J. Matthias Walz
Stephen O. Heard
Immediate Concerns
Major Problems
Progressive dysfunction of multiple organ systems, culminating in the syndrome of multiple organ dysfunction syndrome (MODS), has become a leading cause of death in critically ill and injured patients. MODS is a disease of medical progress. Broader use of intensive care unit (ICU) resources, combined with improvements in single organ–directed therapy, such as mechanical ventilation and renal replacement therapy, has reduced early mortality after major physiologic insults. The result is a longer ICU stay for an increasing number of patients after severe sepsis and trauma, during which inflammation and tissue injury may result in MODS.
MODS represents a systemic disorder of immunoregulation, endothelial dysfunction, and hypermetabolism, with varying manifestations in individual organs. The mortality of MODS will increase as the number of failing organs increases, suggesting that changes in the function of all organs have equal significance in outcome. However, organs differ in their host defense functions and sensitivity to host-derived inflammatory mediators or reductions in oxygen delivery ([D with dot above]O2). Therefore, diagnosis and therapy focus, whenever possible, on preventive measures. Changes in the cellular oxygen (O2) supply and metabolism may cause and complicate MODS. Consequences can include direct hypoxic organ damage, secondary ischemia/reperfusion (I/R) injury mediated by neutrophils and reactive O2species (ROS), and enhanced injury by activation of cytokines, including tumor necrosis factor-α (TNF-α). Initial and subsequent therapy follows a two-tiered approach, targeting systemic factors that contribute to ongoing inflammation and single organ–related problems. Efforts are first directed at stabilizing [D with dot above]O2 while addressing life-threatening derangements in acid-base balance and gas exchange. Prompt correction of hemodynamic instability to defined end points that correlate with resolution of tissue O2 debt minimizes ischemia-related organ damage. The element of time is a critical factor. Delays in completing initial resuscitation, eliminating foci of infection or devitalized tissue, or treating de novo organ-specific problems such as oliguria all worsen outcome. Late-phase (e.g., over 72 hours) problems involve acquired immunosuppression, predisposition to secondary infection, and hypermetabolism, which impairs wound healing and host defense.
Initial Essential Diagnostic Tests and Procedures
Hemodynamic and Metabolic Monitoring
1. Begin assessing the adequacy of initial resuscitation efforts by noninvasive measures including skin color and temperature, arterial blood pressure, pulse rate, respiratory rate, mental status, and urine output; determine if metabolic acidosis is present from arterial blood gas and plasma bicarbonate (NaHCO3) determinations. If acidosis is present, establish whether the anion gap and plasma lactate concentrations are increased.
2. Consider invasive hemodynamic monitoring by arterial and central vascular catheterization. Central venous pressure estimates right heart filling but may not accurately gauge left ventricular preload with tricuspid insufficiency, pre-existing heart disease, pulmonary hypertension, or acute respiratory distress syndrome (ARDS). Exclude myocardial infarction as a cause of hemodynamic instability by electrocardiography, creatine kinase isoenzyme, and troponin I levels.
3. Targeted hemodynamic management can be accomplished by invasive or noninvasive means (e.g., pulmonary artery catheterization, pulse contour analysis, or esophageal Doppler monitoring). Mixed or central venous O2 saturation and lactate concentrations—if the latter are initially elevated—should be monitored to determine the adequacy of resuscitation and assist in the titration of therapy.
4. Hemodynamic instability despite adequate fluid resuscitation in patients with severe sepsis should be treated with inotropes as indicated, and the hemoglobin level should be raised to 10 mg/dL in the early stages of resuscitation according to the principles of early goal-directed therapy (EGDT).
Evaluation for Infection
1. For suspected sepsis upon ICU admission, blood cultures—including fungal cultures where appropriate—should be immediately obtained, as should Gram stains and cultures of urine, an adequate sputum specimen (where “adequate is defined by 25 or more leukocytes per low-power field”) or tracheobronchial washings, and wound discharges before antimicrobial therapy. Suspicious skin lesions should undergo culture by aspiration and biopsy. On discovery of fluid collections, perform thoracentesis and paracentesis within 12 hours or less; determine pH; and perform a Gram stain, culture, cell count, cytologic studies, glucose level, and other chemistries.
2. Evaluate the patient thoroughly for all infectious and potential noninfectious etiologies of MODS.
3. For suspected nosocomial sepsis, reculture blood, urine, and sputum; evaluate all sites of vascular cannulation and remove catheters, if possible; and consider fiberoptic bronchoscopy to obtain protected brush specimen or bronchoalveolar lavage (BAL) samples in patients with pneumonia. Exclude infective endocarditis or endovascular infection by echocardiography and scintigraphic scanning for highgrade or recurrent bacteremia.
4. Serially monitor renal, pancreatic, and hepatic function; exclude acalculous cholecystitis or pancreatitis by abdominal ultrasound. Perform computed tomography of the sinuses, chest, abdomen, and pelvis when appropriate to define fluid collections.
5. Maintain a high index of suspicion for opportunistic fungal infection with Candida sp. despite negative results on blood culture.
Initial Therapy
1. Resuscitation of hemodynamic instability should be rapidly initiated with crystalloid or colloid infusions, followed by replenishment of the red cell mass.
2. Vasopressors—dopamine, norepinephrine, vasopressin—are titrated to a systolic pressure of 90 to 100 mm Hg or a mean arterial pressure of 70 mm Hg or higher.
3. In patients with septic shock and hypotension despite adequate fluid resuscitation, evaluate the patient for evidence of adrenal insufficiency and initiate therapy with low-dose corticosteroids if indicated.
4. Evaluate and treat ionized hypocalcemia and severe metabolic acidosis if the response to catecholamine therapy is inadequate.
5. If shock persists despite rapid and aggressive fluid resuscitation, consider endotracheal intubation and mechanical ventilation, irrespective of arterial blood gas values. Proper titration of ventilatory therapy averts respiratory muscle fatigue and arrest by reducing shock-related increases in the O2 cost of breathing.
6. Evaluate and treat oliguria. Differentiate prerenal causes by obtaining serum and urine Na+, creatinine, and urea nitrogen to calculate the fractional excretion of sodium (FeNa) or urea nitrogen.
7. Stabilize long bone fractures early.
8. Initiate broad-spectrum antimicrobial therapy, including coverage against methicillin-resistant Staphylococcus aureus, Staphylococcus epidermidis, and Pseudomonas aeruginosa. Add coverage for suspected anaerobic intra-abdominal sepsis.
9. Begin antifungal therapy in patients at high risk for fungal sepsis despite negative results on blood culture when clinical findings are suggestive (e.g., extensive colonization by Candida, nonintertriginous skin rash, myositis, or retinitis).
10. Perform prompt re-exploration for suspected intraabdominal sepsis and abscess formation.
Epidemiology of Multiple Organ Dysfunction Syndrome
Significant advances have been made in critical care medicine over the last 30 years, particularly in the last decade. Nonetheless, many critically ill patients often suffer the progressive deterioration in the function of one or more organs, a phenomenon that has been termed multiple organ dysfunction syndrome (1). MODS is the leading cause of death for patients in the intensive care unit. Furthermore, the death rate remains high for patients who survive their ICU admission. In addition, the financial costs are significant, with more than 60% of ICU resources consumed by these patients (2).
Individual organ dysfunction may result from a direct insult, such as pulmonary aspiration of gastric contents (primary MODS), or it can be associated with a systemic process such as shock or pancreatitis (secondary MODS) (1). Alterations in organ function seen during MODS are a continuum rather than a discrete, dichotomous event indicating the failure of an organ. A number of organ dysfunction scores have been developed to predict the clinical outcome of these patients (Table 54.1). These scores not only serve to establish the baseline degree of organ dysfunction, but also enable the clinician to evaluate the progression or resolution of organ dysfunction over time. In general, an increase in the number of dysfunctional organs increases the risk of death. Examples of early scores of organ failure include those published by Goris et al. (3) and Knaus et al. (4). Refinement of these scores led to the development of the multiple organ dysfunction (MOD) score (5) and the sequential organ failure assessment (SOFA) score (6). In principle, these scores are based on parameters for six organ systems: Cardiovascular, respiratory, hematologic, renal, central nervous system (CNS), and hepatic (7). The difference in these scores lies in the parameter to describe cardiovascular dysfunction. The MOD score describes the degree of cardiovascular dysfunction as a composite of heart rate, central venous pressure, and mean arterial pressure (pressure-adjusted heart rate), whereas the SOFA score describes cardiovascular dysfunction by the dose of vasoactive agents administered.
|
Table 54.1 Comparison of the physiologic and biochemical parameters used by four scoring systems for organ dysfunction and failure |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
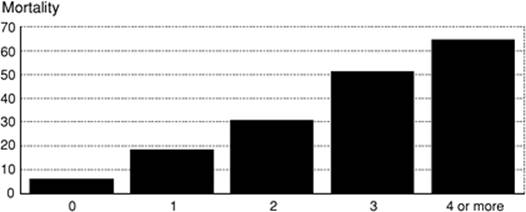
Several trials have evaluated the performance of these scores as descriptors of multiple organ dysfunction and failure, and to assess the incidence of MODS in the intensive care unit. Moreno et al., in a prospective, international multicenter trial composed of 1,449 patients, were able to demonstrate that total maximum SOFA score and change in SOFA score over time (δ) can be used to quantify the degree of organ dysfunction present on ICU admission, the degree of dysfunction or failure that appears during the ICU stay, and the cumulative insult suffered by the patient (7). These findings were subsequently confirmed by Ferreira et al. (8), who demonstrated that changes in the SOFA score were a good indicator of prognosis. In their study of 352 consecutive patients, an increase in SOFA score during the first 48 hours of intensive care predicted a mortality rate of at least 50% (8). In a group of patients with ARDS, the Toronto ARDS Outcomes Group found a significant relationship between the change in MOD score over time of the ICU stay and the distance walked in 6 minutes up to 1 year following discharge from the ICU (9). The recent European Sepsis Occurrence in Acutely Ill Patients (SOAP) multicenter trial analyzed data from 3,147 adult ICU admissions to determine the incidence of MODS and its associated mortality in mixed medical and surgical ICU populations (10). The overall rate of MODS, defined as severe acquired dysfunction in two or more organ systems, was 43% for patients without a diagnosis of sepsis and 73% of those with a diagnosis of severe sepsis, which represents a substantially higher incidence than in some previously published reports (4). Like other investigators, they found a direct relationship between the number of organs failing and the ICU mortality (Fig. 54.1). Single organ failure carried an ICU mortality rate of 6%, whereas patients with four or more failing organs had mortality rates of 65%. While earlier reports have suggested that the increase in mortality associated with an increased number of failed organs is independent of the identity of dysfunctional organ systems (11,12,13), the SOAP investigators found different results. Organ failure in patients with severe sepsis generally carried a higher mortality than in those patients without a diagnosis of severe sepsis. As for individual organ systems in the group of patients with severe sepsis, failure of the coagulation system carried the highest mortality (52.9%), followed by the hepatic (45.1%), CNS (43.9%), cardiovascular (42.3%), and renal system (41.2%). Respiratory failure in this analysis was associated with a mortality risk of 34.5%. Certain subsets of patients admitted to the ICU appear to be at greater risk of MODS: patients older than 65 years (older than 55 years in trauma patients [13]), increased severity of illness as assessed by APACHE II scores (20 or more), and diagnosis of sepsis or acute lung injury (ALI) on admission. Among the patients with severe sepsis, the SOAP investigators found as independent predictors of mortality the following: “Medical” admissions, Pseudomonas species infection, SAPS II score on admission, SOFA score at the onset of sepsis, bloodstream infection, cirrhosis, and cumulative fluid balance within the first 72 hours of the onset of sepsis. The latter variable has not previously been identified as an independent predictor of mortality; further investigations will be necessary to distinguish whether a positive fluid balance in the ICU is simply a marker of severity of illness or is harmful, per se.
|
|
||
|
Figure 54.1. Relationship between the number of failed organs on admission and intensive care unit mortality. (Reproduced from Vincent J-L, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344–353, with permission.) |
||
|
Table 54.2 American College of Chest Physicians/Society of Critical Care Medicine definitions of sepsis and organ failure |
||
|
Pathophysiology
MODS usually occurs in patients who exhibit signs of a generalized inflammatory response (systemic inflammatory response syndrome [SIRS], Table 54.2) (1). Although SIRS is often the result of infection, other conditions such as necrotizing pancreatitis or trauma can also lead to systemic manifestations of inflammation; SIRS due to infection has been defined as sepsis. Recent guidelines (14) have broadened the diagnostic criteria for sepsis that were originally proposed in 1992. For those patients who present with SIRS only, a significant number will progress to sepsis, septic shock, and, ultimately, MODS (15). Although suspected or documented infection is not required for the development of MODS, the syndromes of SIRS, sepsis, and MODS are closely related. Consequently, the review of the pathophysiology of MODS will also include discussions of SIRS and MODS.
Derangements in Oxygen Delivery and Consumption
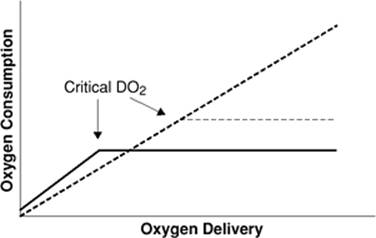
In most tissues, oxygen consumption (VO2) is determined by metabolic demand and is independent of [D with dot above]O2. When [D with dot above]O2 is reduced, VO2 is maintained by increased oxygen extraction by the tissues. If [D with dot above]O2 is reduced to the point where the metabolic need cannot be met, then VO2 becomes “supply dependent” (Fig. 54.2). The point at which VO2 decreases is called the critical [D with dot above]O2. Although one study (16) suggested that the critical [D with dot above]O2 in anesthetized humans is 330 mL/minute/m2, another investigation where life support was withdrawn in critically ill patients demonstrated that the value is substantially lower (17).
|
|
|
Figure 54.2. The relationship between oxygen delivery and consumption. The solid line represents the normal relationship. The dashed line shows pathologic supply dependency. (Reproduced from Heard SO, Fink MP. Multiple-organ dysfunction syndrome. In: Murray MJ, Coursin DB, Pearl RG, et al., eds. Critical Care Medicine. Perioperative Management. Philadelphia: Lippincott Williams & Wilkins; 2002.) |
A number of studies from the 1970s, 1980s, and early 1990s seemed to suggest that systemic VO2 was supply dependent over a wide range of [D with dot above]O2 in patients with sepsis or ARDS. Since VO2and [D with dot above]O2 are independent of each other, the linking of these two variables in patients with ARDS or sepsis was coined pathologic supply dependency. This concept is important, as it implies that inadequate oxygen is being delivered to the tissues and anaerobic metabolism is occurring despite “normal” global perfusion. As a consequence, inadequate production of adenosine triphosphate (ATP) and other high-energy phosphates may contribute to the development of MODS in these patients.
The validity of the concept of pathologic supply dependency was subsequently challenged. Mathematical coupling of data (e.g., VO2 and [D with dot above]O2 both determined by use of a pulmonary artery catheter), pooling of data, and spontaneous changes in metabolic demand (which would increase [D with dot above]O2) can explain many of the results of the studies purporting to show pathologic supply dependency. Clinical investigations that utilized independent means to measure both [D with dot above]O2 and VO2 failed to demonstrate pathologic supply dependency in patients with ARDS or sepsis. Furthermore, critical [D with dot above]O2 in these patients was no higher than in other critically ill patients.
More recently, there has been a recognition that supply dependency may be occurring in patients, but not at the global level. There is an increasing appreciation that the regional circulation and microcirculation—arterioles, capillary bed, and postcapillary venules—play a crucial role in the pathogenesis of organ dysfunction in shock. Heterogeneous microcirculatory abnormalities occur due to changes in the activation state and shape of endothelial cells, alterations in vascular smooth muscle tone, activation of the clotting system, and changes in red and white blood cell deformability (discussed later). The surface receptors and mediators associated with these changes are now being identified, and include oxidants, lectins, proteases, vasoactive products of inducible nitric oxide synthase (iNOS), and altered adrenergic receptor sensitivity. Alterations in microvascular circulation have been demonstrated in congestive heart failure, cardiogenic shock, hemorrhage, and sepsis. Microcirculatory changes of congestive heart failure include reduced conjunctival microvascular density and attenuated nailfold capillary recruitment during postocclusive reactive hyperemia (18). Animal models of hemorrhagic shock demonstrate attenuated functional capillary perfusion—a measure of the number of capillaries that are actively moving blood—of skeletal muscle and the intestinal villi (19,20). Renal and intestinal regional blood flow are reduced despite resuscitation and return of global hemodynamics back to baseline values (21). In patients with severe sepsis, forearm reactive hyperemia measured by air plethysmography is diminished following arterial occlusion, and red blood cell deformability is reduced compared to controls (22). Tissue (skeletal muscle) oxygen saturation in septic patients is no different than that observed in healthy controls or postsurgical patients (23); however, microvascular compliance and skeletal muscle oxygen consumption is reduced, and postischemic reperfusion time is increased compared to controls. Orthogonal polarization spectral (OPS) imaging is a technique by which perfusion of small and large vessels in the microcirculation of mucosal surfaces can be seen and quantified. Clinical studies (24,25) where OPS imaging has been utilized have shown that the fraction of perfused small vessels in patients with severe heart failure, cardiogenic shock, or sepsis is significantly lower than in those critically ill patients without those conditions (Fig. 54.3).
Functional cellular hypoxia—“cytopathic hypoxia”—or metabolic failure is a condition where the cell is incapable of utilizing oxygen to produce ATP despite adequate oxygen delivery. Indeed, in patients with sepsis and in animal models of sepsis, oxygen tension in the intestine, bladder, and skeletal muscle is elevated, suggesting that the problem is not one of inadequate [D with dot above]O2 but one of oxygen utilization (26,27,28). The defect in oxygen utilization likely resides in the mitochondrion. Muscle biopsies from septic patients reveal that skeletal muscle ATP concentrations and respiratory chain activity are lower compared to samples obtained from control patients (29). Furthermore, patients dying from sepsis have even lower ATP concentrations and respiratory chain activity (complex 1) compared to sepsis survivors (29,30). The mechanism by which cytopathic hypoxia or metabolic failure occurs has not been fully elucidated. However, nitric oxide and its metabolite, peroxynitrite, are mediators that are released during sepsis and are inhibitors of the mitochondrial electron transport chain (31). Single-strand breaks in nuclear DNA can occur in sepsis by a variety of endogenously formed oxidants, including peroxynitrite. Poly (ADP-ribose) polymerase (PARP) is an enzyme that is activated by the formation of these DNA breaks. Since the substrate for PARP is NAD+, the activation of PARP can result in a profound reduction in the cellular levels of NAD+, thereby causing cellular energy depletion (32,33).
|
|
|
Figure 54.3. Sublingual microcirculation as assessed by orthogonal polarization spectral imaging in a healthy volunteer (A) and in a patient with early septic shock (B). Normal capillary density is observed in panel A whereas low capillary density is observed in panel B. Real time images may be viewed at http://www.cooperhealthorg/content/gme_fellowship_shock.htm. (Reproduced from De Backer D, Creteur J, Preiser J-C, et al. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002;166:98–104, with permission.) |
Role of Inflammatory and Vasoactive Mediators
Although early clinical series emphasized the implication of uncontrolled infection in the development of MODS, it is clear that MODS can occur with either extensive tissue injury such as that seen with trauma, pancreatitis, or sepsis. A large amount of evidence is available that implicates the release of inflammatory mediators in the pathogenesis of MODS (Table 54.3).
Complement, Neutrophils, and Reactive Oxygen Metabolites
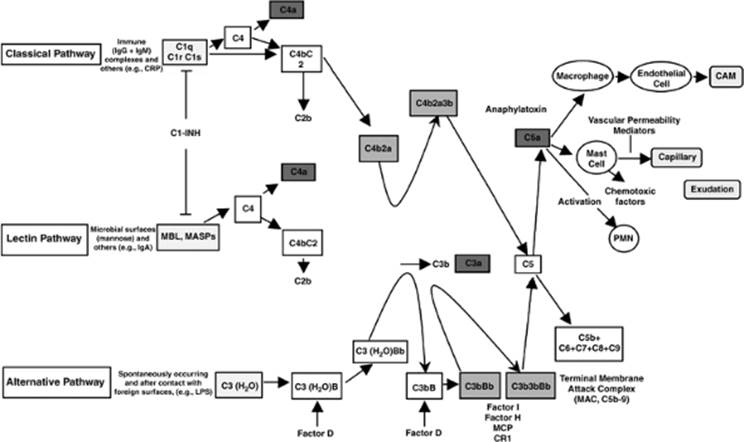
The complement cascade is activated via three pathways (Fig. 54.4). The classical pathway is triggered by antibody-coated targets or antigen–antibody complexes. The alternative pathway is activated by aggregated immunoglobulins, products of tissue trauma, lipopolysaccharide (LPS), and other complex polysaccharides. The lectin-ficolin pathway is initiated by the binding of organisms to mannose binding-lectin (MBL), a protein important in innate immunity (34). Once MBL is bound to a pathogen, an MBL-associated serine protease is produced, which forms a C3 convertase by cleavage of C4 and C2. Products of the complement pathway activate neutrophils, which can obstruct capillaries and release oxygen radicals and lysosomal enzymes—among other mediators, thereby damaging the endothelium. Furthermore, adhesion molecules, which are expressed on both polymorphonuclear leukocytes (PMNs) and vascular endothelium in response to LPS and other inflammatory mediators, facilitate the adherence and diapedesis of PMNs through the endothelium.
|
Table 54.3 Inflammatory mediators important in the pathogenesis of sepsis and the multiple organ dysfunction syndrome |
||
|
A significant amount of evidence exists, suggesting that complement activation is important in the pathophysiology of MODS. In an animal model of generalized inflammation (e.g., with zymosan treatment), C5-deficient mice had a lower mortality compared to wild-type mice; however, late organ failure was not different between the groups (35). An inhibitor of complement, bisbenzylisoquinoline alkaloid, will decrease mortality and the percentage of animals with organ injury in zymosan-treated mice (36). In a rodent model of abdominal aortic aneurysm rupture, a complement C5a receptor antagonist attenuated lung and intestinal permeability indices and lung myeloperoxidase activity compared to controls (37). Clinically, activation of both complement and neutrophils occurs in patients with ARDS or burns (38,39), and circulating plasma levels of C3a correlate with severity of injury and outcome in patients with multiple trauma (40). Evidence of complement and neutrophil activation has also been found in bronchoalveolar lavage (BAL) from patients with ARDS (41). Administration of a C1-inhibitor in patients with severe sepsis and septic shock reduces neutrophil activation (42) and improves renal function and SOFA scores compared to untreated control patients (43).
|
|
|
Figure 54.4. The three complement activation pathways (classic, alternative, and lectin). IgG, immunoglobulin G; IgM, immunoglobulin M; IgA, immunoglobulin A; CRP, C-reactive protein; MBL, mannose-binding lectin protein; MASP, MBL-associated proteases; C1-INH, C1-inhibitor; LPS, lipopolysaccharide; CAM, cell adhesion molecules; MAC, membrane attack complex; PMN, polymorphonuclear leukocytes; MCP, monocyte chemoattractant protein; CR1, complement component receptor 1. (Reproduced from Goldfarb RD, Parrillo JE. Complement. Crit Care Med. 2005;33:S482–S484, with permission.) |
Reactive oxygen species—superoxide anion, hydrogen peroxide, and the hydroxyl radical—are released by activated PMNs and can injure tissues by damaging DNA, cross-linking cellular proteins, and causing peroxidation of membrane lipids (44,45). Lipid peroxidation diminishes membrane fluidity and increases membrane permeability, thereby impairing cellular function. The conclusion that toxic oxygen radicals are important in the pathophysiology of respiratory dysfunction comes from clinical studies of patients with ARDS where plasma levels of lipid peroxides are elevated, levels of hydrogen peroxide are increased in the expiratory condensate (46), and oxidative damage to proteins in BAL fluid is found (47). In addition, patients with ARDS have reduced levels of oxygen radical scavengers (e.g., α-tocopherol, ubiquinone, and glutathione), a sign of “oxidant stress” (48,49). Despite these data, antioxidant therapies have not translated into improved outcome for patients with ARDS or MODS, although such interventions may increase the number of days “free” of acute lung injury (50) or mechanical ventilation (51), decrease the incidence of new organ failures (51), and reduce the oxidative stress during septic shock (52).
The Kallikrein-Kinin System
The kallikrein-kinin system is part of the contact system, and is composed of complement, coagulation, and kallikrein-kinins. Bradykinin, the end product of this cascade, is a potent vasodilator and increases vascular permeability. Some of these effects are mediated by the release of secondary mediators, such as nitric oxide and eicosinoids. Although some experimental and clinical data suggest that the kallikrein-kinin system is important in the pathogenesis of sepsis and MODS, a clinical trial of a bradykinin receptor antagonist (CP-0127) for the adjuvant therapy of sepsis failed to alter the 28-day mortality (53).
|
|
|
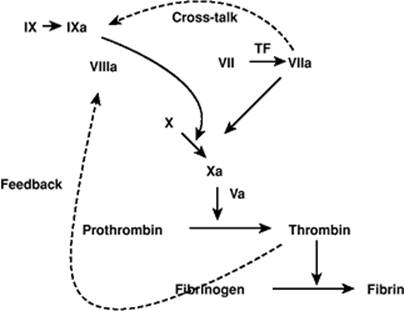
Figure 54.5. Revised coagulation scheme. Coagulation is initiated by the extrinsic pathway and amplified by factor VIIa activation of factor IX, and thrombin activation of factors VIII and XI and platelet surfaces. (Reproduced from Aird WC. Coagulation. Crit Care Med. 2005;33:S485–S487, with permission.) |
The Coagulation System
LPS and many proinflammatory mediators will activate the coagulation system (Fig. 54.5). Coagulation in sepsis or inflammatory states is initiated primarily by the extrinsic tissue factor–dependent pathway, as these mediators induce the expression of tissue factor (TF) on monocytes and endothelial cells (54). Although these same mediators activate the fibrinolytic system, subsequent increases in plasminogen activator inhibitor-1 (PAI-1) and thrombin-activatable fibrinolysis inhibitor (TAFI) effectively suppress fibrinolysis. Important inhibitors of coagulation such as antithrombin, tissue factor pathway inhibitor (TFPI), protein C, protein S, and endothelial-bound modulators—heparan sulfate and thrombomodulin—may be down-regulated (54). Consequently, there is a net procoagulant tendency with the potential for the development of disseminated intravascular coagulation (DIC). DIC can cause microvascular thrombosis and organ failure, and/or bleeding from consumption of platelets and clotting factors (55,56). Recombinant human activated protein C has been shown to reduce the relative risk of death in patients with severe sepsis by over 19%, at least in part by its inhibition of the coagulation cascade (57).
|
|
|
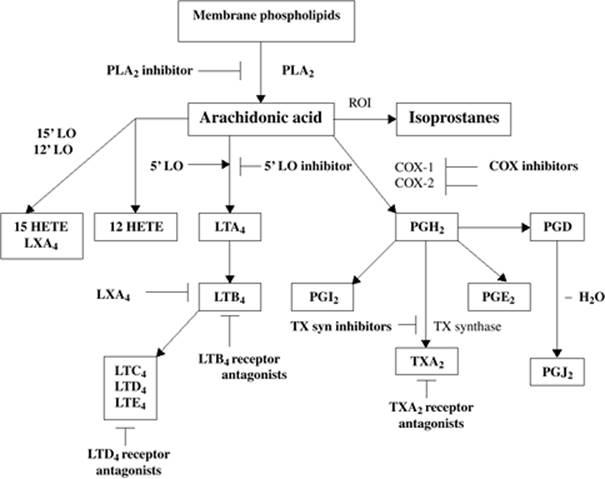
Figure 54.6. The eicosanoid pathway. PLA2, phospholipase A2; ROI, reactive oxygen intermediates; COX, cyclo-oxygenase; LO, lipo-oxygenase; HETE, hydroxyeicosatetraenoic acid; LT, leukotriene; TX, thromboxane; LX, lipoxin; PG, prostaglandin. (Reproduced from Cook JA. Eicosanoids. Crit Care Med. 2005;33:S488–S491, with permission.) |
Prostaglandins, Leukotrienes, and Platelet-Activating Factor
Prostaglandins (PGs), leukotrienes (LTs), and platelet-activating factor (PAF) are potent lipid mediators formed by the stimulation of a membrane-bound enzyme, phospholipase A2 (PLA2), via a variety of mediators including norepinephrine, adenosine, bradykinin, PAF, tumor necrosis factor (TNF), and interleukin (IL)-1β (58). PLA2 catalyzes membrane phospholipids to lyso-PAF and arachidonic acid (Fig. 54.6). Both experimental and clinical studies support the notion that these mediators play a role in the pathophysiology of sepsis and MODS.
Elevated plasma levels of thromboxane B2 (TXB2), the metabolite of the prostaglandin thromboxane A2 (TXA2), are observed in animal models of sepsis and are correlated with organ injury and outcome. Inhibitors of cyclo-oxygenase and thromboxane synthase abrogate the organ injury and improve survival. Clinically, elevated plasma levels of TXB2 and PGI2 have been measured in patients with Gram-negative septic shock and correlate with the severity of organ failure and survival. However, clinical trials of nonselective inhibitors of cyclo-oxygenase (ibuprofen) have failed to show any effect on survival (59), and a recent trial evaluating the effectiveness of a selective inhibitor of a group IIA secretory phospholipase in patients with suspected sepsis and organ failure showed no survival benefit nor effect on organ dysfunction (60). The antifungal agent, ketoconazole, is an imidazole derivative that inhibits thromboxane synthase. Use of ketoconazole in patients at risk for ARDS prevents the development of ARDS (61,62); however, once ARDS is established, this agent is ineffective at reversing the syndrome or improving survival (63). Other clinical trials evaluating inhibitors of prostaglandin production have been underpowered to show any effect.
A significant role of leukotrienes in the pathophysiology of the cardiovascular dysfunction observed in sepsis is unlikely, since the administration of leukotrienes (LTC4 and LTD4) to primates and sheep results in increased systemic vascular resistance and depressed cardiac output. However, intratracheal administration of these leukotrienes in animals results in a significant increase in capillary permeability, leading to a pattern of pulmonary edema not inconsistent with ARDS. Furthermore, elevated concentrations of LTB4, LTC4, and LTD4 have been measured in the BAL fluid recovered from patients with ARDS. Although leukotriene receptor antagonists have improved pulmonary hemodynamics and oxygenation in experimental sepsis studies, no clinical trials using these agents for sepsis or ARDS have been performed.
A large amount of data exists supporting the role of PAF in the pathogenesis of sepsis and MODS. In vitro, incubation of macrophages with PAF will lead to an exaggerated release of TNF and tissue factor by these cells following exposure to LPS. Conversely, LPS-induced release of TNF by macrophages is inhibited by PAF receptor antagonists. PAF expression on the surface of endothelial cells will result in PMN adherence and activation. In addition, stimulation of PAF receptors on the endothelium results in changes in cell shape and cytoskeletal structure. In animal models of endotoxicosis, elevated plasma levels of PAF have been measured and are associated with many of the physiologic abnormalities seen in sepsis: myocardial dysfunction, vasodilatation, and microvascular permeability. Infusion of PAF into animals reproduces many of the findings observed in endotoxicosis or sepsis. In animal models of sepsis, PAF receptor antagonists have had variable effects on outcome; however, most studies have demonstrated an improvement in organ function (58). Clinically, depressed plasma levels of PAF acetylhydrolase (PAF-AH), the enzyme responsible for metabolizing PAF, have been observed in critically ill patients and correlate inversely with organ dysfunction. However, a phase III trial of recombinant PAF-AH failed to improve outcome or prevent organ dysfunction (64). A subsequent study of critically ill patients revealed that plasma levels of PAF-AH were variable over time and with severity of illness (65). Such a finding provides a partial explanation for the lack of efficacy of recombinant PAF-AH.
Cytokines
Cytokines are small proteins that are secreted by nearly all nucleated cells and exhibit autocrine, paracrine, or endocrine activity (66,67); they are generally classified as proinflammatory or anti-inflammatory molecules. This classification, however, is somewhat arbitrary as an individual cytokine may act in either a proinflammatory or anti-inflammatory fashion, depending on the underlying biologic process. Proinflammatory cytokines such as TNF and IL-1 can stimulate the release of other mediators: PAF, nitric oxide, LTs, and PGs.
TNF assumes an important role in the pathogenesis of human sepsis, septic shock, and MODS. TNF is directly cytotoxic to some cell types and will induce the expression of adhesion molecules on neutrophils and endothelial cells to promote the recruitment of these white cells to the site of injury or infection. Furthermore, endothelial permeability is increased. Metabolic effects attributable to TNF include activation of the acute-phase response, fever (along with IL-1), skeletal muscle catabolism, and increased peripheral lipolysis and hepatic lipogenesis (67). When injected into normal volunteers, small doses of LPS or recombinant TNF will reproduce many of the metabolic and hemodynamic changes observed in sepsis (68,69). Similar findings are observed in animal studies, and treatment with anti-TNF antibodies will prevent many of the adverse consequences of endotoxic or live Gram-negative bacterial shock (70). However, in studies of critically ill patients, the correlation of plasma TNF levels and outcome is variable (71,72). Such disparate results may be due to timing and method of the TNF assay, as well as to the acuity, etiology, or treatment of the patient's illness, or genetic differences in patients. Results from multicenter studies of adjuvant therapy with either anti-TNF antibodies or soluble TNF receptors have demonstrated that neither passive immunization nor the soluble receptors reduce mortality from sepsis. However, in one recent trial where patients were stratified according to initial plasma levels of IL-6 (as a marker of severity of illness), outcome was improved, and organ dysfunction was ameliorated with the administration of a monoclonal antibody to TNF (73).
Like TNF, IL-1 has a wide variety of biologic actions and has been implicated in the pathogenesis of sepsis and MODS (74). In addition, TNF and IL-1 often act in a synergistic fashion. IL-1 induces the expression of cyclo-oxygenase (COX)-2 and iNOS expression (75). Furthermore, IL-1 increases the expression of other cytokines—most notably TNF and IL-6—chemokines, adhesion molecules, and a number of tissue proteases and matrix metalloproteases (75). IL-1 also stimulates the release of myeloid progenitor cells, resulting in neutrophilia (75). Animal models suggest a role for IL-1 in sepsis and MODS, and the use of IL-1 receptor antagonist (IL-1RA) is beneficial in several human inflammatory diseases (e.g., rheumatoid arthritis). However, use of IL-1RA in patients with sepsis does not reduce mortality nor reverse organ failure (76,77,78).
Interleukin 6 (IL-6) is another cytokine that has been identified to be important in the response to infection and development of MODS. Small doses of endotoxin administered to normal volunteers will stimulate the release of IL-6 (79). IL-6 will persist for longer periods of time in the blood than other cytokines and may serve as an important marker for the outcome of patients with sepsis or septic shock. Both the IL-6 receptor and the signaling receptor gp130 are required for the biologic activity of IL-6 to be realized (79). Murine models of hemorrhagic shock indicate that IL-6 is important in the development of gut barrier dysfunction (see below) (79). In addition, IL-6 may be important in promoting thrombosis during sepsis. Passive immunization with an anti–IL-6 antibody reduces activation of the coagulation cascade in a primate model of endotoxicosis but has no effect on the coagulation abnormalities associated with low-dose LPS in humans (80).
IL-8 is a chemotactic cytokine (chemokine) and is expressed principally by monocytes and macrophages by stimulation with LPS, bacteria, TNF, and IL-1 (81). IL-8 induces chemotaxis of inflammatory cells, and its presence at sites of inflammation may persist for long periods of time (81). That IL-8 is important in the development of MODS is demonstrated by the observation of high IL-8 levels in BAL fluid from patients with ARDS or pneumonia (82). Although neutralizing IL-8 may reduce cardiac ischemia/reperfusion injury in dogs, a reduction in chemokines increases mortality in animal models of pneumonia (81).
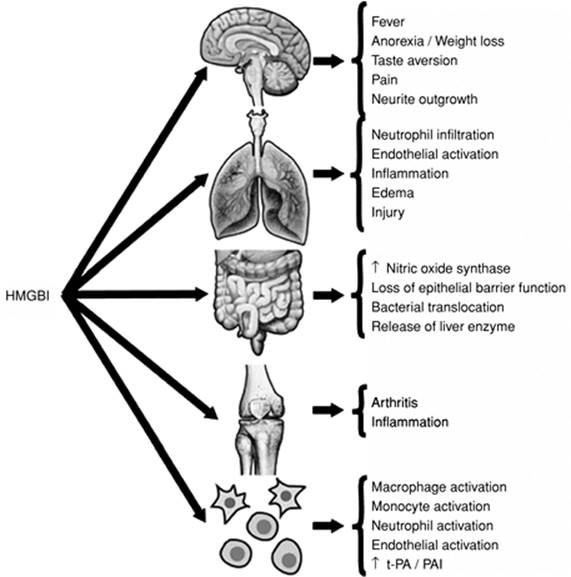
High-mobility group box 1 (HMGB1) is a cytokine that was discovered over 30 years ago, but its importance as an inflammatory mediator was appreciated only recently (Fig. 54.7) (83). HMGB1 is released from a variety of cells in response to LPS or bacteria, and the response is delayed (84). Exposure of the lung to HMGB1 increases neutrophil accumulation, edema, and other proinflammatory cytokines, whereas gastrointestinal exposure results in increased gut permeability and translocation of bacteria to mesenteric lymph nodes (84). Administration of this cytokine to animals causes death as a result of epithelial barrier disruption. Treatment of experimental endotoxicosis or sepsis with antibodies to HMGB1 or HMGB1 antagonists improves survival (83,85). There have been no clinical studies to evaluate the efficacy of anti-HMGB1 therapies in sepsis or other inflammatory conditions.
|
|
|
Figure 54.7. Inflammatory responses in various areas of the host that are medicated by high-mobility group box 1 (HMGB1). tPA, tissue plasminogen activator; PAI, plasminogen activator inhibitor. (Reproduced from Wang H, Yang H, Tracey KJ. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004;255:320–331, with permission.) |
Macrophage migration inhibitory factor (MIF) is a cytokine that is found in macrophages in preformed cytoplasmic pools (86). It is released rapidly in response to bacterial products and works to up-regulate and sustain the activation of a variety of cell types to produce TNF, IL-1, IL-6, and IL-8. The major action of MIF may be the regulation of p53-dependent apoptosis. Anti-MIF therapy in animal models of sepsis is protective (86). Although MIF appears to be an important mediator of sepsis and sepsis-induced MODS (87), it does not appear to play a prominent role in tissue injury resulting from trauma (88).
Nitric Oxide
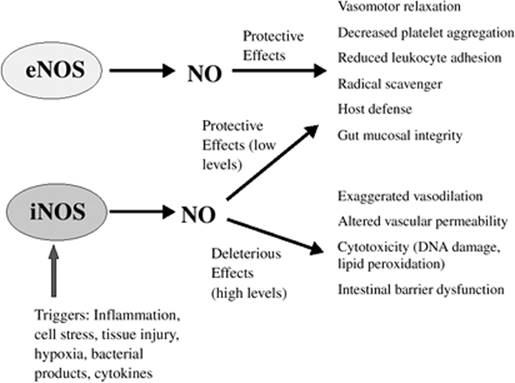
Nitric oxide (NO) is an inorganic free-radical gas, produced by catalysis of one of the terminal guanidine nitrogens of L-arginine by the NO synthase (NOS) group of enzymes (89). Two general classes of NOS have been described: Constitutive (calcium-dependent) NOS (neuronal and endothelial) and inducible (calcium-independent) NOS (90). The production of the latter enzyme is induced by LPS, TNF, and a variety of other inflammatory mediators. A variety of cells and tissues release NO, including endothelium, vascular smooth muscle, neutrophils, and mononuclear, glial, mast, hepatic, and adrenal medullary cells (90). Vasorelaxation, neurotransmission, and microbicidal activity are some of the important functions that NO possesses. The role that NO plays in the host is a function of the rate and timing of its production and the surrounding environment. Normally, NO acts as a direct signaling molecule (e.g., vasorelaxation and neurotransmission), and low levels of NO (produced by constitutive NOS and at times by inducible NOS) have protective effects (Fig. 54.8) (90). Alternatively, it may function as an indirect cytotoxic agent and induce intestinal barrier dysfunction.
|
|
|
Figure 54.8. The divergent effects of nitric oxide (NO) derived from the endothelium (eNOS, endothelial nitric oxide synthase) or as a consequence of inflammation (iNOS, inducible or inflammatory NOS). (Reproduced from Levy RM, Prince JM, Billiar TR. Nitric oxide: a clinical primer. Crit Care Med. 2005;33:S492–S495, with permission.) |
A significant amount of experimental and clinical evidence suggests that NO plays an important role in the pathophysiology of sepsis and MODS. Inducible NOS and NO production increase in animals during both endotoxic and hemorrhagic shock. iNOS-deficient mice are protected from LPS-induced hypotension, and have a higher survival following endotoxicosis (91). NO contributes to TNF-induced cardiac dysfunction in a concentration-dependent fashion (92). Increased urinary excretion of NO metabolites (nitrite and nitrate) has been reported in septic patients and correlates inversely with systemic vascular resistance (SVR) (91).
Excess NO in the presence of superoxide anion results in the formation of peroxynitrite (ONOO-), a reactive oxidant that causes lipid peroxidation, inhibits mitochondrial respiration, inactivates glyceraldehyde-3-phosphate dehydrogenase, inhibits membrane sodium/potassium ATP activity, and triggers DNA single-strand breakage. As mentioned previously, DNA damage activates the nuclear enzyme, PARP, which can lead to cellular energy depletion and death (93). Furthermore, excessive amounts of NO and peroxynitrite can activate the transcription factor, NFκB, and amplify the inflammatory response (94).
The efficacy of inhibitors of NOS in the treatment of sepsis and MODS is unclear, and the use of some inhibitors may actually be detrimental. Although hypotension, vascular leak, and vasopressor requirements can be reduced with the use of nonspecific NOS inhibitors, microcirculatory blood flow can be altered, resulting in organ injury. Indeed, a large randomized, prospective trial evaluating the efficacy of the nonspecific NOS inhibitor, L-NG-monomethylarginine, in patients with septic shock was stopped early because the death rate was higher in the intervention group (95).
The Endothelium
The role of the vascular endothelium is to control the flow of nutrients, blood cells, and a broad array of biologically active molecules to the tissues; this is achieved via membrane-bound receptors for a plethora of molecules and through tight junction proteins and receptors that regulate cell–cell and cell–matrix relationships (96). Endothelial cell injury may well impair the delivery of nutrients to tissues and allow the extravasation of proinflammatory mediators into the interstitial space.
There is a large amount of data supporting the role of an impaired endothelium in the development of MODS. Endothelial cell exposure to LPS will cause anatomic changes including nuclear vacuolization, cytoplasmic swelling and fragmentation, and detachment from the internal elastic lamina (97). In humans with septic shock, elevated levels of circulating endothelial cells can be detected and correlate with outcome. High plasma levels of molecules that are expressed on the surface of endothelial cells (i.e., thrombomodulin [TM], intercellular adhesion molecule [ICAM]-1, and E-selectin) are observed during sepsis and acute lung injury, and are an indirect indication of endothelial damage (98). Furthermore, this injury appears to be sustained, as injection of small doses of LPS into human volunteers will result in high plasma levels of TM that peak at 24 hours and of TF that are still increasing at 48 hours (96).
In the uninjured state, the endothelial cell has important anticoagulant properties. Several heparin-like molecules are expressed on the surface of the cells to accelerate the inactivation of serine proteases of coagulation by antithrombin (96). Thrombomodulin binds thrombin and forms a complex that activates protein C. However, exposure of the endothelial cells to inflammatory or septic mediators will shift the endothelial cells to a procoagulant state by increasing expression of TF and internalization of TM. Furthermore, the endothelial cell will have impaired release of tissue plasminogen activator and an increased release of PAI-1. This procoagulant/antifibrinolytic state is associated with fibrin deposition, platelet consumption, microthrombi, tissue ischemia and necrosis, and an increased risk of death (99).
Before leukocytes and monocytes can migrate into tissues, they must adhere to the endothelium. This process is accomplished by the local synthesis of PAF and cytokines (IL-1, IL-8, and TNF), which stimulates the expression of surface molecules called selectins on leukocytes (L-selectin) and endothelial cells (E-selectin). The interplay between these selectins allows loose binding of the leukocyte to the endothelium. Leukocytes are bound more strongly to the endothelium by the interaction between the CD11/CD18 complex, which is expressed on the leukocyte, and the ICAM-1, which is expressed on the endothelial cell membrane. The role of leukocyte adhesion in the development of MODS is suggested by several lines of evidence. In animal models of sepsis, endotoxicosis, or ischemia/reperfusion, monoclonal antibodies to the CD11/CD18 integrin or to L-selectin will improve organ dysfunction (100). Knockout animals lacking either ICAM-1 or E-selectin have improved survival during experimental sepsis (101). Clinically, plasma ICAM-1 levels are higher in patients with septic shock than in healthy controls or patients with SIRS (98), and the levels correlate with the severity of shock.
Endothelium-derived relaxation is also impaired in sepsis, and such alteration may contribute to MODS. Acetylcholine-induced relaxation of aortic rings obtained from septic animals is attenuated. In animals with chronically overexpressed endothelial cell nitric oxide synthase (ecNOS), resistance to LPS-induced hypotension, lung injury, and death are observed (96). In normal volunteers, small doses of LPS will also impair endothelium-dependent relaxation for days (102). These data help explain the observations that reactive forearm hyperemia is attenuated in patients with sepsis. In addition, the importance of intact ecNOS in these patients is supported by the observation that treatment of patients with septic shock by nonselective NOS inhibitors is associated with no change or an increase in mortality compared to untreated patients (95).
Epithelial Barrier Dysfunction
Epithelial cells help maintain normal function of organs by the maintenance of distinct compartments. The tight junctions between adjacent epithelial cells serve several important functions: (a) differentiation (103) of the cell into apical and basolateral domains, (b) preservation of cellular polarity, (c) generation of distinct internal environments formed by the epithelial layer, and (d) providing a semipermeable barrier that regulates the passive diffusion of solutes between the paracellular pathway and prevents entry of microbes and toxins (e.g., intestine and lung) (103).
Data from studies using cultured epithelial monolayers show that nitric oxide and peroxynitrate, as well as other proinflammatory mediators, increase the permeability of the monolayer. The mechanism for this increase in permeability is incompletely understood; however, some data suggest that there is a decrease in the expression and improper localization of some of the tight junction proteins. Other investigations have determined that inhibition of the epithelial cell membrane Na+-K+-ATPase pump by these mediators results in cellular swelling and an increase in intracellular sodium concentration, which ultimately impairs the expression and localization of the tight junction proteins. Data from murine models of endotoxemia indicate that LPS will decrease the expression and alter the localization of tight junction proteins in the intestine compared to control animals (104). Furthermore, these animals will have increased number of bacteria recovered from regional mesenteric lymph nodes. Nitric oxide appears to be important in the disruption of gut barrier function, as neither intestinal permeability nor bacterial translocation is observed when knockout mice lacking iNOS are treated with LPS. Similar alterations in hepatic (105) and pulmonary (106) epithelial barrier function and tight junction protein expression and localization have been shown during murine endotoxicosis.
Derangements in Gut Barrier Function
Although epithelial barrier function in the intestine may be altered during sepsis and other inflammatory states, thereby allowing bacterial translocation to occur, there are other components of the gut barrier that will prevent the bacteria or bacterial products from gaining access to systemic organs (107). In addition to alterations in the epithelial barrier described previously, other clinical conditions that can contribute to altered gut barrier function include antibiotics, stress ulcer prevention, hypoalbuminemia, vasoactive agents, and use of hyperosmolar feeding preparations. The clinical importance of the disrupted gut barrier function in the pathogenesis of MODS remains ill defined at this point.
Apoptosis
Apoptosis (programmed cell death) is the term used to describe a specific method by which cells die. The event is a well-defined, active, and energy-dependent process. There are two primary pathways involved in apoptosis: The intrinsic (mitochondrial and endoplasmic reticulum) pathway and the extrinsic (“death receptor”) pathway (108). The latter pathway is activated by receptors such as Fas, with the subsequent activation of two enzymes, caspase-8 or -10. The intrinsic pathway stimulates caspase-9 by loss of the mitochondrial membrane potential and movement of cytochrome c into the cytosol. These initiator caspases cleave effector caspases (e.g., caspase-3 and -7), which results in the cleavage of cellular proteins and DNA and, ultimately, apoptosis (109).
The exact role of apoptosis in the development of MODS is unclear. In animal models of infection, lymphocyte, endothelial cell, kidney, lung, and skeletal muscle, apoptosis is increased (110). Data from clinical studies show that up-regulation of apoptotic pathways is increased in patients with ARDS (111). Furthermore, widespread apoptosis occurs in splenic and colonic lymphoid populations in patients who die from sepsis and MODS (112,113). The effect of apoptosis on the immune function includes loss of various immune cells and impairment of immunity by apoptosis-induced immunosuppression of the remaining immune cells (114). Therapies directed against these programmed pathways are under intense investigation and include inhibition of cytochrome c release, use of RNA interference for gene silencing, and caspase inhibitors (114).
Complex Nonlinear Systems
The body may be considered a biologic network that is complex, highly coupled, and nonlinear (115). The host response to trauma, shock, or sepsis—involving metabolic, neural, endocrine, inflammatory, and immune components—is such an example (116). The behavior of such a system cannot be predicted with great reliability; however, the system is “attracted” to specific states or stable configurations: “organized variability” (116,117). A large enough perturbation to an organ or mediator network may have unexpected and significant results elsewhere in the host and ultimately lead to MODS (118). In the healthy individual, there is a high degree of heart rate (beat-to-beat) variability. Several studies have shown a relationship between loss of heart rate variability and increased mortality in critically ill patients (119). In fact, normal volunteers injected with small doses of LPS exhibit loss of heart rate variability (120). Other examples of increased regularity of rhythms associated with disease include Cheyne-Stokes respiration, parkinsonian gait, neutrophil count in chronic myelogenous leukemia, and fever in Hodgkin disease (117). However, several diseases—acromegaly and Cushing disease—are associated with increased complexity (117). These data suggest that health is determined by “distance” from thermodynamic equilibrium: too much or too little variation (low or high entropy) represents pathologic conditions (117). A causal link between perturbations in complex systems and outcome remains elusive. However, following variability over time might allow greater accuracy in patient prognostication or may suggest a medical intervention (121). For example, a low level of complexity in the temperature curve of critically ill patients with MODS portends a poor prognosis (122). More research is needed into nonlinear dynamics to determine its importance and role in the development of MODS.
Genetic Susceptibility
Single base variations in DNA—single nucleotide polymorphisms (SNPs)—are commonly used to discern genetic differences among patient populations (123). Approximately 1 in every 1,000 bases in the human genome is different between two unrelated individuals (123). Although more than 10 million SNPs have been mapped, only 4% of these occur in genes. By comparing healthy individuals to patients, SNPs involved in disease can be identified. Indeed, some—but not—all clinical studies have documented an increased risk of death and organ dysfunction in patients suffering from sepsis or ARDS and who are homozygotes for SNPs (124,125,126,127,128). In the future, patients may be identified in advance as to who should be monitored more closely or who might be benefit from certain interventions (e.g., anticytokine therapies) (123).
Prevention and Treatment of Multiple Organ Dysfunction Syndrome
Although much progress has been made in our understanding in the pathogenesis of MODS, our knowledge remains incomplete. Consequently, the prevention and treatment of MODS are nonspecific and include the goals of maintaining adequate tissue oxygenation, finding and treating infection, providing adequate nutrition support, minimizing iatrogenic complications, and when necessary, providing artificial support (e.g., dialysis or mechanical ventilation) for individual dysfunctional organs.
Resuscitation
An episode of circulatory shock is probably the most common event that occurs before the development of MODS. As a result, timely restoration of intravascular volume and oxygen delivery is important in preventing or abrogating MODS in high-risk patients.
Controversy continues regarding the correct fluid for resuscitation and the optimal circulating hemoglobin concentration. Crystalloid solutions are efficacious and cheaper than colloid solutions. In a prospective study of a diverse population of critically ill patients comparing the efficacy of normal saline to albumin for fluid resuscitation, there was no overall difference in outcome—death, length of stay, or organ dysfunction—between the two groups (129). Subgroup analysis demonstrated an increased relative risk of death for trauma patients who received albumin, whereas the relative risk of death for severe sepsis patients was higher if saline was used (129). Colloids or hypertonic crystalloid solutions—or colloid/hypertonic crystalloid mixtures—may have beneficial effects on the inflammatory response or the development of edema, and may allow for faster resuscitation (130), but these effects have not been proven to be of overall value when these solutions are used clinically.
Assessing the adequacy of tissue oxygenation can often be difficult. The clinical parameters used most often, including arterial blood pressure, skin color, temperature, urine flow, mixed venous oxygen saturation, and blood-lactate concentrations, may be unreliable. Since observational studies have shown that “supranormal” levels of [D with dot above]O2 (660 mL/minute/m2), VO2 (170 mL/minute/m2), and cardiac index (4.5 L/minute/m2) are associated with higher survival rates, some clinicians have advocated resuscitation to such end points in critically ill patients. Although some studies of patients undergoing high-risk surgery or suffering from trauma suggest that such an approach may be of benefit, the majority of studies over the past 15 years clearly show that resuscitation to these end points in critically ill patients is of no benefit, or actually worsens outcome. Likewise, the use of the pulmonary artery catheter to guide therapy has not been shown to be of benefit in a large number of studies (131,132,133) and pulmonary capillary wedge pressure does not accurately reflect ventricular volume, even in normal volunteers (134). Less invasive and probably safer monitors have been developed for monitoring cardiac output that compare favorably with the accuracy of the thermodilution method. Use of systolic blood pressure variation, pulse pressure variation, stroke volume variation, or left ventricular end-diastolic area (as assessed by transesophageal echocardiography) may be of greater benefit to guide volume resuscitation than the use of pulmonary capillary wedge pressure. More recent data suggest that such aggressive resuscitation in the ICU may be too late. Early (i.e., in the emergency department before hospital admission), goal-directed therapy can reduce mortality and organ dysfunction in patients with severe sepsis and septic shock (135). Practice parameters for the hemodynamic support of the patient with sepsis and septic shock have been recently revised and provide useful guidelines for the practitioner (136).
Other largely experimental approaches that may be of value for assessing the adequacy of resuscitation include tonometric determination of subcutaneous tissue PCO2; monitoring of conjunctival PO2; use of near-infrared spectroscopy to assess the oxidation state of cytochrome a, a3; sublingual PCO2 measurements; and tonometric estimation of gastric intramucosal pH (pHi) or PCO2. The latter two techniques are simple and “minimally invasive.” Tonometric evidence of gastric mucosal acidosis (or hypercarbia) has been shown to correlate with both short-term and long-term mortality in critically ill patients, complications after cardiac surgery, and organ dysfunction in patients with pancreatitis (137,138). Sublingual hypercarbia has also been shown to be associated with increased mortality in critically ill patients (139). In addition, prevention of gastric intramucosal acidosis by timely fluid resuscitation and inotropic therapy may reduce mortality (140). However, more recent data have failed to demonstrate the usefulness of using gastric intramural pH as a therapeutic end point for resuscitation (141). Furthermore, the notion that gastric intramucosal acidosis or hypercarbia reflects the hemodynamics of the entire splanchnic bed has been cast into doubt (142).
Mechanical Ventilation
The method by which patients are mechanically ventilated can contribute to organ dysfunction. A plethora of experimental and clinical data indicate that overdistension of the lung through the use of large tidal volumes will cause lung injury, stimulate the release of inflammatory mediators, and effect derangements in organs other than the lung (143,144,145). Use of small tidal volumes (6 mL/kg) in the care of patients with ALI and ARDS will decrease mortality and increase ventilator-free and organ failure–free days (146). It is crucial to note that oxygenation cannot be used as a proxy for efficacy of therapy since patients who are ventilated with small tidal volumes require a longer period of time before PaO2 improves.
Cyclic “opening” and “closing” of collapsed airways during tidal ventilation is also thought to cause lung injury (147). A recent small study of patients with ARDS suggests that the use of positive end-expiratory pressure (PEEP) above the lower inflection point of the respiratory system compliance curve reduces mortality and the number of failed organs compared to control patients (148). However, this concept has been called into question by data demonstrating that efforts to improve recruitment of collapsed lung units with high levels of PEEP do not reduce mortality or duration of mechanical ventilation (149) and a computed tomography (CT) scan investigation of patients with ARDS showing that ventilator-induced hyperinflation rather than cyclic recruitment/derecruitment is associated with a greater release of pulmonary inflammatory mediators (150).
Other methods of improving oxygenation in the patient with ARDS or ALI, such as the recruitment maneuver and the prone position, have been attempted. However, prospective randomized studies (151,152) have failed to demonstrate an outcome benefit to these approaches.
Fluid management is also an important component in the care of the patient with ARDS or ALI. Recent data show that a restrictive fluid strategy where cumulative fluid balance is kept close to zero in these patients improves the oxygenation index and increases the number of ventilator-free days without increasing the number of other organ failures (153).
Acute Renal Failure
Acute tubular necrosis (ATN) accounts for over 75% of the cases of acute renal failure (ARF) in the ICU (154), with a mortality rate ranging from 40% to 80%. The most common insult that predisposes ICU patients to ATN is persistent prerenal azotemia (154). Furthermore, in the critically ill patient, there is often more than one insult to the kidney: sepsis; exposure to aminoglycosides, amphotericin B, or radiocontrast agents; and the administration of nonsteroidal anti-inflammatory agents. Efforts to minimize these insults to the kidneys should be maximized. Timely resuscitation as mentioned previously is very important to prevent renal ischemia (135,155). If aminoglycosides must be used to treat infection, once-daily dosing (156) or the use of drug levels to discern pharmacokinetics (157) appears to reduce the risk of nephrotoxicity. Use of liposomal preparations of amphotericin B reduces the risk of renal damage (156). If patients are to receive contrast agents, hydration with sodium bicarbonate solutions have been shown to reduce the risk of subsequent renal dysfunction (158). Although N-acetylcysteine has been purported to reduce the risk of contrast-induced ARF (159), the observed results may be a reflection of the activation of creatinine kinase or an increase in the tubular secretion of creatinine (156). Medications such as “low-dose” dopamine or fenoldopam, which increase renal blood flow or loop diuretics, have no impact on preserving renal function in high-risk patients and should be avoided (156). The various methods of renal replacement therapy for patients with established renal failure are beyond the scope of this chapter; the reader is referred to Chapter 161.
Debridement of Necrotic Tissue and Fracture Stabilization
The presence of dead or devitalized tissue appears to predispose patients to the development of MODS; timely debridement of dead tissue is an important component in the prevention of the syndrome. Early surgical fixation of major lower extremity fractures will result in a lower incidence of ARDS and pneumonia. However, “damage control” orthopedics has recently gained popularity and is a concept whereby fractures are initially treated with external fixation. Definitive therapy occurs later when the patient is more stable. The inflammatory response appears to be attenuated in these patients, and the incidence of organ dysfunction is no higher compared to patients undergoing definitive therapy (160).
Infection
Sepsis is an important cause (or correlate) of MODS. It is important that the presence of infection is excluded in critically ill patients with signs of deteriorating organ function.
Empiric administration of broad-spectrum antibiotics is often necessary in the patient with suspected infection. Failure to administer the correct antibiotic(s) (161) in a timely fashion (162) can increase the risk of death for the patient.
Intra-abdominal Sepsis
Early and adequate drainage of intra-abdominal sepsis is important to prevent the development of MODS. Some surgeons have advocated multiple planned reoperations or open packing for severe cases of intra-abdominal sepsis or necrotizing pancreatitis. However, recent retrospective studies have questioned the utility of the open abdomen for intra-abdominal sepsis therapy because ICU and hospital length of stay and fistula rates appear higher than in matched controls (163). Randomized, prospective trials will be needed to determine when the open approach is optimal; one such trial is under way for patients with pancreatitis (164). Without clinical or radiologic evidence of intra-abdominal infection, “blind” laparotomy in the patient with worsening MODS is unlikely to be fruitful.
Pulmonary Sepsis
Ventilator-associated pneumonia (VAP) can play a role in the development and course of MODS; this is discussed in more detail in Chapter 111. Proven preventive measures include noninvasive positive pressure ventilation, elevation of the head of the bed (165), continuous subglottic suctioning, weaning protocols, optimization of sedation with daily “wake-ups” for the patient (166), and chlorhexidine oral rinse.
Selective digestive decontamination (SDD) is a technique by which topical nonabsorbable antibacterial and antifungal agents (usually with a concomitant 3- to 5-day course of systemic antibiotic therapy) are applied to the oropharynx and proximal bowel in mechanically ventilated patients to reduce the incidence of nosocomial infections, organ dysfunction, and mortality. A meta-analysis of 57 randomized controlled trials demonstrated a favorable effect on bloodstream infections and mortality (167). Fears concerning the emergence of resistance organisms do not appear to be well founded. SDD may very well reduce the incidence and prevalence of colonization with resistant Gram-negative aerobic bacteria (168). However, the use of SDD in the United States does not enjoy widespread popularity for reasons that are unclear.
Catheter-related Sepsis
Catheter-related bloodstream infections (CRBSIs) may contribute to the development and propagation of MODS. Proven strategies to reduce the risk of CRBSIs include handwashing prior to catheter insertion, use of maximum barrier precautions (cap, mask, sterile gloves and gown, and a large sterile drape that covers the patient), use of an aqueous chlorhexidine skin preparation solution, avoiding the femoral site for catheter insertion, and removing catheters when no longer needed (169). If these measures are ineffective in reducing the risk of infection, catheters with antiseptic surfaces (170,171) or impregnated with antibiotics (172) or the use of chlorhexidine dressing sponges can reduce the risk of infection.
Other Sources of Sepsis
Many other sources of infection in critically ill patients may contribute to the development of MODS. These infections are not always readily apparent, and the practitioner caring for the patient should remain alert to their presence. Some of these sources of infection include purulent sinusitis, suppurative thrombophlebitis, otitis media, perirectal abscess, epididymitis, prostatitis, calculous or acalculous cholecystitis (173), meningitis or brain abscess (particularly after instrumentation of the central nervous system), prosthetic intravascular graft infection, lower or upper urinary tract infection, and endocarditis. Physical examination and appropriate laboratory and radiographic studies should exclude these conditions.
Nutrition Support
Malnutrition can contribute to the morbidity and mortality of sepsis and MODS (see Chapter 64). Proteolysis is a prominent finding in sepsis and, although it cannot be suppressed by infusing amino acids, protein anabolism can be achieved by appropriate nutritional support. Furthermore, catabolism is mediated by endogenous catecholamines, and administration of β-adrenergic blocking agents can reverse the hypermetabolic response and protein catabolism (174). Early nutritional support may be beneficial in patients at risk for developing MODS (175). The consensus among experts is that if nutritional support is started, enteral feeding is preferable to the parenteral route in a variety of critically ill patients (176).
Regardless of the route of feeding, overfeeding should be avoided. The excessive administration of carbohydrates can alter the respiratory quotient with adverse effects on weaning from mechanical ventilation and affect hepatic metabolic function, thereby altering drug clearance and inducing hyperglycemia. Current guidelines for support of hypermetabolic patients with sepsis or MODS include a total caloric intake (exclusive of protein) of 20 to 25 kcal/kg/day—2 to 5 g/kg/day of glucose, plus 0.5 to 1.0 g/kg/day of fat, and 1.2 to 1.5 g/kg/day of protein. The number of calories needed for a given patient can be estimated using the Harris-Benedict equation or refined using indirect calorimetry.
Hyperglycemia can be a difficult problem, even when patients are not being fed. A strategy using a continuous infusion of insulin to maintain a range of serum glucose of 80 to 110 mg/dL in surgical patients can reduce mortality and organ dysfunction (e.g., renal failure and critical-illness polyneuropathy) (177). In medical ICU patients, morbidity, but not mortality, is reduced with such an intensive insulin regimen (178). Such a strategy is cost effective, with average savings of over $1,500 per patient (179). Maintenance of normal serum glucose levels appears to be the factor associated with the favorable outcome rather than the insulin dose (180).
Specialty Formulas
A number of enteral nutritional formulas are available that provide specific nutrients: glutamine, peptides, arginine, omega-3 fatty acids, nucleic acids, and antioxidants (e.g., vitamins E and C, β-carotene). Arginine is the substrate for NO synthase and is important in lymphocyte proliferation and wound healing (181). Omega-3 fatty acids change membrane lipid composition and can alter the inflammatory response (182). Nucleic acids assist in the proliferation of lymphocytes and intestinal crypt cells, as well as DNA and RNA synthesis (181). Several enteral nutrition formulas are available that include combinations of these additives. The Canadian Critical Care Clinical Practice Guideline Committee (183) recommends that arginine and other “select” nutrients not be used for enteral nutrition. However, in patients with ARDS, a formula supplemented with fish oil, borage oil, and antioxidants should be used (183). Although routine use of glutamine is discouraged, in patients with trauma and burns, enteral glutamine should be considered (183).
|
Table 54.4 Suggested strategies for the prevention of multiple organ dysfunction syndrome |
|
|
Summary
Standard therapy for patients with MODS includes adequate cardiovascular resuscitation, identification and timely treatment of infection, early enteral nutrition, “tight” glucose control, individualized support for dysfunctional organs, and minimizing iatrogenic complications by following clinical practice guidelines based on evidence-based medicine for mechanical ventilation and prevention of ventilator-associated pneumonia and catheter-related bloodstream infections (Table 54.4) (184,185). Development of well-functioning ICU teams helps facilitate these paradigms of care. Improved outcome may be realized if patients at high risk for developing the syndrome can be identified earlier so that preventive measures can be instituted when appropriate. Because the pathogenesis of MODS involves numerous mediators, it is doubtful that all patients can be treated with a single agent or mode of therapy.
Pearls
· MODS develops in up to 40% of critically ill patients without a diagnosis of sepsis and up to 70% of those with a diagnosis of severe sepsis. Mortality attributable to MODS rises as the number of failing organ systems increases; mortality rates in patients with one, two, or three failing organs average 30%, 50%, and greater than 70%, respectively, depending on the etiology of MODS and the organ systems involved.
· Population-based, but not individual, risks of mortality can be predicted with high degrees of precision by several severity-of-illness scoring systems and models.
· MODS may result from “single-hit” insults such as severe infection or trauma, or may evolve through several stages, each having characteristic clinical features.
· An uncontrolled focus of infection, ongoing perfusion deficits resulting in diminished tissue [D with dot above]O2, injured or devitalized tissue, and persistent inflammation commonly initiate and sustain MODS.
· Fever or hypothermia and leukocytosis are not always the manifestations of sepsis but may represent systemic inflammation.
· TNF-α, IL-1, IL-6, IL-8, platelet-activating factor, ROS, and NO are pivotal early mediators in the host response to infection and have multiple pathophysiologic effects relevant to MODS.
· Inappropriate regulation of the production of cytokines, eicosanoids, ROS, and NO is thought to be of causal significance in MODS, as are pathologic neutrophil–endothelial interactions and cross-talk among elements of the coagulation, complement, and kinin cascades.
· Alterations in microvascular blood flow play an important role in the pathogenesis of organ dysfunction in shock. The surface receptors and mediators associated with these alterations include oxidants, lectins, proteases, vasoactive products of iNOS, and altered adrenergic receptor sensitivity.
· Clinically occult dysfunction of the gastrointestinal (GI) mucosal barrier in the ICU is common because of splanchnic ischemia from shock, and may result in endogenous endotoxemia and bacterial translocation.
· Neutrophil- and ROS-mediated intestinal I/R injury in the postresuscitation period is a potential mechanism of remote organ damage. This may lead to a domino-like sequence of organ failures.
· The liver plays a pivotal but clinically inapparent role in systemic host defense through four mechanisms. First, mononuclear phagocytic (Kupffer) cell uptake processes control the magnitude and circulating half-life of endotoxin, bacteria, and vasoactive by-products. Second, production and export of TNF-α with other mediators directly modulate lung function and cardiovascular stability. Third, hepatobiliary clearance is important in the metabolic inactivation and detoxification of such mediators. Fourth, the synthesis of acute-phase reactants regulates several key aspects of metabolism and inflammation.
· Reductions in total hepatic blood flow ([Q with dot above]L) and [D with dot above]O2, or its partitioning between portal venous and hepatic arterial flows, may alter the aforementioned mechanisms, thereby influencing systemic immunoregulation.
· Signs of established MODS are manifested differently in each organ (e.g., ARDS, ARF), yet such changes often reflect generalized endothelial injury and inflammation.
· Diverse medical conditions may mimic sepsis-related MODS and should be excluded when appropriate. These include connective tissue diseases, intoxications, and neoplasms.
· Typical metabolic responses in MODS include hyperglycemia from insulin resistance, accelerated Cori cycle activity, and hepatic glucose release from gluconeogenesis and glycogenolysis. Hypertriglyceridemia results from TNF-α–related reductions in lipoprotein lipase activity. Hepatic lipogenesis is enhanced, increasing the respiratory quotient and minute ventilation. Marked protein catabolism from cytokine-mediated muscle proteolysis and urinary nitrogen wasting is typical.
· Early rapid resuscitation from shock, irrespective of its etiology, attenuates injury to regional organs and may decrease the incidence of MODS.
· Goal-oriented hemodynamic therapy should be initiated early (within 6 hours) after presentation and target values achieved within 12 to 24 hours.
Acknowledgments
The authors thank Marguerite Eckhouse and Susan St. Martin for their editorial assistance. This chapter is a revision of the chapter authored by George M. Matuschak, MD, in the third edition of this textbook and relies on material previously coauthored by the senior author (Heard SO, Fink MP. Multiple-organ dysfunction syndrome. In: Murray MJ, Coursin DB, Pearl RG, et al., eds. Critical Care Medicine. Perioperative Management. Philadelphia: Lippincott Williams & Wilkins; 2002).
References
1. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864.
2. Garcia Lizana F, Manzano Alonso JL, Gonzalez Santana B, et al. [Survival and quality of life of patients with multiple organ failure one year after leaving an intensive care unit]. Med Clin (Barc). 2000;114(Suppl 3):99.
3. Goris RJ, te Boekhorst TP, Nuytinck JK, et al. Multiple-organ failure. Generalized autodestructive inflammation? Arch Surg. 1985;120:1109.
4. Knaus WA, Draper EA, Wagner DP, et al. Prognosis in acute organ-system failure. Ann Surg. 1985;202:685.
5. Marshall JC, Cook DJ, Christou NV, et al. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23:1638.
6. Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis- Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22:707.
7. Moreno R, Vincent JL, Matos R, et al. The use of maximum SOFA score to quantify organ dysfunction/failure in intensive care. Results of a prospective, multicentre study. Working Group on Sepsis related Problems of the ESICM. Intensive Care Med. 1999;25:686.
8. Ferreira FL, Bota DP, Bross A, et al. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA. 2001;286:1754.
9. Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348:683.
10. Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344.
11. Bell RC, Coalson JJ, Smith JD, et al. Multiple organ system failure and infection in adult respiratory distress syndrome. Ann Intern Med. 1983;99:293.
12. Knaus WA, Wagner DP, Draper EA, et al. The APACHE III prognostic system. Risk prediction of hospital mortality for critically ill hospitalized adults. Chest. 1991;100:1619.
13. Sauaia A, Moore FA, Moore EE, et al. Early predictors of postinjury multiple organ failure. Arch Surg. 1994;129:39.
14. Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250.
15. Rangel-Frausto MS, Pittet D, Costigan M, et al. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. 1995;273:117.
16. Komatsu T, Shibutani K, Okamoto K, et al. Critical level of oxygen delivery after cardiopulmonary bypass. Crit Care Med. 1987;15:194.
17. Ronco JJ, Fenwick JC, Tweeddale MG, et al. Identification of the critical oxygen delivery for anaerobic metabolism in critically ill septic and nonseptic humans. JAMA. 1993;270:1724.
18. Houben AJ, Beljaars JH, Hofstra L, et al. Microvascular abnormalities in chronic heart failure: a cross-sectional analysis. Microcirculation. 2003;10:471.
19. Vajda K, Szabo A, Boros M. Heterogeneous microcirculation in the rat small intestine during hemorrhagic shock: quantification of the effects of hypertonic-hyperoncotic resuscitation. Eur Surg Res. 2004;36:338.
20. Arslan E, Sierko E, Waters JH, et al. Microcirculatory hemodynamics after acute blood loss followed by fresh and banked blood transfusion. Am J Surg. 2005;190:456.
21. Cryer HM, Gosche J, Harbrecht J, et al. The effect of hypertonic saline resuscitation on responses to severe hemorrhagic shock by the skeletal muscle, intestinal, and renal microcirculation systems: seeing is believing. Am J Surg. 2005;190:305.
22. Astiz ME, DeGent GE, Lin RY, et al. Microvascular function and rheologic changes in hyperdynamic sepsis. Crit Care Med. 1995;23:265.
23. De Blasi RA, Palmisani S, Alampi D, et al. Microvascular dysfunction and skeletal muscle oxygenation assessed by phase-modulation near-infrared spectroscopy in patients with septic shock. Intensive Care Med. 2005;31:1661.
24. De Backer D, Creteur J, Dubois MJ, et al. Microvascular alterations in patients with acute severe heart failure and cardiogenic shock. Am Heart J. 2004;147:91.
25. De Backer D, Creteur J, Preiser J-C, et al. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med. 2002;166:98.
26. VanderMeer TJ, Wang H, Fink MP. Endotoxemia causes ileal mucosal acidosis in the absence of mucosal hypoxia in a normodynamic porcine model of septic shock. Crit Care Med. 1995;23:1217.
27. Rosser DM, Stidwill RP, Jacobson D, et al. Oxygen tension in the bladder epithelium rises in both high and low cardiac output endotoxemic sepsis. J Appl Physiol. 1995;79:1878.
28. Boekstegers P, Weidenhofer S, Kapsner T, et al. Skeletal muscle partial pressure of oxygen in patients with sepsis. Crit Care Med. 1994;22:640.
29. Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219.
30. Svistunenko DA, Davies N, Brealey D, et al. Mitochondrial dysfunction in patients with severe sepsis: an EPR interrogation of individual respiratory chain components. Biochim Biophys Acta. 2006;1757:262.
31. Singer M. Metabolic failure. Crit Care Med. 2005;33(12 Suppl):S539.
32. Szabo C, Zingarelli B, O'Connor M, et al. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93:1753.
33. Szabo C, Zingarelli B, Salzman AL. Role of poly-ADP ribosyltransferase activation in the vascular contractile and energetic failure elicited by exogenous and endogenous nitric oxide and peroxynitrite. Circ Res. 1996;78:1051.
34. Dommett RM, Klein N, Turner MW. Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens. 2006;68:193.
35. Nieuwenhuijzen GA, Meyer MP, Hendriks T, et al. Deficiency of complement factor C5 reduces early mortality but does not prevent organ damage in an animal model of multiple organ dysfunction syndrome. Crit Care Med. 1995;23:1686.
36. Ivanovska N, Hristova M, Philipov S. Complement modulatory activity of bisbenzylisoquinoline alkaloids isolated from Isopyrum thalictroides–II. Influence on C3–9 reactions in vitro and antiinflammatory effect in vivo. Int J Immunopharmacol. 1999;21:337.
37. Harkin DW, Romaschin A, Taylor SM, et al. Complement C5a receptor antagonist attenuates multiple organ injury in a model of ruptured abdominal aortic aneurysm. J Vasc Surg. 2004;39:196.
38. Robbins RA, Russ WD, Rasmussen JK, et al. Activation of the complement system in the adult respiratory distress syndrome. Am Rev Respir Dis. 1987;135:651.
39. Fosse E, Pillgram-Larsen J, Svennevig JL, et al. Complement activation in injured patients occurs immediately and is dependent on the severity of the trauma. Injury. 1998;29:509.
40. Hecke F, Schmidt U, Kola A, et al. Circulating complement proteins in multiple trauma patients–correlation with injury severity, development of sepsis, and outcome. Crit Care Med. 1997;25:2015.
41. Fowler AA, Hyers TM, Fisher BJ, et al. The adult respiratory distress syndrome. Cell populations and soluble mediators in the air spaces of patients at high risk. Am Rev Respir Dis. 1987;136:1225.
42. Zeerleder S, Caliezi C, van Mierlo G, et al. Administration of C1 inhibitor reduces neutrophil activation in patients with sepsis. Clin Diagn Lab Immunol. 2003;10:529.
43. Caliezi C, Zeerleder S, Redondo M, et al. C1-inhibitor in patients with severe sepsis and septic shock: beneficial effect on renal dysfunction. Crit Care Med. 2002;30:1722.
44. Lindsay TF, Luo XP, Lehotay DC, et al. Ruptured abdominal aortic aneurysm, a “two-hit” ischemia/reperfusion injury: evidence from an analysis of oxidative products. J Vasc Surg. 1999;30:219.
45. Saikumar P, Dong Z, Weinberg JM, et al. Mechanisms of cell death in hypoxia/reoxygenation injury. Oncogene. 1998;17:3341.
46. Kietzmann D, Kahl R, Muller M, et al. Hydrogen peroxide in expired breath condensate of patients with acute respiratory failure and with ARDS. Intensive Care Med. 1993;19:78.
47. Lamb NJ, Gutteridge JM, Baker C, et al. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. 1999;27:1738.
48. Richard C, Lemonnier F, Thibault M, et al. Vitamin E deficiency and lipoperoxidation during adult respiratory distress syndrome. Crit Care Med. 1990;18:4.
49. Lang JD, McArdle PJ, O'Reilly PJ, et al. Oxidant-antioxidant balance in acute lung injury. Chest. 2002;122(6 Suppl):314S.
50. Bernard GR, Wheeler AP, Arons MM, et al. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest. 1997;112:164.
51. Gadek JE, DeMichele SJ, Karlstad MD, et al. Effect of enteral feeding with eicosapentaenoic acid, gamma-linolenic acid, and antioxidants in patients with acute respiratory distress syndrome. Enteral Nutrition in ARDS Study Group. Crit Care Med. 1999;27:1409.
52. Ortolani O, Conti A, De Gaudio AR, et al. Protective effects of N-acetylcysteine and rutin on the lipid peroxidation of the lung epithelium during the adult respiratory distress syndrome. Shock. 2000;13:14.
53. Fein AM, Bernard GR, Criner GJ, et al. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. JAMA. 1997;277:482.
54. Levi M. Disseminated intravascular coagulation: what's new? Crit Care Clin. 2005;21:449.
55. Gando S, Iba T, Eguchi Y, et al. A multicenter, prospective validation of disseminated intravascular coagulation diagnostic criteria for critically ill patients: comparing current criteria. Crit Care Med. 2006;34:625.
56. Ten Cate H. Trombocytopenia: one of the markers of disseminated intravascular coagulation. Pathophysiol Haemost Thromb. 2003;33:413.
57. Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699.
58. Bulger EM, Maier RV. Lipid mediators in the pathophysiology of critical illness. Crit Care Med. 2000;28(4 Suppl):N27.
59. Bernard GR, Wheeler AP, Russell JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997;336:912.
60. Abraham E, Naum C, Bandi V, et al. Efficacy and safety of LY315920Na/S-5920, a selective inhibitor of 14-kDa group IIA secretory phospholipase A2, in patients with suspected sepsis and organ failure. Crit Care Med. 2003;31:718.
61. Slotman GJ, Burchard KW, D'Arezzo A. Ketoconazole prevents acute respiratory failure in critically ill surgical patients. J Trauma. 1988;28:648.
62. Yu M, Tomasa G. A double-blind, prospective, randomized trial of ketoconazole, a thromboxane synthetase inhibitor, in the prophylaxis of the adult respiratory distress syndrome. Crit Care Med. 1993;21:1635.
63. Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. The ARDS Network. JAMA. 2000;283:1995.
64. Opal S, Laterre PF, Abraham E, et al. Recombinant human platelet-activating factor acetylhydrolase for treatment of severe sepsis: results of a phase III, multicenter, randomized, double-blind, placebo-controlled, clinical trial. Crit Care Med. 2004;32:332.
65. Claus RA, Russwurm S, Dohrn B, et al. Plasma platelet-activating factor acetylhydrolase activity in critically ill patients. Crit Care Med. 2005;33:1416.
66. Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112(6 Suppl):321S.
67. Gosain A, Gamelli RL. A primer in cytokines. J Burn Care Rehabil. 2005;26:7.
68. Michie HR, Manogue KR, Spriggs DR, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318:1481.
69. Michie HR, Spriggs DR, Manogue KR, et al. Tumor necrosis factor and endotoxin induce similar metabolic responses in human beings. Surgery. 1988;104:280.
70. Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662.
71. Martins GA, Da Gloria Da Costa Carvalho M, Rocha Gattass C. Sepsis: a follow-up of cytokine production in different phases of septic patients. Int J Mol Med. 2003;11(5):585–591.
72. Oberholzer A, Souza SM, Tschoeke SK, et al. Plasma cytokine measurements augment prognostic scores as indicators of outcome in patients with severe sepsis. Shock. 2005;23:488.
73. Panacek EA, Marshall JC, Albertson TE, et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab′)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004;32:2173.
74. Oberholzer A, Oberholzer C, Moldawer LL. Cytokine signaling–regulation of the immune response in normal and critically ill states. Crit Care Med. 2000;28(4 Suppl):N3.
75. Dinarello CA. Interleukin-1beta. Crit Care Med. 2005;33(12 Suppl):S460.
76. Vincent JL, Slotman G, Van Leeuwen PA, et al. IL-1ra administration does not improve cardiac function in patients with severe sepsis. J Crit Care. 1999;14:69.
77. Opal SM, Fisher CJ Jr, Dhainaut JF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25:1115.
78. Fisher CJ Jr, Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836.
79. Song M, Kellum JA. Interleukin-6. Crit Care Med. 2005;33(12 Suppl):S463.
80. Derhaschnig U, Bergmair D, Marsik C, et al. Effect of interleukin-6 blockade on tissue factor-induced coagulation in human endotoxemia. Crit Care Med. 2004;32:1136.
81. Remick DG. Interleukin-8. Crit Care Med. 2005;33(12 Suppl):S466.
82. Rodriguez JL, Miller CG, DeForge LE, et al. Local production of interleukin-8 is associated with nosocomial pneumonia. J Trauma. 1992;33:74.
83. Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248.
84. Yang H, Tracey KJ. High mobility group box 1 (HMGB1). Crit Care Med. 2005;33(12 Suppl):S472.
85. Yang H, Ochani M, Li J, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296.
86. Leng L, Bucala R. Macrophage migration inhibitory factor. Crit Care Med. 2005;33(12 Suppl):S475.
87. Calandra T, Echtenacher B, Roy DL, et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164.
88. Joshi PC, Poole GV, Sachdev V, et al. Trauma patients with positive cultures have higher levels of circulating macrophage migration inhibitory factor (MIF). Res Commun Mol Pathol Pharmacol. 2000;107:13.
89. Liaudet L, Soriano FG, Szabo C. Biology of nitric oxide signaling. Crit Care Med. 2000;28(4 Suppl):N37.
90. Levy RM, Prince JM, Billiar TR. Nitric oxide: a clinical primer. Crit Care Med. 2005;33(12 Suppl):S492.
91. Ochoa JB, Udekwu AO, Billiar TR, et al. Nitrogen oxide levels in patients after trauma and during sepsis. Ann Surg. 1991;214:621.
92. Horton JW, Maass D, White J, et al. Nitric oxide modulation of TNF-alpha-induced cardiac contractile dysfunction is concentration dependent. Am J Physiol Heart Circ Physiol. 2000;278:H1955.
93. Szabo C, Billiar TR. Novel roles of nitric oxide in hemorrhagic shock. Shock. 1999;12:1.
94. Zingarelli B. Nuclear factor-kappaB. Crit Care Med. 2005;33(12 Suppl):S414.
95. Lopez A, Lorente JA, Steingrub J, et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med. 2004;32:21.
96. Vallet B. Bench-to-bedside review: endothelial cell dysfunction in severe sepsis: a role in organ dysfunction? Crit Care. 2003;7:130.
97. Lee MM, Schuessler GB, Chien S. Time-dependent effects of endotoxin on the ultrastructure of aortic endothelium. Artery. 1988;15:71.
98. Sessler CN, Windsor AC, Schwartz M, et al. Circulating ICAM-1 is increased in septic shock. Am J Respir Crit Care Med. 1995;151:1420.
99. Vincent JL, De Backer D. Does disseminated intravascular coagulation lead to multiple organ failure? Crit Care Clin. 2005;21:469.
100. Gardinali M, Borrelli E, Chiara O, et al. Inhibition of CD11-CD18 complex prevents acute lung injury and reduces mortality after peritonitis in rabbits. Am J Respir Crit Care Med. 2000;161:1022.
101. Xu H, Gonzalo JA, St Pierre Y, et al. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med. 1994;180:95.
102. Bhagat K, Moss R, Collier J, et al. Endothelial “stunning” following a brief exposure to endotoxin: a mechanism to link infection and infarction? Cardiovasc Res. 1996;32:822.
103. Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care. 2003;9:143.
104. Han X, Fink MP, Yang R, et al. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 2004;21:261.
105. Han X, Fink MP, Uchiyama T, et al. Increased iNOS activity is essential for hepatic epithelial tight junction dysfunction in endotoxemic mice. Am J Physiol Gastrointest Liver Physiol. 2004;286:G126.
106. Han X, Fink MP, Uchiyama T, et al. Increased iNOS activity is essential for pulmonary epithelial tight junction dysfunction in endotoxemic mice. Am J Physiol Lung Cell Mol Physiol. 2004;286:L259.
107. Magnotti LJ, Deitch EA. Burns, bacterial translocation, gut barrier function, and failure. J Burn Care Rehabil. 2005;26:383.
108. Perl M, Chung CS, Ayala A. Apoptosis. Crit Care Med. 2005;33(12 Suppl):S526.
109. Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796.
110. Mahidhara R, Billiar TR. Apoptosis in sepsis. Crit Care Med. 2000;28(4 Suppl):N105.
111. Hashimoto S, Kobayashi A, Kooguchi K, et al. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:237.
112. Hotchkiss RS, Schmieg RE Jr, Swanson PE, et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med. 2000;28:3207.
113. Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230.
114. Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813.
115. Godin PJ, Buchman TG. Uncoupling of biological oscillators: a complementary hypothesis concerning the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 1996;24:1107.
116. Seely AJ, Christou NV. Multiple organ dysfunction syndrome: exploring the paradigm of complex nonlinear systems. Crit Care Med. 2000;28:2193.
117. Seely AJ, Macklem PT. Complex systems and the technology of variability analysis. Crit Care. 2004;8(6):R367.
118. Aird WC. Endothelial cell dynamics and complexity theory. Crit Care Med. 2002;30(5 Suppl):S180.
119. Haji-Michael PG, Vincent JL, Degaute JP, et al. Power spectral analysis of cardiovascular variability in critically ill neurosurgical patients. Crit Care Med. 2000;28:2578.
120. Godin PJ, Fleisher LA, Eidsath A, et al. Experimental human endotoxemia increases cardiac regularity: results from a prospective, randomized, crossover trial. Crit Care Med. 1996;24:1117.
121. Papaioannou VE, Maglaveras N, Houvarda I, et al. Investigation of altered heart rate variability, nonlinear properties of heart rate signals, and organ dysfunction longitudinally over time in intensive care unit patients. J Crit Care. 2006;21:95.
122. Varela M, Churruca J, Gonzalez A, et al. Temperature curve complexity predicts survival in critically ill patients. Am J Respir Crit Care Med. 2006;174:290.
123. Villar J, Maca-Meyer N, Perez-Mendez L, et al. Bench-to-bedside review: understanding genetic predisposition to sepsis. Crit Care. 2004;8:180.
124. Stuber F, Petersen M, Bokelmann F, et al. A genomic polymorphism within the tumor necrosis factor locus influences plasma tumor necrosis factor-alpha concentrations and outcome of patients with severe sepsis. Crit Care Med. 1996;24:381.
125. Lorenz E, Mira JP, Frees KL, et al. Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch Intern Med. 2002;162:1028.
126. Gong MN, Zhou W, Williams PL, et al. Polymorphisms in the mannose binding lectin-2 gene and acute respiratory distress syndrome. Crit Care Med. 2007;35:48.
127. Walley KR, Russell JA. Protein C-1641 AA is associated with decreased survival and more organ dysfunction in severe sepsis. Crit Care Med. 2007;35:12.
128. Lin MT, Albertson TE. Genomic polymorphisms in sepsis. Crit Care Med. 2004;32:569.
129. Finfer S, Bellomo R, Boyce N, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247.
130. Wills BA, Nguyen MD, Ha TL, et al. Comparison of three fluid solutions for resuscitation in dengue shock syndrome. N Engl J Med. 2005;353:877.
131. Richard C, Warszawski J, Anguel N, et al. Early use of the pulmonary artery catheter and outcomes in patients with shock and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2003;290:2713.
132. Harvey S, Harrison DA, Singer M, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): a randomised controlled trial. Lancet. 2005;366:472.
133. Gattinoni L, Brazzi L, Pelosi P, et al. A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 Collaborative Group. N Engl J Med. 1995;333:1025.
134. Kumar A, Anel R, Bunnell E, et al. Pulmonary artery occlusion pressure and central venous pressure fail to predict ventricular filling volume, cardiac performance, or the response to volume infusion in normal subjects. Crit Care Med. 2004;32:691.
135. Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368.
136. Hollenberg SM, Ahrens TS, Annane D, et al. Practice parameters for hemodynamic support of sepsis in adult patients: 2004 update. Crit Care Med. 2004;32:1928.
137. Heard SO. Gastric tonometry: the hemodynamic monitor of choice (Pro). Chest. 2003;123(5 Suppl):469S.
138. Kovacs GC, Telek G, Hamar J, et al. Prolonged intestinal mucosal acidosis is associated with multiple organ failure in human acute pancreatitis: gastric tonometry revisited. World J Gastroenterol. 2006;12:4892.
139. Marik PE, Bankov A. Sublingual capnometry versus traditional markers of tissue oxygenation in critically ill patients. Crit Care Med. 2003;31:818.
140. Gutierrez G, Palizas F, Doglio G, et al. Gastric intramucosal pH as a therapeutic index of tissue oxygenation in critically ill patients. Lancet. 1992;339:195.
141. Splanchnic hypoperfusion-directed therapies in trauma: a prospective, randomized trial. Am Surg. 2005;71:252.
142. Creteur J, De Backer D, Vincent JL. Does gastric tonometry monitor splanchnic perfusion? Crit Care Med. 1999;27:2480.
143. Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:109.
144. Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282:54.
145. Ranieri VM, Giunta F, Suter PM, et al. Mechanical ventilation as a mediator of multisystem organ failure in acute respiratory distress syndrome. JAMA. 2000;284:43.
146. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301.
147. Rouby JJ, Brochard L. Tidal recruitment and overinflation in acute respiratory distress syndrome: yin and yang. Am J Respir Crit Care Med. 2007;175:104.
148. Villar J, Kacmarek RM, Perez-Mendez L, et al. A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: a randomized, controlled trial. Crit Care Med. 2006;34:1311.
149. Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327.
150. Terragni PP, Rosboch G, Tealdi A, et al. Tidal hyperinflation during low tidal volume ventilation in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med. 2007;175:160.
151. Oczenski W, Hormann C, Keller C, et al. Recruitment maneuvers after a positive end-expiratory pressure trial do not induce sustained effects in early adult respiratory distress syndrome. Anesthesiology. 2004;101:620.
152. Gattinoni L, Tognoni G, Pesenti A, et al. Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med. 2001;345:568.
153. Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354:2564.
154. Gill N, Nally JV Jr, Fatica RA. Renal failure secondary to acute tubular necrosis: epidemiology, diagnosis, and management. Chest. 2005;128:2847.
155. Lin SM, Huang CD, Lin HC, et al. A modified goal-directed protocol improves clinical outcomes in intensive care unit patients with septic shock: a randomized controlled trial. Shock. 2006;26:551.
156. Venkataraman R, Kellum JA. Prevention of acute renal failure. Chest. 2007;131:300.
157. Rybak MJ, Abate BJ, Kang SL, et al. Prospective evaluation of the effect of an aminoglycoside dosing regimen on rates of observed nephrotoxicity and ototoxicity. Antimicrob Agents Chemother. 1999;43:1549.
158. Merten GJ, Burgess WP, Gray LV, et al. Prevention of contrast-induced nephropathy with sodium bicarbonate: a randomized controlled trial. JAMA. 2004;291:2328.
159. Duong MH, MacKenzie TA, Malenka DJ. N-acetylcysteine prophylaxis significantly reduces the risk of radiocontrast-induced nephropathy: comprehensive meta-analysis. Catheter Cardiovasc Interv. 2005;64:471.
160. Harwood PJ, Giannoudis PV, van Griensven M, et al. Alterations in the systemic inflammatory response after early total care and damage control procedures for femoral shaft fracture in severely injured patients. J Trauma. 2005;58:446.
161. Micek ST, Heuring TJ, Hollands JM, et al. Optimizing antibiotic treatment for ventilator-associated pneumonia. Pharmacotherapy. 2006;26:204.
162. Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34:1589.
163. Adkins AL, Robbins J, Villalba M, et al. Open abdomen management of intra-abdominal sepsis. Am Surg. 2004;70:137.
164. van Santvoort HC, Besselink MG, Cirkel GA, et al. A nationwide Dutch study into the optimal treatment of patients with infected necrotising pancreatitis: the PANTER trial. Ned Tijdschr Geneeskd. 2006;150:1844.
165. Drakulovic MB, Torres A, Bauer TT, et al. Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial. Lancet. 1999;354:1851.
166. Schweickert WD, Gehlbach BK, Pohlman AS, et al. Daily interruption of sedative infusions and complications of critical illness in mechanically ventilated patients. Crit Care Med. 2004;32:1272.
167. Silvestri L, van Saene HK, Milanese M, et al. Selective decontamination of the digestive tract reduces bacterial bloodstream infection and mortality in critically ill patients. Systematic review of randomized, controlled trials. J Hosp Infect. 2007;65:187.
168. de Jonge E, Schultz MJ, Spanjaard L, et al. Effects of selective decontamination of digestive tract on mortality and acquisition of resistant bacteria in intensive care: a randomised controlled trial. Lancet. 2003;362:1011.
169. O'Grady NP, Alexander M, Dellinger EP, et al. Guidelines for the prevention of intravascular catheter-related infections. Infect Control Hosp Epidemiol. 2002;23:759.
170. Maki DG, Stolz SM, Wheeler S, et al. Prevention of central venous catheter-related bloodstream infection by use of an antiseptic-impregnated catheter. A randomized, controlled trial. Ann Intern Med. 1997;127:257.
171. Ranucci M, Isgro G, Giomarelli PP, et al. Impact of oligon central venous catheters on catheter colonization and catheter-related bloodstream infection. Crit Care Med. 2003;31:52.
172. Darouiche RO, Raad II, Heard SO, et al. A comparison of two antimicrobial-impregnated central venous catheters. Catheter Study Group. N Engl J Med. 1999;340:1.
173. Laurila J, Laurila PA, Saarnio J, et al. Organ system dysfunction following open cholecystectomy for acute acalculous cholecystitis in critically ill patients. Acta Anaesthesiol Scand. 2006;50:173.
174. Herndon DN, Hart DW, Wolf SE, et al. Reversal of catabolism by beta-blockade after severe burns. N Engl J Med. 2001;345:1223.
175. Perel P, Yanagawa T, Bunn F, et al. Nutritional support for head-injured patients. Cochrane Database Syst Rev. 2006;(4):CD001530.
176. McClave SA, Chang WK, Dhaliwal R, et al. Nutrition support in acute pancreatitis: a systematic review of the literature. JPEN J Parenter Enteral Nutr. 2006;30:143.
177. van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359.
178. Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449.
179. Krinsley JS, Jones RL. Cost analysis of intensive glycemic control in critically ill adult patients. Chest. 2006;129:644.
180. Van den Berghe G, Wouters PJ, Bouillon R, et al. Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Crit Care Med. 2003;31:359.
181. Cerra FB, Benitez MR, Blackburn GL, et al. Applied nutrition in ICU patients. A consensus statement of the American College of Chest Physicians. Chest. 1997;111:769.
182. Endres S, Ghorbani R, Kelley VE, et al. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N Engl J Med. 1989;320:265.
183. Canadian Critical Care Clinical Practice Guidelines. Summary of Topics and Recommendations. 2007. http://www.criticalcarenutrition.com. Accessed January 30, 2007.
184. Le Gall JR, Klar J, Lemeshow S, et al. The Logistic Organ Dysfunction System. A new way to assess organ dysfunction in the intensive care unit. ICU Scoring Group. JAMA. 1996;276:802.
185. Russell JA, Singer J, Bernard GR, et al. Changing pattern of organ dysfunction in early human sepsis is related to mortality. Crit Care Med. 2000;28:3405.