to leak out into the interstitium. The general principle of fluid resuscitation is that intravascular hypovolemia should be replaced with isotonic fluid, which tends to distribute in the ECF (3:1) intravascular:interstitium. Hypotonic fluid will distribute between all body compartments with only a small amount remaining in the intravascular space (since water is freely permeable). Maintaining intravascular blood volume is of primary importance to deliver nutrition (via plasma) and oxygen (via red cells) to the tissues. Frequently used isotonic solutions are 0.9% normal saline (154 mEq/L of Na+ and Cl each) and lactated Ringer solution (130 mEq/L Na+, 109 mEq/L Cl-, 4 mEq/L K+, 3 mEq/L Ca2+, 28 mEq/L lactate).
The end point of fluid resuscitation continues to be an area of great debate since routine measurement of intravascular (plasma) volume is not the norm. Surrogate markers are used to assess adequate fluid resuscitation: blood pressure, heart rate, urine output, and parameters of perfusion and cardiac function. Although every clinician wants to treat patients to “euvolemia,” there are only a few centers measuring intravascular volume using radioisotope studies (see Blood Volume chapter). Debate will continue on how much fluid to give until an easy method of measuring intravascular volume is available.
Disorders of Water Metabolism
Overview of Water Balance
Dysnatremias (hyponatremia or hypernatremia) are among the most common electrolyte disturbances and usually are associated with poor outcomes. This problem persists due to failure to promptly recognize a life-threatening condition and initiate appropriate treatment. This chapter will focus on the pathogenesis, diagnosis, treatment, and prevention of dysnatremias.
Regulation of Water Balance
Extracellular fluid (ECF) tonicity is generally reflected in the concentration of sodium in the serum. The serum sodium is proportional to the total body exchangeable sodium (Nae) plus the total body exchangeable potassium (Ke). This critical relationship is shown mathematically in Equation 41.1. Equation 41.1 is not used clinically; rather, it illustrates the relationship between total body solutes and total body water. Decreases in potassium often accompany hyponatremia, and replacement of intracelllular solute losses can also be an important part of treating hyponatremia. As water intake and excretion are tightly regulated to maintain near-constant plasma osmolality (Fig. 41.1), disturbances in serum sodium indicate disorders in water balance, not gains or loss of sodium. This is a crucial point in understanding dysnatremias. The actions of antidiuretic hormone (ADH), also called arginine vasopressin (AVP) (1) on the kidney tightly regulates water excretion. To maintain water balance, an intact thirst mechanism and the ability of the kidneys to vary urinary concentration are required.
![]()
|
|
|
Figure 41.1. Water intake and excretion regulation. ADH, antidiuretic hormone. |
Renal Water Handling
The kidney can vary urinary concentration significantly and either excrete a large water load in very dilute urine or conserve water significantly such that the daily solute load is excreted in a small volume of urine. When the urinary filtrate passes into the cortical collecting duct, it is very dilute (as low as 50 mOsm/kg). As the urine moves through the cortical collecting duct into the collecting tubule, water reabsorption occurs in the presence of ADH. In the absence of ADH activity (as in diabetes insipidus), urine concentration will remain very low (50–80 mOsm/kg). When ADH activity is maximal, urinary concentration can be as high as 1,200 mOsm/kg. This ability to excrete very dilute or a very concentrated urine allows the body to achieve water balance across a very wide range of water intake (between approximately 0.8 L per day and 15 L per day). Impairments of this hormonal system that links perturbations in serum osmolality as detected by osmoreceptors in the hypothalamus to variations in urinary concentration can lead to impairments in water balance. Administration of water to a patient with impaired water excretion (Table 41.1) can lead to hyponatremia.
Electrolyte Free Water
Electrolyte free water is a useful concept in the approach to the patient with a disturbance in water balance. Electrolyte free water is a conceptual volume of a body fluid (usually urine) that represents the volume of that fluid that would be required to dilute the electrolytes contained within total volume of the fluid to the same tonicity as plasma electrolytes (Fig. 41.2). The remainder of the volume (total volume minus electrolyte free water) can be thought of as containing the nonelectrolyte osmoles. This nonelectrolyte water excretion is the amount of water excreted above the excretion of electrolyte solutes, and thus if it is not replaced, will have an effect on the plasma sodium concentration. In other words, the osmolality of a solution is not important in determining if it contains “free water”; rather, it is the concentration of electrolytes that is important. An example of this is that the administration of dextrose solutions provides the same amount of free water as an equal volume of deionized water, whereas 0.45% normal saline contains approximately 50% less. Electrolyte free water clearance can be calculated as a convenient clinical tool in assessing water need in a patient. The amount of electrolyte free water in a body fluid (e.g., urine, sweat, nasogastric [NG] aspirate) is calculated by the following formula:
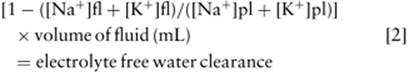
where fl is body fluid and pl is plasma.
|
Table 41.1 States of impaired water excretion in the ICU resulting in hyponatremia |
||||||||||||||
|
Clinical Application of Electrolyte Free Water Clearance
The most important conceptual point to understand is that the urine electrolytes and not the urine osmolality determine the degree of free water excretion in the urine. It is not always necessary to calculate an exact value for the electrolyte free water clearance if the relationship between the plasma electrolytes and the urine electrolytes is understood. If the concentration of electrolytes in the urine is greater than the concentration of electrolytes in the plasma, then free water is not being excreted in the urine. If the concentration of electrolytes in the urine is less than that in the plasma, then the patient is excreting free water in the urine. This relationship is illustrated in Figure 41.3. This is a simple test that can allow for a quick assessment of the ongoing losses of water in the urine.
|
|
|
Figure 41.2. Electrolyte free water. |
Hyponatremia
Hyponatremia commonly occurs in hospital settings and especially in the ICU setting. Often the condition is asymptomatic, but hyponatremic encephalopathy (brain dysfunction due to cerebral edema in turn due to hyponatremia) can result (2,3,4). Hyponatremic encephalopathy is a life-threatening medical emergency, and it must be recognized and promptly treated as it can often lead to death or devastating neurologic complications (5,6). Differentiating between these two spectrums of the disease presentation is critical. Among risk factors for life-threatening hyponatremia are female gender of premenopausal age (7), children (5), and hypoxia (7). Research over the last decade has elucidated the pathogenetic mechanisms that underlie these risk factors, and this has prompted new thinking and mandated shifts in the clinical approach to hyponatremia.
Pathogenesis of Hyponatremia
Hyponatremia is defined as a serum sodium of <135 mEq/L. The ability of the kidney to dilute the urine and thus excrete free water is the body's primary defense against the development of hyponatremia. Excess ingestion of water as the sole cause of hyponatremia is rare since the typical adult with normal renal function can excrete a massive free water load (15 L of free water per day). The combination of factors necessary for the development of hyponatremia are free water intake in the setting of an underlying condition that impairs free water excretion (Table 41.1). The states that impair water excretion are usually states where ADH release is a physiologic response to a stimulus such as volume depletion, pain, nausea, postoperative state, or congestive heart failure (due to decreased circulating blood volume). In other instances, pathologic release of ADH occurs in syndrome of inappropriate ADH release (SIADH) and with certain medications such as thiazide diuretics and anticonvulsants.
|
|
|
Figure 41.3. Relationship of electrolyte concentration in urine and plasma to the amount of free water excreted. |
Brain Defenses against Cerebral Edema
Hyponatremia induces an osmotic gradient that favors water movement into the brain leading to cerebral edema and neurologic injury. However, the brain is contained within a specialized compartment separated from the systemic circulation by the blood–brain barrier that impedes entry of water and has specialized mechanisms for handling water fluxes (8,9,10). The blood–brain barrier is a specialized structure with tight junctions between vascular endothelial cells (11,12,13) that interface with glial cells (astrocytes) on the brain side of the blood–brain barrier. Astrocytes form an important part of the microvascular compartment in the brain and project foot processes that abut the endothelial cells of the brain capillaries (14). This highly specialized cell performs many supporting functions in maintaining the fluid environment and the electrolyte milieu of the extracellular space of the brain (15,16). Among these functions is shunting of potassium from the microenvironment by uptake and release of potassium with water accompanying, in the perivascular space away from neurons. This function is accomplished through a concentration of aquaporin 4 (AQ4) water channels and Kir4.1 potassium channel located at the end-feet around the perivascular space (8). There is increasing evidence that glial cells also have an important role in brain water handling. The observation that glial cells (but not neurons) selectively swell following hypotonic stress presaged the existence of a glial-specific water pore, which has now been shown to be aquaporin 4 (AQ4). Mice lacking AQ4 do not develop cerebral edema in response to hyponatremia, suggesting that these channels may have an important role not only in normal water regulation in the brain, but also in the pathogenesis of hyponatremia-induced cerebral edema (17). During states of cytotoxic brain edema, water is shunted through the astrocyte, which swells, and the neuron is protected from this influx of water. Therefore, astrocytes are the principal regulator of the brain water content as they comprise the bulk of the intracellular space, and the response of these cells following osmolar stress is an important determinant of the changes in brain volume during hyponatremia.
The brain has several defenses against the development of cerebral edema. The first response is the shunting of cerebrospinal fluid from within the brain, but this mechanism has a limited capacity to buffer volume changes (18). Immediately after a volume stress, cell volume regulatory mechanisms in the cerebral astrocytes play an important role in reducing brain volume through reduction in cellular osmolyte (mainly electrolyte) content. These are adaptive mechanisms that are used by multiple cell types to counteract an increase in cell volume; however, the astrocyte responds to cellular swelling differently than many other cells (19). In erythrocytes, white blood cells, and epithelial cells, swelling occurs due to a hypotonic environment and calcium influx that begins a series of events termed the regulatory volume decrease (RVD) mediated by activation of K+ and Cl- channels that allow these ions to be released into the extracellular environment. In glial cells this is not the predominant response. The glial cell uses ATP-dependent mechanisms (19) that require the Na+/K+ ATPase during which ions are extruded from the glial cell and water obligatorily follows the extruded ions, reducing brain volume and protecting from the development of cerebral edema. This response is ongoing, and in animal models of acute hyponatremia, brain water content is close to the baseline value 6 hours after induction of acute hyponatremia (20). The Na+/K+ ATPase is ubiquitous and plays an essential role in cellular ion homeostasis. In the brain, this enzyme is very important in the response of the cell to volume stress and hypotonic insult (19). The enzyme has binding domains for sodium, potassium, and cardiac glycosides and requires the hydrolysis of ATP to ADP to provide energy for moving these ions against concentration gradients (21). Therefore, in vitro evidence suggests that the actions of the sodium potassium ATPase are the important immediate responses in determining the brain's response to hypo-osmolar stress (Fig. 41.4).
In summary, during times of systemic hypo-osmolality, water enters the brain through aquaporin 4 channels located in the glial cell end-feet surrounding brain capillaries. This osmotic swelling may also allow sodium into the glial cell, and expansion of the glial cell quickly ensues. Immediate shunting of cerebrospinal fluid (CSF) accommodates some of this expansion, but this is a limited mechanism. The glial cell reduces the intracellular volume by energy-dependent extrusion of solutes via the Na+/K+ ATPase pump. Several clinical factors have been shown to impair these glial cell adaptive responses, resulting in poor patient outcomes.
Clinical Manifestations
Symptoms of hyponatremia are due to osmotic swelling of the brain that accompanies the decrease in plasma osmolality. Manifestations are varied, and they depend on the degree of central nervous system adaptation to hypo-osmolality. Significant degrees of hyponatremia can be asymptomatic, such as chronic hyponatremia secondary to cirrhosis or heart failure. Conversely, hyponatremic encephalopathy is the clinical term for symptomatic cerebral edema secondary to hyponatremia, and this condition can have a fulminant presentation. Early signs are usually nonspecific—nausea, vomiting, headaches (22)—and often go unrecognized and are thought to be due to cerebral edema. When pressure is exerted on the skull by the brain, seizures may occur and if uncorrected, brainstem herniation with respiratory failure and death will follow (23).
Risk Factors for Hyponatremic Encephalopathy
The time to development of hyponatremia, i.e., acute versus chronic, has previously been presumed to be an important risk factor in determining severity of symptoms. In vitro studies have shown that full brain adaptation to hypo-osmolar stress occurs over a period of days. However, epidemiologic studies have not demonstrated time to development of hyponatremia to be predictive of death (7,24). There is disparity in patient outcomes even when the time to development of hyponatremia is similar. For an example of the perplexing problem presented by hyponatremia, consider that an elderly male following transurethral resection of the prostate may acutely develop a serum sodium of 110 or a mentally ill patient may ingest large volumes of water to develop hyponatremia of similar degree, and encephalopathy does not result. However, young female patients following surgery may develop respiratory arrest and die due to brainstem herniation with serum sodium as high as 128 mEq/L (7). Although a component of acuity of the insult is important in affecting the outcomes, other factors appear to affect outcomes independent of the time course. A predilection for hyponatremic encephalopathy to affect females is a clue that patient-specific factors may be important in determining patient outcomes (7,25).
|
|
|
Figure 41.4. The brain's response to hypo-osmolar stress based on action of the sodium potassium ATPase. A: Normal glial cell. B: Glial cell swelling following water influx under hyponatremic conditions. C: Restoration of glial cell volume through extrusion of solutes via sodium-potassium ATPase. ADP, adenosine diphosphate; ATP, adenosine triphosphate. |
Neurologic symptoms of hyponatremic encephalopathy are due to pressure of the brain on the rigid skill. Brain size is determined by the incremental change in cell size due to osmotic influx of water, minus the regulatory volume decrease in response to hypo-osmolality. There are three major risk factors for poor outcomes following hyponatremic encephalopathy, which are discussed below. Patient factors play an important role in outcome.
In an epidemiologic study, being female of premenopausal age was shown to be the most important factor in predicting poor outcomes among postsurgical patients developing hyponatremia (7). Although male and female patients were equally likely to develop hyponatremia and hyponatremic encephalopathy postoperatively, permanent brain damage or death was 25 times more likely to occur in female patients (7). The degree of reduction in serum sodium and time to development of hyponatremia did not influence outcome. Comorbid conditions (coronary artery disease, chronic obstructive pulmonary disease, and peripheral vascular disease) were all more prevalent in male survivors than in female nonsurvivors with hyponatremic encephalopathy. Hypoxia at disease presentation was also identified as an important risk factor for hyponatremic encephalopathy. These findings have subsequently been verified in a general inpatient population (24). Therefore, three patient-related clinical factors—gender, age, and the presence of hypoxia—are more important in determining outcomes than the rate of development or the severity of hyponatremia (7). Children are another risk group for poor outcomes due to a high brain-to–cranial vault ratio as brain development is complete around age 6 years, but the skull is not fully grown until adulthood. The identification of risk factors for poor outcomes has been an important step leading to aggressive therapy and a high degree of vigilance.
Gender in Hyponatremic Encephalopathy
There are no significant anatomic differences that are known to exist between males and females that could explain this disparity in outcomes, and therefore it was hypothesized that differences in brain adaptation to hypo-osmolar stress may exist. Estrogens have a similar core steroidal structure to ouabain and cardiac glycosides (such as digoxin), which are among the best known inhibitors of the sodium potassium ATPase (26). Ouabains bind to the α subunit of the Na+/ K+ ATPase and inhibit the catalytic activity of the enzyme, and estrogen likely acts in a similar mechanism to reduce the activity of the sodium potassium ATPase. In diverse tissues such as mammalian heart, diaphragm, red blood cells, and liver, female sex hormones have been shown to inhibit the activity of the Na+/K+ ATPase pump (27), and female rats have increased morbidity from hyponatremia (28,29). The uptake of sodium by isolated synaptosomes from hyponatremic animals is increased in female rats compared with male rats suggesting an impairment in sodium extrusion (28,30). Regulatory volume decrease is inhibited by the presence of estrogen/progesterone in rat astrocytes treated in vitro (31), demonstrating that female sex hormones can impair the critical energy-dependent astrocyte cell volume regulation that actively extrudes ions from the intracellular space of the edematous astrocytes.
|
|
|
Figure 41.5. The role of hypoxia. |
Female rats undergo more intense vasoconstriction than male rats in response to vasopressin (29). This intense vasoconstriction may precipitate tissue hypoxia in the brain.
Role of Hypoxia in Hyponatremic Encephalopathy
Epidemiologic studies have shown that hypoxic patients fare much worse than patients without hypoxia, after adjustment for comorbid conditions in patients with hyponatremic encephalopathy (7,24). Recall that the glial cell is the primary cell involved in volume regulation in the brain. As estrogens had been shown to impair brain adaptive mechanisms through inhibition of the Na+/K+ ATPase, hypoxia may also impair brain adaptation as tissue hypoxia will impair energy-dependent mechanisms such as astrocyte cell volume regulation. In fact, impairment of energy use in the brain alone can lead to diffuse cerebral edema termed cytotoxic cerebral edema seen after asphyxiation or cardiac arrest. This impairment in volume regulatory mechanisms through hypoxia can lead to more severe cerebral edema and lead to worse patient outcomes.
The proposed mechanism for the association of hypoxia and poor outcomes is an impairment in brain adaptation due to insufficient regulatory volume decrease (32). Brain hypoxia can occur in two major settings: ischemia or systemic hypoxemia. Although the effect on tissue oxygenation is the same in these settings, cerebral blood flow is significantly different because brain blood flow increases in response to hypoxia. In response to hypoxia, there are many cellular adaptations that are aimed at preserving the levels of ATP (33). Cells activate pathways such as glycolysis that can produce ATP independent of cellular respiration, and there is down-regulation of ATP-consuming processes. Activity of the Na+/K+ ATPase is down-regulated through increased targeting of the enzyme for endocytosis; thus the cell favors maintenance of intracellular ATP levels over cell volume regulatory mechanisms. This may contribute to the glial cell impairment in volume regulation. Subsequent animal studies have shown that in brain hypoxia induced by either tissue ischemia or by systemic hypoxia, brain adaptation and survival are significantly impaired (32,34).
Hypoxia has been shown to develop in patients with hyponatremic encephalopathy through two mechanisms: hypercapnic respiratory failure and neurogenic pulmonary edema (35,36). Hypercapnic respiratory failure usually develops as a consequence of central respiratory depression and is a first sign of impending brain herniation. Neurogenic pulmonary edema is a well-described complication of cerebral edema from other causes as well as in hyponatremic encephalopathy (35,36). Neurogenic pulmonary edema is a complex disorder characterized by increased vascular permeability and increased catecholamine release (37) that occurs secondary to elevated intracranial pressure. Hypoxemia plays the role of both a risk factor and pathogenetic mechanism in severe cerebral edema. Whether hypoxia is present initially or develops as a consequence of hyponatremia through hypercapnic respiratory failure and/or neurogenic pulmonary edema (35,36), poor outcomes ensue (Fig. 41.5).
Approach to Hyponatremic Patient
The first step in working up the hyponatremic patient is to exclude hyperosmolar hyponatremia (Fig. 41.6). An osmotically active substance that is confined to the extracellular fluid (usually glucose or mannitol) will remove water from the intracellular space and will dilute the serum sodium concentration (translocational hyponatremia). To assess for a sodium disturbance in the setting of hyperglycemia, one must correct the serum sodium by adding 1.6 mEq/L for every 100 mg/dL increase of the serum glucose above 100 mg/dL. Significant hyperosmolality can exist in the setting of a normal or low serum sodium in cases of hyperglycemia. For example, a patient has a serum glucose of 650 mg/dL and a serum sodium of 130 mEq/L. Correct the serum sodium as follows:

Thus the corrected serum sodium in this patient is 139 mEq/L.
The entity of pseudohyponatremia should also be kept in mind. Hyperproteinemia and hyperlipidemia can lead to spuriously low serum sodium measurements when samples are diluted prior to measurement of the serum sodium. If a potentiometric method is used, then this is not a concern. In pseudohyponatremia, the measured serum osmolality will be normal.
The remainder of the diagnostic approach is based on the history, urinary electrolytes, and clinical assessment of the patient's intravascular volume status. Physical examination can be unreliable in the clinical assessment of volume status and thus should be interpreted with caution (Fig. 41.6). A difficult group of patients not emphasized in Figure 41.6 are those with total body fluid overload (with edema, weight gain) due to shock and loss of fluid into the interstitial space, but who are intravascular volume depleted. Despite edema, they would be categorized as the “volume depletion” group, and the intravascular volume may best be assessed by blood volume measurements.
|
|
|
Figure 41.6. Diagnostic approach to hyponatremia. DDAVP, desmopressin; GI, gastrointestinal; SIADH, syndrome of inappropriate antidiuretic hormone. |
Treatment of Hyponatremic Encephalopathy
Use of hypertonic saline to correct hyponatremia is reserved for patients with signs of hyponatremic encephalopathy and is not appropriate in asymptomatic patients regardless of serum sodium (6). Also, the serum osmolality should be measured prior to beginning therapy with hypertonic saline to verify that a hypotonic state exists (22). Fluid restriction alone is never appropriate to manage a patient with hyponatremic encephalopathy, especially in intravascular hypovolemic states. Early recognition of the problem and prompt therapy are the most important factors associated with successful intervention and good neurologic outcomes (22).
The rationale for treatment with hypertonic saline can be summarized as follows: (i) Remove patients with severe manifestations of cerebral edema from immediate danger, (ii) correct serum sodium to a mildly hyponatremic level, and (iii) maintain this level of serum sodium to allow for the brain to adapt to the change in serum osmolality. Table 41.2 gives an overview of the approach to therapy. Prompt therapy should be instituted in all patients with hyponatremic encephalopathy. In patients with severe manifestations (active seizures or respiratory failure), a bolus of 100 mL 3% saline (given over 10 minutes) can be given to promptly change the serum osmolality. The goal is to raise the serum sodium by about 2 to 4 mEq/L. If seizures or respiratory failure persist, the bolus may be repeated. Following the bolus, an infusion of hypertonic saline should be given with the goal of raising the serum sodium to mildly hyponatremic levels. However, the total change in serum sodium should not exceed 15 to 20 mEq/L over 48 hours (6). For patients with hyponatremic encephalopathy without active seizures or respiratory arrest, an infusion of 3% saline without a bolus is appropriate. Again, the goal is also to raise the serum sodium to mildly hyponatremic levels; the total change in serum sodium should not exceed 15 to 20 mEq/L over 48 hours (6).
|
Table 41.2 Treatment of hyponatremia |
|||||||||||
|
Additional precautionary steps are necessary to prevent therapy-induced brain injury. The serum sodium should never be corrected to normonatremic or hypernatremic levels for a few days following hyponatremic encephalopathy. This maintenance period will allow the patient to adjust to the new plasma tonicity. In patients with impaired cardiac output in whom pulmonary edema may develop with vigorous volume expansion, intravenous furosemide should be given in addition to hypertonic saline to prevent volume overload.
There are formulas that have been advocated to calculate the infusion rate when giving hypertonic saline; however, we do not endorse their use. This practice can lead to patient injury when a calculated rate is used as a substitute for proper patient monitoring. Any patient receiving 3% saline should have the serum sodium checked at least every 2 hours until the patient and the serum sodium values are stable. All patients with severe manifestations must be placed in an intensive care setting. The rationale for close monitoring is that ongoing water losses cannot be predicted, and equations that calculate infusion rates assume a closed system (i.e., no ongoing water losses) and significant overcorrection can occur. To guide initial therapy, an infusion of 3% saline of 1 mL/kg will estimate an increase in serum sodium by approximately 1 mEq/L. Initial infusion rates should be adjusted based on repeated serum sodium values and the patient's clinical response.
During the treatment of hyponatremia, the clinician needs to be vigilant to be sure that a free water diuresis does not occur. Clinical scenarios where this is more of concern are interruption of desmopressin (DDAVP) therapy, psychogenic polydipsia, and drug-induced hyponatremia when the offending agent is stopped. To prevent ongoing water losses in the urine and “autocorrection” of the serum sodium, DDAVP can be given to increase urinary concentration and reduce free water losses. This must be done carefully with the patient strictly fluid restricted or kept with no enteral intake in an intensive care unit setting. If unrestricted fluid intake occurs during DDAVP administration, significant hyponatremia can develop. Consultation with an expert experienced in treating sodium disorders is mandatory in such cases. An increase in urine output is the first sign that a water diuresis is ensuing, and therefore hourly urine output needs to be followed in all patients with hyponatremic encephalopathy. The difficulty in treating cases where autocorrection can occur is illustrated in the special case studies below.
Risk Factors for the Development of Cerebral Demyelination
Cerebral demyelination is a rare, serious complication associated with the correction of severe hyponatremia. When symptoms manifest, it is usually a delayed phenomenon occurring days to weeks following correction of hyponatremia and can manifest as a pseudocoma with a locked-in stare. This condition can also be asymptomatic and therefore magnetic resonance imaging (MRI) (which is sensitive for the detection of demyelinating lesions) is necessary for the diagnosis. It is important to understand that the rate of correction of serum sodium alone does not predict the development of cerebral demyelination; rather, the absolute change in serum sodium over 48 hours is predictive (6). Other clinical factors such as liver disease and hypoxia increase the risk of demyelination, and care must be exercised in these groups (6). As noted above, the serum sodium can be quickly be corrected in an acutely symptomatic patient without increasing the risk of demyelination as long as the absolute change over 48 hours does not exceed 15 to 20 mEq/L and the patient is not corrected to normonatremic levels. Patients with liver disease are particularly susceptible to cerebral demyelination, and caution should be exercised in this setting; the safe degree of correction over 48 hours in this group is not known (6).
Management of Asymptomatic Hyponatremia
Regardless of the cause and regardless of the absolute level of the serum sodium, asymptomatic hyponatremia does not require aggressive therapy, but the diagnostic approach to hyponatremia is similar (Fig. 41.6), and treatment should be based on the cause. Fluid restriction can be used alone if hypovolemia is not suspected, but this will typically not result in resolution of the hyponatremia, especially in cases of SIADH where electrolyte free water excretion is negative. Precipitating medications such as thiazide diuretics and anticonvulsants should be stopped. If hyponatremia persists despite fluid restriction, demeclocycline can be used to lower osmolality and increase free water excretion. Vasopressin receptor (V2) blockers are new agents that show promise for the treatment of hyponatremia (38). There is currently limited experience with these medications. However, they may become the mainstay of therapy for asymptomatic hyponatremia in the future as demeclocycline has potential side effects.
Approach to Fluid Therapy and Prevention of Hospital-Acquired Hyponatremia
Until proven otherwise, a patient in a critical care setting should be assumed to have an impairment in free water excretion. In patients with intact kidney function, ADH levels are likely to be high. In patients with renal failure, free water excretion is also impaired. Common situations where water balance is impaired include the following: cases of effective circulating volume depletion (cirrhosis, heart failure, and third spacing of fluid), gastrointestinal fluid losses, diuretic use (especially thiazides), renal failure (acute and chronic), SIADH, cortisol deficiency, and hypothyroidism (Table 41.1). As noted above, hypotonic intravenous fluids should not be used except in the setting of replacement of a water deficit (i.e., hypernatremia). Isotonic solution normal saline (0.9% NaCl) containing 154 mEq/L of Na+and Cl- is the most appropriate parenteral fluid when intravenous fluids are indicated for the maintenance of intravascular volume in the postoperative period (39). Also, any patient receiving fluid therapy should have the serum sodium measured at least daily.
Hospital-acquired hyponatremic encephalopathy occurs most commonly when hypotonic fluids are administered to a patient with an impairment of free water excretion. A clinical setting that merits specific discussion is the postoperative state. Approximately 1% of patients develop a serum sodium of <130 mEq/L following surgery, and clinically important hyponatremia complicates 20% of these cases (7). The postoperative state commonly includes multiple stimuli for ADH release including pain, stress, nausea, vomiting, narcotic medications, and volume depletion (7). Administration of a hypotonic intravenous fluid in the postoperative state or in other clinical settings characterized by impaired free water excretion can have disastrous consequences. The use of hypotonic fluids is reserved for treatment of a free water deficit, such as exists in the setting of hypernatremia.
Special Case Studies
Postoperative Hyponatremia
A 32-year-old female with no significant past medical history undergoes elective laparoscopic bilateral tubal ligation at 8:00 a.m.; 5% dextrose, 1/4 normal saline is started by the anesthesiologist and maintained at 125 mL per hour. The patient remains in recovery until late in the afternoon and is kept because she is too sedated to leave. Intravenous meperidine is given, with adequate pain relief. She does not tolerate oral intake, and the IV fluids are continued at the current rate. At 2:45 a.m. the following day, the patient complains of headache and she is given Vicodin by the on-call physician. At 9:00 a.m., a nurse notifies the surgeon of a sodium of 129 mEq/L; no new orders are received, and intravenous fluids are continued. Later that morning the patient is noted to be lethargic, and the surgeon is notified by the nursing staff; an order is received to hold pain medications. At 3:30 p.m., the patient has a generalized seizure and goes into respiratory failure. She is intubated and mechanical ventilation is initiated. Serum sodium at this time is 124 mEq/L.
Key Points:
· This patient has multiple stimuli for ADH release and thus impaired free water excretion. Administration of a hypotonic fluid was not appropriate and placed the patient at risk for hyponatremia.
· The most important measure to prevent postoperative hyponatremia is to avoid the use of a hypotonic fluid in a postsurgical patient and administer 0.9% sodium chloride when parenteral fluids are indicated.
· In this case the clinicians failed to recognize the early signs of hyponatremic encephalopathy (headache, nausea, and vomiting), which occurred when the patient's sodium was 129 mEq/L. The presence or absence of symptoms of hyponatremic encephalopathy and not the absolute level of the serum sodium determines whether or not a life-threatening condition exists.
Exercise-induced Hyponatremia
A 21-year-old woman collapses 30 minutes after completing a marathon and is brought to the emergency room. She is disoriented and significantly short of breath on arrival. Physical exam reveals a normal cardiac exam, crackles in all lung fields, and a nonfocal neurologic exam with depressed mental status. Chest radiograph reveals pulmonary edema. Serum electrolytes include sodium of 126 mEq/L and potassium of 3.1 mEq/L.
Key Points:
· Exercise-associated hyponatremia has been described in marathon runners. Those who develop this problem consume large amounts of water throughout the race, in excess of the water lost through sweating (36,40). The proposed mechanism is that significant portions of consumed water remains sequestered in the gut as there is divergence of blood flow from the splanchnic circulation during the race. Additionally, ADH is released secondary to the extreme physical exertion of the race. Following completion of the marathon, the ingested water is absorbed and acute hyponatremia ensues, which can be fatal.
· Noncardiogenic pulmonary edema induced by cerebral edema has been described in association with exercise-induced hyponatremia. Paradoxically, treatment with 3% saline leads to resolution of the pulmonary edema by treating the underlying cerebral edema (41).
· Limiting fluid intake is necessary as hypotonic electrolyte sports drinks or salt consumption during the race do not appear to be effective in prevention of this condition (40).
DDAVP Withdrawal
A 76-year-old nursing home patient with a history of urinary incontinence, following transurethral resection of the prostate, is treated with intranasal DDAVP, 10 µg each night. On a routine chemistry panel 1 week prior to admission, his sodium was 138 mEq/L and the patient was doing quite well. Two days prior to presentation he was started on DDAVP, 10 µg in the late morning. On the day of admission, the patient is found to be lethargic and unresponsive and is transported to an emergency room. His serum sodium is 104 mEq/L and serum potassium is 4.0 mEq/L. Urine sodium is 95 mEq/L and urine potassium is 45 mEq/L. He is treated with a 75-mL bolus, then infusion of 3% saline, and his neurologic status improves. Infusion of 3% saline is held when the serum sodium is 120 mEq/L. DDAVP has also been withheld because it is noted to have caused the hyponatremia. Urine output increases significantly over the ensuing night and on the following morning his serum sodium is 139 mEq/L, urine sodium is 17 mEq/L, and urine potassium is 11 mEq/L.
Key Points:
· DDAVP alone will not induce hyponatremia. DDAVP will cause retention of free water, and thus the dosing needs to be titrated in conjunction with the patient's fluid intake. Proper patient instruction and close monitoring are essential during dose changes.
· If DDAVP is withheld, which is commonly done in cases of DDAVP-associated hyponatremia, a free water diuresis will ensue and dangerous overcorrection of the serum sodium hypernatremia may occur.
· The best approach to a patient with hyponatremic encephalopathy due to DDAVP-associated hyponatremia is to continue DDAVP and restrict all enteral fluid intake. Three percent NaCl should be used to correct the patient to the desired serum sodium level, then should be discontinued. A slow infusion of 0.9% can be continued if necessary to support volume status, and absolutely no hypotonic fluids should be administered. This will prevent overcorrection of the serum sodium secondary to a water diuresis. Consultation with a specialist is recommended in these complex cases.
Hypernatremia
Hypernatremia, a serum sodium greater than 145 mEq/L, is commonly encountered in the intensive care unit. Thirst is a powerful protective mechanism, and therefore, restricted access to water is nearly always necessary for the development of hypernatremia. Restricted access to water occurs in various settings, often at the extremes of age: patients who are debilitated by an acute or chronic illness, in neurologic impairment such as dementia, in infants, in moribund patients, or in those on mechanical ventilation with inability to drink water, or in any situation without free access to water. Hypernatremia can develop in essentially any critically ill patient with altered mental status or intubated for mechanical ventilation, because these patients have restricted access to fluids. In most patients in the ICU with hypernatremia, there is a combination of impaired access to water and ongoing free water losses. Renal water losses may occur secondary to solute diuresis (typically urea or glucose), renal concentrating defects (such as loop diuretics), excess hypertonic sodium bicarbonate administration and gastrointestinal fluid losses (especially nasogastric suction and lactulose administration).
Pathogenesis of Hypernatremia
The thirst mechanism and the kidney's urinary concentrating ability that minimize water losses in the urine are the body's defenses against the development of hypernatremia. The common causes of hypernatremia in the ICU usually involve states of impaired water access in conjunction with excessive free water losses (Fig. 41.7). Hypernatremia is a relatively uncommon diagnosis on admission to the intensive care unit but frequently develops during critical illness, and the cause is nearly always iatrogenic. Failure to recognize significant electrolyte free water losses in the urine and to provide adequate replacement in either parenteral or enteral solutions is a common cause of hypernatremia in the intensive care setting.
|
|
|
Figure 41.7. Common causes of hypernatremia in the ICU. |
During hypernatremic states, the brain is subject to osmolar stress that favors the movement of water out of the brain and can lead to significant brain damage. In vivo studies have shown that the brain adapts to hypernatremia through increases in osmotically active ions and de novo generation of osmotically active idiogenic osmoles. The osmotically active cations that are increased in the brain during hypernatremia are sodium and potassium. Idiogenic osmoles refer to a heterogeneous group of osmotically active substances such as myoinositol, glycerophosphocholine, choline, and sorbitol (42,43). This response is seen in the acute setting, and no further changes in brain osmolality are observed after 1 week of hypernatremia (44). These defenses preserve brain volume during elevations in the plasma osmolality and prevent significant decrease in brain size due to osmotic water losses in the brain. In correction of chronic hypernatremia, idiogenic osmoles do not dissipate quickly, and rapid correction of chronic hypernatremia over 24 hours can lead to cerebral edema (44).
Approach to the Hypernatremic Patient
A detailed history focusing on fluid intake and losses is the first step in the evaluation of the hypernatremic patient. Various sources of water losses in the critically ill patient are (Fig. 41.8) water losses in the urine, from the gastrointestinal tract (diarrhea and nasogastric suction), and insensible losses (fever, sepsis, massive diaphoresis, burns). These amounts should be calculated (when accurate counts are available) or estimated. In assessing urinary water losses, both the urine osmolality and electrolytes should be measured in evaluation of urinary concentrating ability and to assess urinary water losses. Caution should be exercised in the interpretation of the urine osmolality as this is an area where error is frequently made. The urine osmolality alone cannot be used to determine whether or not there is free water loss in the urine. This is because water is excreted with nonelectrolyte osmoles (which under physiologic conditions is typically urea) and with electrolyte osmoles. Both contribute to the osmolality of the urine but have differential effects on water balance. Water that is excreted with nonelectrolyte osmoles is water that is lost in excess of electrolyte loss. Recall that the serum sodium is proportional to total body electrolytes relative to total body water (Equation 41.1). Therefore loss of water with the nonelectrolyte osmoles will raise the serum sodium. By contrast, water that is excreted with electrolyte osmoles will not affect the serum sodium (as long as the concentration of electrolytes in the urine and serum are similar). This is because both the numerator and denominator of Equation 41.1 is decreasing and therefore the proportion is unchanged. In cases where there is a high urea or glucose load, significant amounts of water can be lost in the urine despite maximal urinary concentration. Failure to concentrate the urine in the face of hypernatremia should raise suspicion of a urinary concentrating defect. Renal failure, loop diuretics, tubulointerstitial renal disease, and diabetes insipidus are the main causes of urinary concentrating defects. In summary, all sources of water intake and water loss should be considered in assessing the water needs of a critically ill patient (Fig. 41.8) as an imbalance favoring water loss over water intake will lead to hypernatremia.
|
|
|
Figure 41.8. Sources of water intake and loss in the ICU. GI, gastrointestinal; IVF, intravenous fluid; TPN, total parenteral nutrition. |
Clinical Manifestations of Hypernatremia
Hypernatremia leads to an efflux of fluid from the intracellular space to the extracellular space to maintain osmotic equilibrium across the cell membranes. The primary clinical manifestations are due to central nervous system depression as cerebral dehydration and cell shrinkage occurs. Hypernatremia carries an overall mortality between 40% and 70% in children (45). The elderly and patients with end-stage liver disease are at particular risk for complications from hypernatremia. Treatment of hepatic encephalopathy with lactulose frequently causes an osmotic diarrhea that leads to water losses in the stool. As a result, hypernatremia can quickly develop and lead to severe morbidity. Patients with liver disease are at high risk for cerebral demyelination in the setting of changes in serum sodium (6).
Central Diabetes Insipidus
Central diabetes insipidus (CDI) is a unique cause of hypernatremia that can be seen in the ICU and is most commonly in the setting of head injury, pituitary surgery, and cerebral hemorrhages. Specific therapy is indicated for this condition and therefore it needs to be recognized early. With polyuria secondary to water diuresis, severe hypernatremia can rapidly develop in an individual who has restricted access to fluids such as an ICU patient. Sodium-retentive mechanisms are intact in patients with CDI, and therefore clinical volume depletion is not a characteristic feature. The diagnosis of CDI should be suspected if the urine is not concentrated in the setting of hypernatremia (46). In general, the plasma osmolality typically exceeds the urine osmolality in CDI. Formal diagnostic testing for diabetes insipidus is beyond the scope of this text and is usually undertaken in consultation with a nephrologist. A simple and reliable clinical test that can be used to distinguish CDI from nephrogenic diabetes insipidus is to administer a V2 receptor agonist, such as DDAVP. A 50% increase in urine osmolality following DDAVP administration strongly suggests CDI. Once the diagnosis is established, DDAVP can be given either subcutaneously or intranasally. When DDAVP is administered, water intake should be adjusted appropriately to avoid precipitation of significant hyponatremia, and serial serum electrolytes should be monitored during dose titration (23).
Treatment of Hypernatremia
The goal of treatment of hypernatremia is to achieve normal circulatory volume as patients typically have circulatory volume depletion and then to correct the serum sodium with free water replacement (Table 41.3). The first step is to assess the ongoing water losses in the urine to determine if the water losses are renal in origin, or if the kidneys are appropriately conserving water. A simple method for assessing free water loss in the urine is displayed in Equation 41.3, which is Equation 41.2 but looking specifically at the urine:

The degree of ongoing water losses in the urine will assist in determining the rate of fluid administration. In cases of extrarenal fluid losses, the fluid loss will need to be estimated. Fluid resuscitation with normal saline or colloid to replenish the circulating volume should precede correction of the water deficit (Table 41.3). In hypernatremic states, insulin resistance has also been observed (47). This can lead to severe hyperglycemia and potential worsening of hyperosmolality during therapy with glucose-containing solutions. For this reason, glucose-containing solutions are potentially harmful and should be avoided if possible. If intravenous glucose solutions must be used (for example, 5% dextrose in water), hourly measurement of the plasma glucose should be made and an insulin drip considered if plasma glucose becomes elevated.
Oral hydration is preferable to parenteral and should be used when possible. Serial measurement of electrolytes every 2 hours is necessary until the patient is neurologically stable. In patients without evidence of hypernatremic encephalopathy, the serum sodium should not be corrected more quickly than 1 mEq/hour or 15 mEq/24 hours. In severe cases (>170 mEq/L), sodium should not be corrected to below 150 mEq/L in the first 48 to 72 hours (36).
|
Table 41.3 Treatment of Hypernatremia |
||
|
Case Scenario: Solute Diuresis from Excess Urea Load
A 46-year-old man is admitted with severe necrotizing pancreatitis. He has a history of alcohol abuse, hepatitis C, and chronic liver disease. The patient weighs 76 kg. Admission labs are listed below. The patient is kept without enteral intake overnight and volume expanded with 6 L of normal saline. Twenty-four hours after admission, abdominal pain worsens and he is continued without enteral intake. Serum sodium is 145. Over the next 24 hours, urine output increases and isotonic saline is continued at 100 mL per hour. Total parenteral nutrition is initiated with a total volume of 2 L, 120 mEq of sodium, and high amino acid content. The chemistry and urine studies 48 hours after admission are listed.
|
Admission |
48 hours after admission |
|
|
Sodium (mEq/L) |
137 |
155 |
|
Potassium (mEq/L) |
3.5 |
3.1 |
|
Chloride (mEq/L) |
103 |
112 |
|
Bicarbonate (mmol/L) |
21 |
24 |
|
Blood urea nitrogen (mg/dL) |
23 |
53 |
|
Creatinine (mg/dL) |
1.3 |
1.1 |
|
Urine output (mL per hour) |
40 |
200 |
|
Urine sodium (mEq/L) |
— |
45 |
|
Urine potassium (mEq/L) |
— |
22 |
|
Urine osmolality (mOsm/kg) |
— |
610 |
What is the cause of the polyuria in this patient? Answer: Solute diuresis.
Learning Point:
· Solute diuresis secondary to a high urea load is a common cause of hypernatremia in the critical care setting.
This patient presents a typical example of a solute diuresis leading to hypernatremia in the critical care setting. By taking the ratio of the sodium plus potassium in the urine over the sodium plus potassium in the serum, urinary water losses can be assessed. The sodium plus potassium in the urine is lower than that in the blood. In this case, the ratio is 70/156 = 0.45. This means that 45% of his urine is electrolyte containing and conversely that 55% of the urine is electrolyte free water. At his current urine output, he is losing (0.55 × 200 mL/hour) = 110 mL of water per hour in the urine. Water replacement must be at least equal to this to replace his ongoing water losses in the urine. The urine osmolality is high due to ADH secretion, and this is increasing the urine concentration. The low urine sodium and potassium, at a time when the urine osmolality is high, signifies that there is a nonelectrolyte osmole in the urine that is obligating water loss. This is a classic presentation of an osmotic diuresis secondary to urea. The high urea load in this case is probably multifactorial, being secondary to the hypercatabolic state with muscle breakdown in addition to the necessarily high protein in the total parenteral nutrition.
Prevention of Hypernatremia in the ICU
The prevention of hypernatremia is best accomplished by recognition of patients at risk for this disorder and at risk of a poor outcome. It is not necessary to memorize a list of conditions but to understand that hypernatremia requires at least one of the following to occur: impaired access to water (dementia, mental illness, encephalopathy, child/ infant, critically ill patient, patient who is restricted in enteral intake or using a feeding tube) or a massive sodium load (improper infant formula mixture, administration of large amounts of hypertonic sodium solutions such as sodium bicarbonate or sodium phosphate) (23).
Disorders of Potassium Balance
Physiology of Potassium Homeostasis
Potassium is present mainly in the intracellular space (approximately 4,000 mEq of total body stores) and is sequestered in the intracellular space through the action of the Na+/K+ ATPase pump. The serum potassium level is tightly regulated so that the potential differences across membranes, especially cardiac, are not affected by alterations in potassium level. As the extracellular space contains very little of the total body potassium, shifting of potassium from one space to another is a major cause of both hyperkalemia and hypokalemia. The major influences that favor potassium movement into cells are insulin and β2-adrenergic stimulation. Metabolic acidosis favors potassium movement out of cells as H+ is exchanged for K+ when H+ moves intracellularly. However, this may have less importance in settings where an organic anion is generated as a result of the acidosis (such as lactic acidosis) as the organic ions may also move intracellularly and thus negate the electrogenic stimulus for potassium to move out of the cell.
Potassium excretion is tightly regulated, and the excretion of potassium can be varied such that potassium balance is maintained across a wide range of potassium intake. There are two main determinants of potassium excretion of clinical significance: flow of tubular filtrate into the distal nephron and secretion of potassium into the electronegative filtrate through epithelial potassium channels. The electronegativity of the tubular filtrate is determined by reabsorption of sodium from the filtrate at a faster rate than reabsorption of Cl-. This reabsorption occurs through the epithelial sodium channel ENaC (47,48). The effect of aldosterone with sodium retention is the most important influence increasing the action of ENaC and therefore stimulates potassium excretion. Medications such as amiloride, triamterene, and trimethoprim (49) block ENaC and lead to decreased K+ secretion and can lead to clinically important hyperkalemia. Decrease in the rate of tubular flow into the distal nephron stimulates the renin angiotensin system, which in turn stimulates release of aldosterone. The renin angiotensin axis, in addition to stimulating the release of aldosterone, also affects hemodynamic changes at the glomerular level, which act to maintain glomerular filtration rate and therefore maintain distal flow into the nephron with the ability to excrete potassium. In summary, the renal excretion of potassium is enhanced during states of high tubular flow and also under the actions of aldosterone. Therefore, inhibition of either of these through a decrease in glomerular filtration rate (of any cause) or inhibition of the action of aldosterone (e.g., adrenal insufficiency or pharmacologic blockade) can lead to potassium retention.
Hyperkalemia
Pathophysiology
Hyperkalemia occurs through two major mechanisms: shifting of potassium from intracellular compartment to the extracellular fluid compartment or through total body excess (Table 41.4). Total body potassium excess nearly always implies a deficiency in renal potassium excretion as the kidney. It is difficult to overcome a normal kidney's ability to increase potassium excretion by increasing potassium intake except in cases of a massive potassium load (rhabdomyolysis, tumor lysis syndrome). Chronic renal failure alone typically does not lead to hyperkalemia until the glomerular filtration rate (GFR) falls below approximately 15 mL/minute. Other mitigating factors will exacerbate hyperkalemia in the setting of chronic renal failure such as medications (Table 41.5). Hyperkalemia commonly complicates acute renal failure and is less tolerated in the acute setting. Hyperkalemia is most often encountered in unexpected development of acute renal failure such as in postsurgery (especially after cardiac or vascular surgery), post–intravenous contrast study, administration of nephrotoxic antibiotics (especially aminoglycosides), post–cardiac catheterization. Other settings include end-stage renal disease, adrenal insufficiency, type IV renal tubular acidosis (RTA) (usually seen in diabetics), crush injury, diabetes, and massive blood transfusion. Medications are a very important risk factor for the development of hyperkalemia (Table 41.5).
Management of Hyperkalemia
The first step in the management of hyperkalemia is to differentiate life-threatening hyperkalemia from less urgent cases and then to identify the diagnosis. The absolute levels of the potassium cannot be reliably used to determine if a life-threatening condition exists, and the effect of elevated potassium on the cardiac membrane must be determined through an electrocardiogram. The management of emergent hyperkalemia is detailed in Table 41.6. As a temporizing measure, shifting of potassium intracellularly with insulin or beta-agonists can be used until definitive removal therapy is instituted. If normal renal function is present, diuretics and intravenous saline can be used to remove potassium. Cation exchange resins can be used as an adjunctive therapy. In patients with renal failure, especially with oliguria, emergency dialysis is often necessary. Hyperkalemia not associated with electrocardiographic changes can sometimes be managed with discontinuation of the offending agent. Some medications causing hyperkalemia such as spironolactone can have a long half-life, and the effect can last for up to 1 to 2 weeks. Close follow-up is mandatory with serial measurements of serum potassium and renal function (50). If managed properly, hyperkalemia is usually associated with good long-term prognosis.
|
Table 41.4 Causes of Hyperkalemia |
||||||||||||
|
||||||||||||
|
Table 41.5 Medications That Cause Hyperkalemia |
||||||||||||
|
Case Scenario: Heparin-induced Hyperkalemia
A 60-year-old female patient is 5 days post–total hip replacement for osteoarthritis. She has a history of diabetes, hypertension, and a seizure disorder and no known kidney disease. Her outpatient medications include insulin, metformin, atenolol, hydrochlorothiazide, and Dilantin. The surgery was uneventful, but on postsurgical day 2, she develops severe shortness of breath and chest pain. A pulmonary embolism is diagnosed, and she is placed on unfractionated heparin and transferred to the intensive care unit. Following the surgery, she has additionally been receiving promethazine, 12.5 mg as needed (prn) for nausea and vomiting, and meperidine, 25 mg prn for pain. Her preoperative labs were all normal; today's chemistry panel (on postoperative day 4) is listed below:
|
Sodium (mEq/L) |
138 |
|
Potassium (mEq/L) |
5.9 |
|
Chloride (mEq/L) |
102 |
|
Bicarbonate (mEq/L) |
27 |
|
Blood urea nitrogen (BUN) (mg/dL) |
18 |
|
Creatinine (mg/dL) |
1.1 |
|
Glucose (mg/dL) |
153 |
Which of the following best explains the development of hyperkalemia? Answer: Use of heparin.
Key Point:
· Heparin can lead to hyperkalemia by decreasing aldosterone levels (51).
Heparin is a frequent cause of hyperkalemia in the hospital setting in both patients with normal renal function and in those with renal insufficiency. In this case, renal function appears to be normal. Heparin inhibits the synthesis of aldosterone and leads to impairment of renal potassium secretion in the distal tubule. Two major factors are necessary for potassium excretion to occur: distal flow of urine to the distal nephron (this is impaired in acute renal failure) and aldosterone action. The necessity for distal secretion of potassium to maintain potassium balance is because of the very low filtered load of potassium due to the low concentration of potassium in the blood. Aldosterone acts by promoting the exchange of potassium for sodium in the distal tubule. The glomerular filtration rate is normal in this case, but due to decreased distal secretion of potassium, potassium excretion is impaired.
|
Table 41.6 Treatment of Hyperkalemia |
|||||||||||||
|
Hypokalemia
Pathophysiology
Hypokalemia can occur either through shifting from ECF to ICF or through total body potassium depletion via gastrointestinal losses (distal to the stomach) or urinary losses (Table 41.7). Gastric secretion contains only 10 mEq/L of K+, but hypovolemia associated with vomiting/NG suction stimulates aldosterone with sodium retention and potassium excretion. Shifting of potassium into the intracellular compartment is a physiologic process that normally occurs in response to insulin secretion. Without this process, a meal could lead to a dangerously high potassium level since the total amount of potassium contained within the extracellular space is quite low. β2-adrenergic stimulation is another avenue for cellular uptake of potassium. This is clinically significant as β2 agonists can be used to transiently treat hyperkalemia and possibly lead to hypokalemia in certain clinical settings.
|
Table 41.7 Common Causes of Hypokalemia |
||||||||
|
Clinical Manifestations
The effects of hypokalemia are related to the effect of low potassium on neuromuscular transmission and conduction in the heart. A decrease in extracellular potassium makes the membrane less excitable and muscle weakness results. Hypokalemia can lead to arrhythmias (especially in patients on digitalis or with heart disease). Other effects include ileus, muscular cramps, augmented ammonia production in the kidney (which can potentiate hepatic encephalopathy), and rhabdomyolysis. Hypokalemia is often associated with hypomagnesemia, and serum magnesium status should be assessed in all patients with hypokalemia.
Management of Hypokalemia
Potassium can be replaced with potassium chloride (KCl). For severe hypokalemia, potassium chloride should be given intravenously (IV) at a rate of 10 to 20 mEq per hour. Hypomagnesemia should also be corrected if present. Total body potassium deficits are typically large (200–300 mmol for serum potassium of 3.0 mEq/L, i.e., for each 1 mEq below normal), and repeated replacements may be needed with serial measurements (50).
Since K+ is a major intracellular cation, serum levels may not reflect the severity of deficiency.
Case Scenario: Hypokalemia Associated with Metabolic Acidosis
A 21-year-old woman is admitted for a 1-week history of muscular weakness and shortness of breath. Her past medical history is insignificant except for a history of dental caries and dry eyes. Her admission laboratories are listed below:
|
Sodium (mEq/L) |
137 |
|
Potassium (mEq/L) |
1.8 |
|
Chloride (mEq/L) |
115 |
|
Bicarbonate (mEq/L) |
9 |
|
BUN (mg/dL) |
16 |
|
Creatinine (mg/dL) |
0.9 |
|
Glucose (mg/dL) |
110 |
|
Arterial pH |
7.09 |
|
PCO2 |
14 |
|
PO2 |
115 |
The patient is placed in the intensive care unit on a cardiac monitor.
What is the most likely diagnosis? This is a dramatic presentation of distal renal tubular acidosis with profound hypokalemia. Her history is suggestive of Sjögren syndrome, which can be complicated by distal renal tubular acidosis (52).
What is the next appropriate step in electrolyte and acid-base management? Replete potassium with intravenous potassium chloride. It is important not to treat this patient initially with alkali therapy; this can lead to shifting of potassium intracellularly and fatal hypokalemia. Once her potassium has been corrected, she can be treated with intravenous sodium bicarbonate.
Disorders of Divalent Ions
The divalent ions phosphorus, calcium, and magnesium play important roles in cellular function such as regulation of enzymes and energy metabolism. In critical illness, the homeostasis of these ions is often altered, and abnormal levels are commonly encountered. The best treatment of these conditions requires knowledge of the pathophysiology of the alterations in the levels in these ions.
Phosphate Homeostasis
Phosphate is important for many basic processes of cellular metabolism and bone metabolism. Clinically in the intensive care setting, the need for phosphate for proper cellular energy metabolism through high-energy phosphate compounds (e.g., adenosine triphosphate) is among the most important. Phosphate is obtained in the diet primarily through protein intake. The typical Western diet contains sufficient phosphate to meet metabolic demands; however, in patients with poor nutritional status, often seen in alcoholics, hypophosphatemia may develop. The phosphate excretion in the kidney is regulated by several factors that favor tubular phosphate reabsorption (such as insulin, prostaglandin E2, and thyroid hormone) and factors that inhibit phosphate reabsorption (such as parathyroid hormone and the newly characterized phosphaturic hormone FGF-23 that plays a role in hereditary and acquired phosphate-wasting disorders). Phosphate reabsorption is handled principally in the proximal tubule through the NPT2 sodium phosphate cotransporter. Therefore, disorders of the proximal tubule, such as Fanconi syndrome, may lead to renal phosphate wasting.
Hypophosphatemia
Pathophysiology
There are three major mechanisms of hypophosphatemia (Table 41.8): shifting of phosphate from the extracellular to the intracellular space, renal phosphate losses, and gastrointestinal losses. Very often the disorder occurs as a multifactorial disease with acid-base disturbances, decreased intake, and renal losses all contributing. Phosphate, like potassium and magnesium, is mainly an intracellular ion. Therefore, low serum phosphate usually represents significant total body phosphate depletion. Hypophosphatemia commonly occurs in alcoholics and in hospitalized patients. The diagnosis is usually evident from the history; however, if the cause is in doubt, renal phosphate losses can be assessed by calculating the fractional excretion of phosphorus.
![]()
where P is phosphorus level, u is urine, pl is plasma, and Cr is creatinine level.
This will help discern if the phosphate losses are gastrointestinal (GI) in origin or if the losses are in the urine. The fractional excretion of phosphorus should be measured at a time when serum phosphorus is low, and if the fractional excretion of phosphorus is below 5%, the kidney is appropriately conserving phosphate and therefore the losses are extrarenal. In critically ill patients, there are many common conditions and circumstances that set the patient up to be at risk for hypophosphatemia. Alcoholism, nasogastric suction, malnutrition, extensive bowel resection, malignancy, low sun exposure, and diuretic use are among the conditions seen in ICU patients that are associated with hypophosphatemia.
|
Table 41.8 Causes of Hypophosphatemia |
||||
|
Clinical Manifestations
Symptoms are often absent in mild cases. In severe depletion, muscular weakness (especially large muscle groups, proximal extremity weakness), arrhythmias, rhabdomyolysis, hypotension, hypoventilation, seizures, coma, and hemolytic anemia may occur. The most life threatening is cardiac and ventilatory failure. Other undesirable effects of hypophosphatemia are the following: shift of oxygen dissociation curve to the left from deficiency of 2,3,DPG; depressed immune function (chemotaxis, phagocytosis, bacteriocidal activity); platelet dysfunction (adhesion and aggregation); metabolic acidosis (impaired bicarbonate resorption and ammonia production); and abnormal glucose metabolism from decreased entry into the cell to make high-energy phosphates.
Treatment
Phosphorus replacement in asymptomatic patients should be oral and can be given as a potassium or sodium salt. Parenteral phosphorus administration should be undertaken in symptomatic patients or in patients with severe depletion (<1.0 mg/dL) or when the oral route is not appropriate. Parenteral phosphate preparations are generally ordered in millimoles. One millimole (mmol) of phosphate equals 31 mg of phosphorus. Since phosphate is administered as either a sodium salt or a potassium salt, intolerance to either of these should be considered in settings such as renal failure or congestive heart failure. Phosphate replacement should be judicious in patients with renal failure as significant hyperphosphatemia can occur.
Case Scenario: Refeeding Syndrome
A 37-year-old man with a long history of alcoholism is admitted to the intensive care unit with severe pancreatitis. His friends state that for the last several weeks he has been drinking beer all day and has been eating very little except for salty snacks. Over the course of the next 3 days, his ICU course is complicated by development of sepsis and a pancreatic abscess that has required drainage. Five days into his hospital course he remains on mechanical ventilation and vasopressor support. On this day, his chemistry panel is given below:
|
Serum |
|
|
Sodium (mEq/L) |
136 |
|
Potassium (mEq/L) |
4.3 |
|
Chloride (mEq/L) |
105 |
|
Bicarbonate (mEq/L) |
22 |
|
BUN (mg/dL) |
6 |
|
Creatinine (mg/dL) |
0.6 |
|
Calcium (mg/dL) |
7.0 |
|
Albumin (g/dL) |
2.7 |
|
Phosphorus (mg/dL) |
2.1 |
|
Magnesium (mg/dL) |
2.0 |
|
Glucose (mg/dL) |
98 |
He is started on total parenteral nutrition with 2,000 total calories, 20% from fat. There is 120 mEq of sodium and 80 mEq of potassium, and the total volume is 2 L a day.
This patient is at high risk for which complications following the institution of total parenteral nutrition? Answer: Refeeding syndrome and rhabdomyolysis.
Learning Points:
· Refeeding syndrome is a potential complication in patients who are malnourished and suddenly given a large calorie load.
· Rhabdomyolysis is a potentially severe complication of the refeeding syndrome.
This patient is at high risk for the refeeding syndrome, and therefore the potential for rhabdomyolysis is a concern (53). His history and laboratory values (low BUN and albumin) suggest malnutrition, and thus phosphate depletion is likely present despite the close-to-normal value for serum phosphate. The phosphate is sequestered in the intracellular compartment, and it is the intracellular depletion of phosphate that is critical. Once this patient is supplied with calories, especially carbohydrates, oxidative phosphorylation and generation of ATP will quickly deplete the intracellular phosphate and lead to rhabdomyolysis. The best means of preventing this complication from occurring is recognizing patients who are at high risk and monitoring serum phosphate closely.
Hyperphosphatemia
Pathophysiology
Hyperphosphatemia occurs primarily in the setting of impaired renal function and is not typically seen outside of iatrogenic causes in patients with normal kidney function. Significant phosphate loading can occur due to exogenous phosphate administration or as part of the tumor lysis syndrome (however, renal insufficiency also commonly complicates this condition). Occult sources of phosphate loading include sodium phosphate solutions (either orally or as an enema), which can produce significant and, in some rare cases, fatal outcomes. In renal failure, elevated serum phosphorus is associated with cardiovascular disease and cardiovascular mortality and should be avoided.
Clinical Manifestations
There are few overt symptoms of hyperphosphatemia, and the syndrome may be insidious. Nephrocalcinosis can complicate hyperphosphatemia and manifest as renal insufficiency. On rare occasions, acute dialysis may be indicated to treat hyperphosphatemia. Extended dialysis session (>4 hours) may be necessary as phosphorus mobilization is time dependent and phosphorus removal in short dialysis sessions is limited (54).
Case Scenario: Hyperphosphatemia as a Complication of Sodium Phosphate Administration
An 85-year-old man with a history of hypertension is admitted to the intensive care unit with refractory hypotension. His wife states that he had been complaining of constipation and had used several enemas at home over the past 48 hours. His wife then stated that he has become progressively lethargic throughout the day, and she called an ambulance because he fell after trying to get up from a chair and was severely confused. He takes only hydrochlorothiazide for hypertension and is otherwise without significant medical history. The physical exam is significant for blood pressure of 85/45 and pulse of 58; respirations are 8 breaths per minute, and he is afebrile. The physical exam is otherwise significant only for Chvostek sign.
The chemistry panel on admission is given below:
|
Serum |
|
|
Sodium (mEq/L) |
149 |
|
Potassium (mEq/L) |
4.5 |
|
Chloride (mEq/L) |
109 |
|
Bicarbonate (mEq/L) |
20 |
|
BUN (mg/dL) |
23 |
|
Creatinine (mg/dL) |
1.7 |
|
Calcium (mg/dL) |
4.8 |
|
Albumin (g/dL) |
3.8 |
|
Phosphorus (mg/dL) |
8.2 |
|
Magnesium (mg/dL) |
2.0 |
|
Glucose (mg/dL) |
98 |
What risk factor for hyperphosphatemia did this patient have? Answer: Renal insufficiency put this patient at risk. The creatinine level of 1.7 in a patient of his age is indicative of significant decrease in GFR (estimated at 41 mL/minute). The use of hyperosmolar phosphate enemas is a risk in this population. The time that the solution is allowed to dwell determines the amount of phosphate absorbed. Fatal cases have been reported even in patients without renal failure (55). This patient is exhibiting signs of hypocalcemia, hypotension, and impending cardiovascular collapse that can occur with acute hyperphosphatemia.
Overview of Calcium Balance
Calcium concentration in plasma is tightly regulated. Intracellular calcium levels are very low whereas the total body calcium stores in the bone are plentiful. Calcium balance and the calcium concentration in the blood are maintained by regulatory processes that promote calcium influx into the extracellular fluid (mainly intestinal calcium absorption and release of calcium from bone demineralization) and remove calcium from the extracellular fluid (urinary calcium excretion and bone formation). Disturbances in serum calcium occur when there is an imbalance between these processes regulating calcium movement. The hormonal influences that regulate these processes are complex but involve the actions of two principal hormones: parathyroid hormone and calcitriol. Calcium exists in the plasma as protein-bound calcium and ionized calcium, usually measured in mmol/L. Measurement of the total serum calcium reflects the aggregate amount of these two pools of plasma calcium. Ionized calcium is the active component and the more important physiologically.
The total measured serum calcium needs to take into account changes in the amount of albumin, which is the predominant calcium-binding protein in the circulation. A simple correction for the total calcium based on perturbations of the serum albumin is the following: a reduction in plasma albumin of a 1 g/L will decrease the serum total calcium by 0.8 mg/dL (56). If the decrease in total serum calcium is within the range as would be expected based on the decrease in albumin, then it can be surmised that the ionized serum calcium is probably normal. The validity of this correction in critical illness has been questioned, possibly due to factors that affect binding of calcium to albumin. Measurement of ionized calcium levels in critically ill patients is a good practice, and results may be available within minutes of using point-of-care testing.
Hypocalcemia
Pathophysiology
The causes of hypocalcemia can be divided into either efflux of calcium out of the extracellular space or by decreased entry of calcium into the extracellular space (Table 41.9) (57). Under normal circumstances, homeostatic mechanisms can maintain serum calcium levels despite low intake by increasing mobilization from the bone. Hypocalcemia can result from decreased calcium absorption and decreased calcium mobilization from bone stores in hypoparathyroidism and vitamin D deficiency. Increased calcium removal from the serum occurs in severe pancreatitis through saponification of fats and in osteoblastic metastatic disease (Table 41.9). The ionized calcium is the important factor for determining severity, and conditions that alter protein binding of calcium in the blood may precipitate symptomatic hypocalcemia. Alkalemia will increase the affinity of albumin for calcium (through altering the net charge of the protein) and will lower the ionized calcium. Acute increases in total albumin may lower ionized calcium. Risk factors for hypocalcemia include the following: neck irradiation, parathyroid surgery, renal transplantation in patients with tertiary hyperparathyroidism, malignancy, alcoholism, low sun exposure, and malnutrition.
Clinical Manifestations
The clinical manifestations include altered mental status and tetany (Chvostek and Trousseau signs). The association between hypocalcemia and sepsis is not well understood although it is now recognized that elevated intracellular calcium levels are a common mechanism for cell death. Possible mechanisms for the association of sepsis and hypocalcemia are parathyroid hormone (PTH) insufficiency, altered vitamin D metabolism, hypomagnesemia, chelation of calcium by lactate and also calcitonin precursors (58,59,60,61,62). Hypocalcemia in critical illness is a common finding and is often asymptomatic.
|
Table 41.9 Causes of Hypocalcemia |
||||||||||
|
Management of Hypocalcemia
One of the first questions to be addressed in approaching a patient with altered levels of serum calcium is to what degree is the ionized calcium altered and why? If the ionized calcium is decreased due to chelation from hyperphosphatemia, treatment of the hypocalcemia can be dangerous as it can precipitate vascular calcification. In sepsis, hypocalcemia is common and treatment is not usually necessary unless cardiovascular collapse with ionized calcium levels of less than 0.8 mmol/L is present. Data from animal models suggest that it may be harmful to treat hypocalcemia in the setting of sepsis (63,64). There are no randomized clinical studies to guide treatment of hypocalcemia in sepsis, and treatment should be reserved for those with symptomatic hypocalcemia and/or severe hypocalcemia. Reflex treatment of hypocalcemia without consideration of the underlying cause (especially in the setting of hyperphosphatemia) can be detrimental: therefore, it is important to determine the cause and degree of symptoms before deciding to initiate therapy.
For acute symptomatic hypocalcemia, administer intravenous calcium chloride or calcium gluconate (56). Calcium gluconate is advantageous because it is less caustic to veins. Calcium should not be infused more rapidly than 2.5 to 5 mmol in 20 minutes because of the risk of cardiac abnormalities and asystole. Intravenous calcium will transiently normalize the calcium level, and an infusion should be started following the bolus, especially in settings where there is an ongoing process such as the hungry bone syndrome following parathyroidectomy. Magnesium deficiency, by inducing resistance to the actions of parathyroid hormone, can be an important cause of hypocalcemia in the critical care setting. If magnesium deficiency is the cause, calcium levels should return to normal once magnesium is replaced.
Case Scenario: Hypocalcemia Associated with Sepsis
A 43-year-old man with end-stage liver disease secondary to chronic hepatitis C infection presents with shortness of breath and abdominal pain. He has significant ascites and has decreased urine output for the last week. His appetite has been very poor over the last week although he has no other complaints. His blood pressure is 71/50, pulse is 86, and temperature 98.4°F. Admission labs are given below:
|
Sodium (mEq/L) |
131 |
|
Potassium (mEq/L) |
4.9 |
|
Chloride (mEq/L) |
108 |
|
Bicarbonate (mEq/L) |
20 |
|
BUN (mg/dL) |
24 |
|
Creatinine (mg/dL) |
1.6 |
|
Albumin (mg/dL) |
2.4 |
|
Calcium (mg/dL) |
6.8 |
|
Phosphorus (mg/dL) |
2.8 |
|
Glucose (mg/dL) |
78 |
He is admitted to the intensive care unit and treated with broad-spectrum antibiotics and vasopressor for presumed septic shock.
What considerations should be taken in determining if this hypocalcemia should be treated? Answer: The cause is an important factor. This is likely a case of mild hypocalcemia due to sepsis. Check the ionized calcium level, serum magnesium level, and assess the degree of symptoms including electrocardiogram. If the serum magnesium level is decreased, this should be treated. If the ionized calcium is <0.8 mmol/L or if the patient appears to be symptomatic, the patient may benefit from treatment with intravenous calcium.
Key Point:
· Sepsis is a common cause of hypocalcemia. Check the level of ionized calcium and magnesium in critically ill patients with hypocalcemia.
Hypercalcemia
Pathophysiology
Hypercalcemia is caused by entrance of calcium into the intravascular space in excess of renal excretion and calcium incorporation into bone. Two sources of calcium influx into the intravascular space are intestinal absorption and bone reabsorption. The most common causes are primary hyperparathyroidism, malignancy, and granulomatous diseases. Other causes are listed in Table 41.10. Additionally, hypercalcemia can be caused by decreased renal excretion as occurs in primary hyperparathyroidism and thiazide diuretics. Total calcium must be corrected for serum albumin because of protein binding, and ionized calcium should be measured in hypercalcemic patients.
Clinical Manifestations
Hypercalcemia produces a decrease in neuromuscular excitability and decreased muscular tone. Clinical manifestations of hypercalcemia include: lethargy, confusion, coma, nausea, constipation, polyuria, and hypertension. Hypercalcemia can lead to volume depletion, nephrolithiasis, and nephrogenic diabetes insipidus.
Approach to Diagnosis
The causes of hypercalcemia are listed in Table 41.10. The first step is to measure PTH level. The normal response of the parathyroid gland in the presence of elevated ionized calcium levels is to decrease secretion of parathyroid hormone. Failure to suppress parathyroid hormone secretion in the face of hypercalcemia denotes hyperparathyroidism. Primary hyperparathyroidism is among the most common causes of hypercalcemia in the general population but typically does not result in severe, symptomatic hypercalcemia. In hypercalcemia of malignancy, parathyroid hormone is usually suppressed (this is the normal response of the parathyroid gland). PTH-related peptide (PTHrp) should also be ordered if malignancy is suspected as many cancers express this protein product that stimulates PTH receptors (65). Additionally, 1,25-hydroxyvitamin D and 25-hydroxyvitamin D levels can be helpful if abnormal vitamin D metabolism is suspected. Certain granulomatous conditions lead to increased 1,25 vitamin D (66), and in cases of vitamin D intoxication, high levels of 25-OH vitamin D reflective of total body vitamin D stores may be seen (67).
|
Table 41.10 Causes of Hypercalcemia |
|||
|
Management of Hypercalcemia
In most patients with symptomatic hypercalcemia, volume depletion occurs and one of the first steps is volume repletion with intravenous saline (0.9% saline is the treatment of choice). This replacement of the extracellular volume will increase calcium excretion in the urine. Caution is advised in infusing large amounts of saline to an oliguric patient. After fluid resuscitation, normal saline should be continued and furosemide can also be given to increase urinary calcium excretion. A common mistake is to administer furosemide at the same time as starting fluid resuscitation. Diuretics should not be given until the patient is completely volume resuscitated. The action of loop diuretics is distinct from that of thiazide diuretics. Loop diuretics act in the ascending limb of Henle and will promote calcium excretion in the urine. Thiazide diuretics act more distally in the nephron and will decrease calcium excretion. Bisphosphonates are good long-term treatment for hypercalcemia of malignancy by impairing bone reabsorption, but these agents must be used with caution in patients with renal insufficiency (Table 41.11). Calcitonin is usually effective in the short term, but tachyphylaxis develops. It is best used early in the management while waiting for diuresis to remove calcium from the body. In patients with renal failure, dialysis is usually necessary to treat symptomatic hypercalcemia.
Case Scenario: Hypercalcemia of Malignancy
A 46-year-old man with a 1-year history of pain in his legs and back is brought to the emergency room for decreased mental status. His wife states that he is very active and works as an attorney, but he has been easily fatigable over the last several months. Over the last 12 hours, he has gotten progressively more lethargic and is difficult to arouse.
|
Sodium (mEq/L) |
136 |
|
Potassium (mEq/L) |
4.8 |
|
Chloride (mEq/L) |
108 |
|
Bicarbonate (mEq/L) |
16 |
|
BUN (mg/dL) |
51 |
|
Creatinine (mg/dL) |
5.8 |
|
Albumin (mg/dL) |
3.8 |
|
Calcium (mg/dL) |
16.8 |
|
Magnesium (mg/dl) |
2.8 |
|
Phosphorus (mg/dL) |
4.6 |
|
Glucose (mg/dL) |
98 |
A Foley catheter is inserted, and the urine output is 50 mL. In the emergency room, he is administered 2 L of 0.9% NaCl intravenously, and over the next 2 hours there is no urine output.
What is the next appropriate step in management of hypercalcemia in this patient? Answer: This patient should undergo emergency hemodialysis, which can be used in cases of renal failure and severe hypercalcemia (68). From history, this patient likely has multiple myeloma with complications of this disorder: hypercalcemia and renal failure. It is appropriate to give a volume challenge with normal saline to treat prerenal azotemia. However, with no urine output after 2 L of saline, acute renal failure is likely, and hemodialysis offers the best treatment for severe hypercalcemia. The patient should be dialyzed on a hypocalcemic bath in an intensive care setting, with cardiac monitoring and very close monitoring of the ionized calcium.
|
Table 41.11 Treatment of Severe Hypercalcemia |
|
|
Hypomagnesemia
Pathophysiology
Magnesium depletion occurs in many conditions (Table 41.12) and is common in critical illness (69). Hypomagnesemia, because of its effect on the myocardial membrane, is important to identify, especially in those with known (or at risk for) cardiac arrhythmias. Sources of magnesium loss can be through urinary losses or also in the GI tract. Often, inadequate intake in malnourished patients is the principal cause of the magnesium deficiency. High-risk patients at particular risk for hypomagnesemia include hospitalized patients, alcoholics, and intensive care unit patients. Magnesium depletion has effects on the homeostasis of other ions, and therefore, magnesium depletion often occurs in the presence of hypokalemia and hypocalcemia. Magnesium depletion causes a potassium-wasting state, and patients with hypokalemia should be assessed for magnesium depletion. Hypomagnesemia induces a state of reduced parathyroid hormone secretion and PTH resistance. Tissue magnesium depletion can be present in the absence of decreased serum magnesium levels (since most magnesium is in the intracellular space), and that tissue depletion can lead to adverse outcomes (70).
|
Table 41.12 Causes of Hypomagnesemia |
|||
|
Decreased total serum magnesium levels are common in critically ill patients. This is often due to a decrease in serum albumin, which is the principal binding protein of magnesium, and many of these patients do not have decreased ionized magnesium levels (69). The correlation of ionized hypomagnesemia and mortality in ICU patients is controversial (69). Magnesium is not actively mobilized from a body pool (unlike calcium), and therefore, serum levels are very dependent on intake. If magnesium intake is exceeded by magnesium loss (e.g., urinary losses), then the serum magnesium levels will be decreased.
Clinical Manifestations
The most serious consequences of hypomagnesemia are its cardiovascular effects. Hypomagnesemia is associated with poor outcomes in acute myocardial infarction and may predispose to arrhythmias (71). Magnesium treatment may decrease arrhythmias after myocardial infarction, but studies conflict on whether outcomes are improved with magnesium treatment (72,73,74). Magnesium depletion increases the risk of torsades des pointes, a form of polymorphic ventricular tachycardia. Intravenous magnesium is regarded as treatment for torsades des pointes and refractory ventricular tachycardia even in the absence of documented hypomagnesemia. Additionally, hypomagnesemia may contribute to atrial arrhythmias following cardiopulmonary bypass (75) Other manifestations include altered mental status, seizures, and muscular weakness. Magnesium is an important cofactor for multiple enzyme function (including ATPase) and an essential electrolyte.
Treatment of Hypomagnesemia
For acutely symptomatic patients, parenteral administration should be undertaken. In asymptomatic patients, the oral route should be used although absorption is variable. Parenterally administered magnesium inhibits magnesium reabsorption in the ascending limb of Henle, and much of a parenterally administered dose may be wasted in the urine. For patients with severe manifestations, 8 to 16 mEq should be administered intravenously in 5 to 10 minutes followed by 48 mEq over the next 24 hours.
Case Scenario: Hypomagnesemia and Associated Electrolyte Disturbances
A 56-year-old man is admitted to the surgical intensive care unit after an emergency coronary artery bypass performed following the dissection of his left anterior descending artery during an attempted percutaneous coronary intervention. He has a history of hypertension, smoking, and alcohol abuse. His medications are atenolol and aspirin. His surgery was uncomplicated, and his urine output was 70 mL per hour. His chemistry panel on admission to the intensive care unit is given:
|
Sodium (mEq/L) |
138 |
|
Potassium (mEq/L) |
3.1 |
|
Chloride (mEq/L) |
106 |
|
Bicarbonate (mEq/L) |
24 |
|
BUN (mg/dL) |
22 |
|
Creatinine (mg/dL) |
1.4 |
|
Albumin (mg/dL) |
3.7 |
|
Calcium (mg/dL) |
7.3 |
|
Magnesium (mg/dL) |
1.3 |
|
Phosphorus (mg/dL) |
2.4 |
|
Ionized calcium (mmol/L) |
0.8 |
|
Glucose (mg/dL) |
98 |
Treatment of hypomagnesemia in this patient is likely to improve which other electrolyte disorders in this patient? Answer: magnesium deficiency due to poor dietary intake is the most likely cause of both the hypokalemia and hypocalcemia. Treatment of the hypomagnesemia will likely lead to resolution of the hypocalcemia. Potassium replacement should be given as well, as hypomagnesemia has lead to renal potassium wasting.
Hypermagnesemia
Pathophysiology
Hypermagnesemia is not a common condition, and it is often iatrogenic. Magnesium is readily excreted by the kidney in patients with normal renal function, and this disorder is uncommon except in cases of large ingestions (Epsom salt and magnesium-containing cathartics). In the presence of gastrointestinal disease such as peptic ulcer disease, magnesium absorption can be enhanced, and magnesium-containing antacids in this setting can lead to toxic hypermagnesemia (76). Patients with renal failure are at highest risk for developing hypermagnesemia, and the administration of magnesium-containing cathartics to these patients can be fatal (77). Clinical manifestations of hypermagnesemia include hypotension, bradycardia, respiratory depression, decreased mental status, and ECG abnormalities.
Management
Removal of the offending agent may be sufficient in mild cases with normal renal function. In symptomatic hypermagnesemia, the neuromuscular membrane can be stabilized by administration of 1 g of calcium chloride intravenously over 5 to 10 minutes. Dialysis is usually necessary in the setting of renal insufficiency or severe toxicity.
Case Scenario: Hypermagnesemia and Risk Factors for Development
An 84-year-old man is brought in from a nursing home with decreased level of consciousness. He has a past medical history significant for hypertension and peptic ulcer disease. He was recently diagnosed with a gastric ulcer and erosive esophagitis by esophagogastroduodenoscopy, and he has been taking lansoprazole. He has had continuous heartburn and symptoms, and he has been taking antacids frequently according to his wife. His physical exam is significant for blood pressure of 85/40 and pulse of 72. Respiratory rate is 8 breaths per minute.
|
Sodium (mEq/L) |
138 |
|
Potassium (mEq/L) |
4.0 |
|
Chloride (mEq/L) |
106 |
|
Bicarbonate (mEq/L) |
22 |
|
BUN (mg/dL) |
23 |
|
Creatinine (mg/dL) |
1.5 |
|
Albumin (mg/dL) |
3.9 |
|
Calcium (mg/dL) |
7.3 |
|
Magnesium (mg/dL) |
6.8 |
|
Phosphorus (mg/dL) |
2.2 |
|
Glucose (mg/dL) |
91 |
Why did this patient develop severe hypermagnesemia? Answer: this patient has two risk factors that predisposed to the development of hypermagnesemia. He has chronic renal dysfunction, which was not appreciated by the treating physicians. He also has gastrointestinal disease, which can increase the absorption of magnesium from the gut, especially in magnesium-containing antacids.
References
1. Dunn FL, Brennan TJ, Nelson AE, et al. The role of blood osmolality and volume in regulating vasopressin secretion in the rat. J Clin Invest. 1973;52(12):3212–3219.
2. Arieff AI, Ayus JC. Endometrial ablation complicated by fatal hyponatremic encephalopathy. JAMA. 1993;270(10):1230–1232.
3. Arieff AI, Ayus JC. Pathogenesis of hyponatremic encephalopathy. Current concepts. Chest. 1993;103(2):607–610.
4. Ayus JC, Levine R, Arieff AI. Fatal dysnatraemia caused by elective colonoscopy. BMJ. 2003;326(7385):382–384.
5. Arieff AI, Ayus JC, Fraser CL. Hyponatraemia and death or permanent brain damage in healthy children. BMJ. 1992;304(6836):1218–1222.
6. Ayus JC, Krothapalli RK, Arieff AI. Treatment of symptomatic hyponatremia and its relation to brain damage. A prospective study. N Engl J Med. 1987;317(19):1190–1195.
7. Ayus JC, Wheeler JM, Arieff AI. Postoperative hyponatremic encephalopathy in menstruant women. Ann Intern Med. 1992;117(11):891–897.
8. Nielsen S, Nagelhus EA, Amiry-Moghaddam M, et al. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17(1):171–180.
9. Agre P, Preston GM, Smith BL, et al. Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol. 1993;265(4 Pt 2):F463–476.
10. Nielsen S, Smith BL, Christensen EI, et al. Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci U S A. 1993;90(15):7275–7279.
11. Simard M, Arcuino G, Takano T, et al. Signaling at the gliovascular interface. J Neurosci. 2003;23(27):9254–9262.
12. Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4(12):991–1001.
13. Dolman D, Drndarski S, Abbott NJ, et al. Induction of aquaporin 1 but not aquaporin 4 messenger RNA in rat primary brain microvessel endothelial cells in culture. J Neurochem. 2005;93(4):825–833.
14. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53.
15. Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129(4):877–896.
16. Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237(4817):896–898.
17. Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6(2):159–163.
18. Reulen HJ, Tsuyumu M, Tack A, et al. Clearance of edema fluid into cerebrospinal fluid. A mechanism for resolution of vasogenic brain edema. J Neurosurg. 1978;48(5):754–764.
19. Olson JE, Sankar R, Holtzman D, et al. Energy-dependent volume regulation in primary cultured cerebral astrocytes. J Cell Physiol. 1986;128(2):209–215.
20. Melton JE, Patlak CS, Pettigrew KD, et al. Volume regulatory loss of Na, Cl, and K from rat brain during acute hyponatremia. Am J Physiol. 1987;252(4 Pt 2):F661–669.
21. Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275(5 Pt 2):F633–650.
22. Moritz ML, Ayus JC. The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant. 2003;18(12):2486–2491.
23. Achinger SG, Moritz ML, Ayus JC, et al. Why are patients still dying? South Med J. 2006;99(4):1–12.
24. Nzerue CM, Baffoe-Bonnie H, You W, et al. Predictors of outcome in hospitalized patients with severe hyponatremia. J Natl Med Assoc. 2003;95(5):335–343.
25. Arieff AI. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women. N Engl J Med. 1986;314(24):1529–1535.
26. Chen JQ, Contreras RG, Wang R, et al. Sodium/potassium ATPase (Na(+), K (+)-ATPase) and ouabain/related cardiac glycosides: a new paradigm for development of anti-breast cancer drugs? Breast Cancer Res Treat. 2006;96(1):1–15.
27. Davis RA, Kern F Jr, Showalter R, et al. Alterations of hepatic Na+,K+-atpase and bile flow by estrogen: effects on liver surface membrane lipid structure and function. Proc Natl Acad Sci U S A. 1978;75(9):4130–4134.
28. Fraser CL, Kucharczyk J, Arieff AI, et al. Sex differences result in increased morbidity from hyponatremia in female rats. Am J Physiol. 1989;256(4 Pt 2):R880–885.
29. Arieff AI, Kozniewska E, Roberts TP, et al. Age, gender, and vasopressin affect survival and brain adaptation in rats with metabolic encephalopathy. Am J Physiol. 1995;268(5 Pt 2):R1143–1152.
30. Fraser CL, Sarnacki P. Na+-K+-ATPase pump function in rat brain synaptosomes is different in males and females. Am J Physiol. 1989;257(2 Pt 1):E284–289.
31. Fraser CL, Swanson RA. Female sex hormones inhibit volume regulation in rat brain astrocyte culture. Am J Physiol. 1994;267(4 Pt 1):C909–914.
32. Vexler ZS, Ayus JC, Roberts TP, et al. Hypoxic and ischemic hypoxia exacerbate brain injury associated with metabolic encephalopathy in laboratory animals. J Clin Invest. 1994;93(1):256–264.
33. Hochachka PW, Buck LT, Doll CJ, et al. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A. 1996;93(18):9493–9498.
34. Ayus JC, Armstrong D, Arieff A. Hyponatremia with hypoxia: effects on brain adaptation, perfusion and histology in rodents. Kidney Int 2006;69(8):1318–1325.
35. Ayus JC, Arieff AI. Pulmonary complications of hyponatremic encephalopathy. Noncardiogenic pulmonary edema and hypercapnic respiratory failure. Chest. 1995;107(2):517–521.
36. Ayus JC, Varon J, Arieff AI. Hyponatremia, cerebral edema, and noncardiogenic pulmonary edema in marathon runners. Ann Intern Med. 2000;132(9):711–714.
37. McClellan MD, Dauber IM, Weil JV. Elevated intracranial pressure increases pulmonary vascular permeability to protein. J Appl Physiol. 1989;67(3):1185–1191.
38. Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099–2112.
39. Moritz ML, Ayus JC. Prevention of hospital-acquired hyponatremia: a case for using isotonic saline. Pediatrics. 2003;111(2):227–230.
40. Hew-Butler T, Almond C, Ayus JC, et al. Consensus statement of the 1st International Exercise-Associated Hyponatremia Consensus Development Conference, Cape Town, South Africa 2005. Clin J Sport Med. 2005;15(4):208–213.
41. Ayus JC, Arieff AI. Noncardiogenic pulmonary edema in marathon runners. Ann Intern Med. 2000;133(12):1011.
42. Heilig CW, Stromski ME, Blumenfeld JD, et al. Characterization of the major brain osmolytes that accumulate in salt-loaded rats. Am J Physiol. 1989;257(6 Pt 2):F1108–1116.
43. Lien YH, Shapiro JI, Chan L. Effects of hypernatremia on organic brain osmoles. J Clin Invest. 1990;85(5):1427–1435.
44. Ayus JC, Armstrong DL, Arieff AI. Effects of hypernatraemia in the central nervous system and its therapy in rats and rabbits. J Physiol. 1996;492 (Pt 1):243–255.
45. Moritz ML, Ayus JC. The changing pattern of hypernatremia in hospitalized children. Pediatrics. 1999;104(3 Pt 1):435–439.
46. Moritz ML, Ayus JC. Dysnatremias in the critical care setting. Contrib Nephrol. 2004;144:132–157.
47. Ayus JC, Krothapalli RK. Hyperglycemia (HG) associated with non-diabetic hypernatremia (NDH) patients. J Am Soc Nephrol. 1990;1:317A.
48. Halperin ML, Kamel KS. Potassium. Lancet. 1998;352(9122):135–140.
49. Schlanger LE, Kleyman TR, Ling BN. K(+)-sparing diuretic actions of trimethoprim: inhibition of Na+ channels in A6 distal nephron cells. Kidney Int. 1994;45(4):1070–1076.
50. Gennari FJ. Disorders of potassium homeostasis. Hypokalemia and hyperkalemia. Crit Care Clin. 2002;18(2):273–288, vi.
51. Leehey D, Gantt C, Lim V. Heparin-induced hypoaldosteronism. Report of a case. JAMA. 1981;246(19):2189–2190.
52. al-Jubouri MA, Jones S, Macmillan R, et al. Hypokalaemic paralysis revealing Sjögren syndrome in an elderly man. J Clin Pathol. 1999;52(2):157–158.
53. Marinella MA. Refeeding syndrome and hypophosphatemia. J Intensive Care Med. 2005;20(3):155–159.
54. Achinger SG, Ayus JC. The role of daily dialysis in the control of hyperphosphatemia. Kidney Int Suppl. 2005(95):s28–32.
55. Pitcher DE, Ford RS, Nelson MT, et al. Fatal hypocalcemic, hyperphosphatemic, metabolic acidosis following sequential sodium phosphate-based enema administration. Gastrointest Endosc. 1997;46(3):266–268.
56. Body JJ, Bouillon R. Emergencies of calcium homeostasis. Rev Endocr Metab Disord. 2003;4(2):167–175.
57. Tohme JF, Bilezikian JP. Hypocalcemic emergencies. Endocrinol Metab Clin North Am. 1993;22(2):363–375.
58. Zaloga GP, Chernow B. The multifactorial basis for hypocalcemia during sepsis. Studies of the parathyroid hormone-vitamin D axis. Ann Intern Med. 1987;107(1):36–41.
59. Robertson GM Jr., Moore EW, Switz DM, et al. Inadequate parathyroid response in acute pancreatitis. N Engl J Med. 1976;294(10):512–516.
60. Weir GC, Lesser PB, Drop LJ, et al. The hypocalcemia of acute pancreatitis. Ann Intern Med. 1975;83(2):185–189.
61. Hersh T, Siddiqui DA. Magnesium and the pancreas. Am J Clin Nutr. 1973;26(3):362–366.
62. Cooper DJ, Walley KR, Dodek PM, et al. Plasma ionized calcium and blood lactate concentrations are inversely associated in human lactic acidosis. Intensive Care Med. 1992;18(5):286–289.
63. Zaloga GP, Sager A, Black KW, et al. Low dose calcium administration increases mortality during septic peritonitis in rats. Circ Shock. 1992;37(3):226–229.
64. Malcolm DS, Zaloga GP, Holaday JW. Calcium administration increases the mortality of endotoxic shock in rats. Crit Care Med. 1989;17(9):900–903.
65. Moseley JM, Kubota M, Diefenbach-Jagger H, et al. Parathyroid hormone-related protein purified from a human lung cancer cell line. Proc Natl Acad Sci U S A. 1987;84(14):5048–5052.
66. Adams JS, Singer FR, Gacad MA, et al. Isolation and structural identification of 1,25-dihydroxyvitamin D3 produced by cultured alveolar macrophages in sarcoidosis. J Clin Endocrinol Metab. 1985;60(5):960–966.
67. Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289(1):F8–28.
68. Camus C, Charasse C, Jouannic-Montier I, et al. Calcium free hemodialysis: experience in the treatment of 33 patients with severe hypercalcemia. Intensive Care Med. 1996;22(2):116–121.
69. Soliman HM, Mercan D, Lobo SS, et al. Development of ionized hypomagnesemia is associated with higher mortality rates. Crit Care Med. 2003;31(4):1082–1087.
70. Ryzen E, Elkayam U, Rude RK. Low blood mononuclear cell magnesium in intensive cardiac care unit patients. Am Heart J. 1986;111(3):475–480.
71. Dyckner T. Serum magnesium in acute myocardial infarction. Relation to arrhythmias. Acta Med Scand. 1980;207(1–2):59–66.
72. Woods KL, Fletcher S, Roffe C, et al. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet. 1992;339(8809):1553–1558.
73. ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group. Lancet. 1995;345(8951):669–685.
74. Early administration of intravenous magnesium to high-risk patients with acute myocardial infarction in the Magnesium in Coronaries (MAGIC) Trial: a randomised controlled trial. Lancet. 2002;360(9341):1189–1196.
75. Maslow AD, Regan MM, Heindle S, et al. Postoperative atrial tachyarrhythmias in patients undergoing coronary artery bypass graft surgery without cardiopulmonary bypass: a role for intraoperative magnesium supplementation. J Cardiothorac Vasc Anesth. 2000;14(5):524–530.
76. Clark BA, Brown RS. Unsuspected morbid hypermagnesemia in elderly patients. Am J Nephrol. 1992;12(5):336–343.
77. Onishi S, Yoshino S. Cathartic-induced fatal hypermagnesemia in the elderly. Intern Med. 2006;45(4):207–210.







