Aimée C. LeClaire
Dale H. Whitby
Pharmacotherapy is an essential component in the successful treatment of the critically ill patient. Thus, critical care practitioners must possess a good working knowledge and understanding of pharmacokinetic and pharmacodynamic principles as well as altering or confounding factors. Clinical application of these principles in the clinical setting is equally important.
Critical care therapeutics often involves the use of multiple pharmacologic agents, each having a therapeutic purpose, toxicity, and side effects. Many agents are affected by acute or chronic impairment of metabolic organs such as the liver, kidney, and lungs; changes in fluid balance; drug–drug and drug–nutrient interactions; and other factors.
The goal of pharmacotherapy is the attainment of a desired therapeutic response without untoward toxicity. The goal of this chapter is to present principles that will be clinically useful in developing a practical approach to pharmacotherapy in the critically ill patient. Pharmacokinetic principles, special population considerations, drug–drug interactions, adverse drug reactions, and the role of the clinical pharmacist are reviewed.
Pharmacokinetics and Pharmacodynamics
Pharmacokinetics can be defined as the quantitative study of the processes of absorption, distribution, metabolism, and elimination of a drug in the body (1). The use of mathematical models describing these processes allows predictions to be made about drug concentrations in various parts of the body as a function of dosage, route of administration, clearance, and time.
Pharmacodynamics is the study of the relationship between the concentration of a drug and the biochemical or physiologic response obtained by that drug in a given patient (2). Some drugs exhibit a linear dose–response relationship through the entire range of clinically used doses; that is, doubling the dose doubles the response. Others may exhibit a linear dose–response relationship to a response ceiling, where increases in drug dose do not elicit any additional response. Some agents do not behave in a linear fashion at all. A decrease in heart rate during beta-antagonist or calcium channel antagonist therapy and decrease in ectopy during antiarrhythmic therapy are examples of quantifiable pharmacodynamic measurements. Some pharmacodynamic responses are more difficult to measure, such as the response to corticosteroid or anticonvulsant therapies.
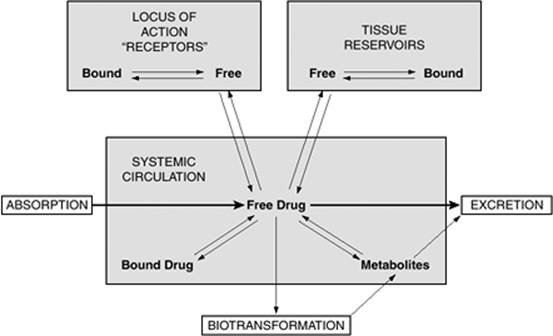
A pharmacokinetic-pharmacodynamic relationship exists in most cases, as demonstrated in Figure 62.1. In general, because it crosses tissues and elicits a pharmacodynamic response, free or unbound drug in the plasma is considered to be the pharmacologically active component in achieving efficacy or producing toxicity. The free drug fraction is also the only portion available to be metabolized and eliminated.
Pharmacokinetics
As stated, pharmacokinetics is the study of the absorption, distribution, metabolism, and elimination of drugs. Much of our knowledge of this subject has resulted from the development of sensitive and specific assays for determining drug concentrations in biologic fluids. Conceptual models and mathematical equations have been devised to describe these behaviors (3); however, the pharmacokinetic parameters described in the literature often are based on data from small numbers of patients or normal volunteers, which may not accurately reflect the behavior of a drug in a critically ill patient. In individualizing a patient's drug therapy, appropriate interpretation of accurately obtained plasma level measurements of a given drug in a specific patient provides significantly more information than any empiric calculation could provide.
The simplest pharmacokinetic model describes the body as a singular “compartment” or one-compartment model. Drugs enter the compartment at rates determined by routes of administration (e.g., intravenous bolus, intravenous infusion, intramuscular, oral, or transdermal) and leave the compartment at rates determined by routes of elimination (e.g., renal, hepatic, or pulmonary).
A two-compartment model reasonably describes the behavior of most drugs in humans (4). In this model, a small central compartment consists of the rapidly perfused organs (heart, lungs, kidney, and endocrine glands), and a larger peripheral compartment consists of the less rapidly perfused organs (skin, muscle, bone, and fat). After administration, drugs initially distribute into the central compartment and then redistribute into the peripheral compartment, reaching equilibrium between compartments at a rate dependent on the perfusion of peripheral compartment tissues and the tissue affinity for the drug.
The two-compartment model assumes that the drug obeys first-order or linear kinetics, where a constant proportion of the drug is removed per unit of time. Rate of elimination of the drug is proportional to the serum concentration and diminishes logarithmically over time. However, the fraction of drug removed per unit of time remains constant and independent of dose. In first-order elimination, both clearance and volume of distribution also remain constant. Thus, serum concentration can be affected by changing the dose in relation to the desired change in concentration; in simplest terms, doubling the dose doubles the concentration.
|
|
|
Figure 62.1. Schematic representation of the interrelationship of the absorption, distribution, binding, metabolism, and excretion of a drug and its concentration at its locus of action. Possible distribution and binding of metabolites are not depicted. (From Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: the dynamics of drug absorption, distribution and elimination. In: Hardin JG, Limbird LE, Gilman AG, et al., eds. Goodman and Gilman's The Pharmacologic Basis of Therapeutics. 9th ed. New York, NY: McGraw-Hill; 1996:3, with permission.) |
However, some drugs follow zero-order kinetics, where a constant amount of drug is eliminated per unit of time, irrespective of serum concentration. Other medications obey nonlinear or saturable kinetics. Drugs having saturable kinetics exhibit capacity-limited metabolism, and elimination may not be proportional to serum concentration. For example, phenytoin exhibits Michaelis-Menten kinetics and demonstrates linear elimination to a point where the patient's hepatic enzyme system is functioning at the maximum rate. Subsequently, phenytoin serum levels increase out of proportion to dose, meaning an increase in dose from 300 to 400 mg/day may result in an exponentially greater increase in serum level.
In first-order kinetics, a plot of the logarithm of serum concentration after initial distribution versus time is a straight line. As seen in Figure 62.2, the intravenous injection of a drug results in an initially high serum drug concentration, followed by a rapid decrease because of drug distribution. After the distribution (or alpha) phase, the serum concentration further decreases because of the drug elimination (or beta) phase. At any point in time, the serum drug concentration (Cs) can be calculated from the biexponential disappearance function:
|
|
|
Figure 62.2. Schematic graph of serum drug concentrations (Cs) plotted on a logarithmic scale versus time after a single intravenous bolus injection. (From Greenblatt DJ, Koch-Weser J. Clinical pharmacokinetics. N Engl J Med. 1975;293:703, with permission.) |
Cs = Ae-αt + Be-βt
where α and β are the first-order rate constants for the alpha and beta phases, respectively (5).
Pharmacokinetic parameters of particular value in the application of pharmacokinetic principles to clinical practice include the volume of distribution, clearance, half-life, and bioavailability.
Volume of distribution (Vd) is the apparent volume of fluid in which a given dose would have to be distributed to achieve the observed serum concentration as mathematically described by the following equations:
Cs = dose/Vd or Vd = dose/Cs
Clinically, the volume of distribution is useful for estimating the initial loading dose required to achieve a desired serum concentration or for calculating an incremental bolus dose required to raise a serum level by a desired amount. For instance, theophylline has a volume of distribution of approximately 0.5 L/kg in most patients; thus a 60-kg person would have an apparent volume of distribution of 30 L. If the desired serum concentration is 15 mg/L, the loading dose required to achieve this goal may be calculated as follows:
Dose = (Vd) (Cs) = (30 L) (15 mg/L) = 450 mg
Clearance (Cl) is defined as the volume of blood or serum from which all drug is removed per unit of time. Clearance can be affected by alterations in distribution, metabolism, or excretion. Specific issues relating to changes in clearance from impairment of renal or hepatic function are discussed later in this chapter. At steady-state, the drug administration rate equals the drug elimination rate (6). Based on this principle, drug clearance can be useful in determining the amount of drug required to maintain a therapeutic drug level. With an intravenously administered or completely absorbed oral agent, the steady-state serum concentration (Css) can be determined by the following equations:
![]()
Half-life (t1/2) is a function of both drug volume of distribution and clearance:
![]()
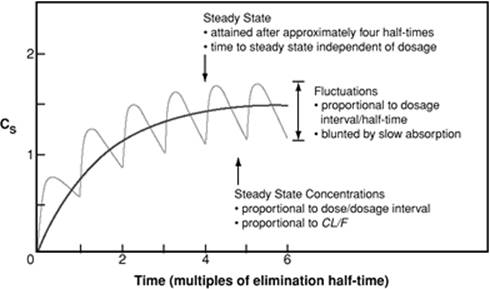
In most cases, this variable refers to elimination half-life, meaning the time required to reduce the initial serum concentration by 50% after the initial distribution phase. The drug half-life can be useful in determining the amount of time required to reach steady-state serum concentration. Four elimination half-lives are required to achieve approximately 94% of steady state. The time required to reach steady-state serum concentration is independent of dose and dosage interval. As seen in Figure 62.3, drug accumulation occurs with repeated dosing until equilibrium or steady state is reached at approximately four to five half-lives. If, in this example, the desired pharmacologic response is observed at a serum concentration of 1 to 2 units, a period of time after the initiation of therapy exists during which the serum concentration is subtherapeutic. To ascertain therapeutic levels at the initiation of therapy, a loading dose may be given, followed by maintenance doses.
|
|
|
Figure 62.3. Fundamental pharmacokinetic relationships for repeated administration of drugs. (From Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: the dynamics of drug absorption, distribution and elimination. In: Hardin JG, Limbird LE, Gilman AG, et al., eds. Goodman and Gilman's The Pharmacologic Basis of Therapeutics. 9th ed. New York, NY: McGraw-Hill; 1996:23, with permission.) |
For example, digoxin has an elimination half-life of 36 hours in a patient with normal renal function. Therefore, a period of 6 days is required to achieve a steady-state drug level with daily dosing. If a loading dose of 1 mg is incrementally given over 24 hours, therapeutic digoxin concentrations may be achieved more rapidly.
The bioavailability of a drug is defined as the fraction of a drug dose that reaches the systemic circulation. Bioavailability is a consideration primarily with orally administered agents. Absorption can be affected by many factors relating to the pharmaceutical dosage form (e.g., tablet, capsule, oral liquid, suspension, or sustained-release product), gastric pH and emptying time, intestinal motility, and drug complex formation with other drugs or nutrients (7). Many orally absorbed drugs demonstrate a first-pass effect with significant metabolism in the liver before the drug enters the systemic circulation. Examples are propranolol, hydralazine, verapamil, and lidocaine. Lidocaine's first-pass effect is so great that it must be administered by the parenteral route. For other drugs like propranolol and verapamil, oral doses need to be substantially higher than parenteral doses to account for this effect. For these drugs, the fraction of drug available to the systemic circulation or bioavailability (F) must be included in the calculation of steady-state serum concentration as follows:
![]()
Pharmacodynamics
Whereas pharmacokinetics is a detailed, often mathematical, approach to the way that the body handles a drug, pharmacodynamics deals with the mechanism of action of drugs and their biochemical and physiologic effects on the patient (8). Although frequently discussed separately, in reality, pharmacokinetics and pharmacodynamics are closely interconnected. For example, along with the dosing regimen, the pharmacokinetic properties of a drug determine the concentration of that drug in the body, and in many situations, the drug concentration is directly related to the effects experienced by the patient. In addition, the degree of protein binding that a particular drug exhibits is typically considered a pharmacokinetic property. However, in many cases, it is only the unbound drug that is pharmacologically active and therefore responsible for the pharmacodynamic effects.
Drugs produce desired effects through various types of mechanisms of action. Some drugs demonstrate a relatively direct mechanism of action, such as mannitol administered for osmotic diuresis. When mannitol is administered intravenously, the osmolarity of the blood and other body fluids is increased, causing shifts in the distribution of water. Other medications, such as epinephrine and beta-blockers, act by agonizing or antagonizing physiologic receptors. The effect of the drug is, therefore, directly related to the physiologic action of the associated receptor. Continued stimulation of some receptors with agonists frequently results in a state of down-regulation. In the intensive care unit (ICU), where exogenous catecholamines are commonly used, receptor down-regulation is important given that the continued or subsequent exposure to the same concentration of a particular agonist may have a lesser clinical effect over time. Other drugs, such as steroids, have very complex mechanisms of action involving crossing the cell membrane, regulating the transcription of specific genes, and ultimately altering protein synthesis. A practitioner's knowledge of the mechanisms of action and physiologic effects of drugs will help in determining the appropriate therapy, monitoring treatment, and anticipating pharmacodynamic drug interactions.
Special Populations
Renal Impairment
Whether a pre-existing disease state or a new condition developed in the ICU, renal dysfunction is often present in critically ill patients. Reduced renal function may be related to a disease state, either iatrogenic or age related. Acute and chronic renal impairment can have profound effects on the pharmacokinetics and pharmacodynamics of many commonly used agents. In addition, the degree of renal dysfunction and resulting degree of fluid retention and uremia determine the degree of pharmacokinetic and pharmacodynamic changes. For example, a drug may exhibit no clinically significant changes in a patient with mild renal dysfunction, or the medication may be contraindicated in a patient with renal failure. The practitioner must evaluate each drug individually in relation to the degree of renal dysfunction present.
Renal dysfunction has been shown to alter bioavailability, protein binding, volume of distribution, and excretion of certain compounds (9). There are relatively few data providing details regarding the alteration of bioavailability of most drugs in patients with renal dysfunction. However, certain conditions that are more common among uremic patients, including a higher gastric pH, altered gastric emptying time, vomiting, and intestinal edema, are known to alter the absorption and bioavailability of enterally administered drugs.
The distribution of some drugs may also be significantly altered in renal dysfunction. Because patients with significant renal dysfunction have a higher percentage of their body mass as water, the apparent volume of distribution for water-soluble drugs is often increased. In addition, the protein binding of drugs is often altered in the presence of uremia, which changes the apparent volume of distribution. In general, the plasma protein binding of acidic drugs, like phenytoin, is decreased (10). Because there is a larger percentage of unbound (free) drug available for pharmacologic activity, uremic patients may experience toxicity with total drug concentrations in the therapeutic range. For this reason, some practitioners prefer to measure free phenytoin concentrations instead of total phenytoin concentrations in patients with renal failure. Altered tissue binding also may affect the apparent volume of distribution of a drug. For example, digoxin's volume of distribution has been reported to be significantly reduced in patients with renal disease (11). As a result, in patients with renal dysfunction, digoxin loading doses should be used cautiously and serum concentrations should be monitored carefully.
The major pharmacokinetic effect seen in renal dysfunction is the impaired elimination of drugs and their metabolites by the kidney. Renal clearance of pharmacologic agents is complex, involving one or more of the processes of filtration, tubular secretion, and reabsorption. Few drugs are excreted solely by glomerular filtration. Additionally, tubular secretion and reabsorption for many compounds may vary with the type of renal dysfunction and the administration of other agents (e.g., probenecid).
Another complicating factor is changing renal dysfunction. Multiorgan system dysfunction in critically ill patients is a very dynamic process. As a consequence, renal function can deteriorate very quickly when changes in clinical condition such as hypotension or poor cardiac output occur. In addition, many of the agents that are frequently used in critically ill patients may cause renal dysfunction; radiographic contrast nephropathy and aminoglycoside nephrotoxicity are common examples (12). Changes in renal function may be acute or progressive; therefore, drug clearance may be highly variable in the intensive care setting and requires constant reassessment of the patient's clinical condition.
In evaluating medication use in renal impairment, the first step is to estimate the degree of dysfunction. Most medications that require dosage adjustment in patients with renal dysfunction are adjusted based on creatinine clearance. Although many hospital laboratories now report estimated GFR (eGFR) along with serum creatinine values, The National Kidney Disease Education Program recommends the use of equations, such as the Cockcroft-Gault equation, which is designed to estimate creatinine clearance for drug dosage adjustments (13).
When evaluating a medication profile, the intensivist must evaluate each drug individually and determine if a dosage adjustment or additional monitoring is necessary. Although for most medications, initial or loading dose does not need to be altered in renal dysfunction, maintenance dosing regimens may need to be adjusted by prolonging dosing intervals, reducing the standard maintenance dose, or both, depending on the particular agent and degree of renal dysfunction. In cases of drugs with a narrow therapeutic index, serum concentrations should be monitored carefully to avoid potential toxicity. In general, drug administration steady state should be achieved before a serum concentration is obtained, unless the patient rapidly progresses to renal failure, in which case an earlier sample may be warranted prior to reaching steady state to assess the appropriateness of redosing at current interval. For example, in patients with normal renal function, a vancomycin trough is typically obtained before the third or fourth dose; however, in the setting of renal dysfunction, a trough level prior to the second dose may be beneficial to help determine the patient's ability to clear the drug before further dosing.
In addition to the parent drug, practitioners also have to consider the route of elimination of any metabolites. For example, the active metabolite of morphine, morphine-6-glucuronide, is renally excreted and may exert important clinical effects when it accumulates in patients with renal failure (14). Although this does not prohibit the use of morphine in patients with renal dysfunction, practitioners may want to consider another agent, such as fentanyl, that does not have an active metabolite (15). If morphine is the preferred drug, a longer duration of action should be expected in a patient with renal impairment when compared to the same dose in a patient with normal renal function. Frequent assessment of morphine use by pain score and sedation level should be planned.
Critical care practitioners may also have to consider the effect of renal replacement therapy on drug clearance. The degree of drug clearance is determined by both drug-specific factors and the type of renal replacement therapy used. Drug-specific factors include molecular weight, solubility, degree of ionization, protein binding, and volume of distribution (9). For example, standard modes of dialysis are relatively ineffective in significantly clearing phenytoin, a highly protein-bound drug, from the body. However, more complicated techniques such as high-flux dialysis with charcoal hemoperfusion have been reported to remove considerable amounts of drug in a patient with phenytoin toxicity (16). Dialysis-specific factors that can affect the degree of drug clearance include the mode of dialysis, type of filter used, filter pore size, ultrafiltration rate, and blood and dialysate flow rates (17). As a general rule, peritoneal dialysis is less efficient in drug removal compared to intermittent hemodialysis and continuous renal replacement therapies (such as continuous venovenous hemodiafiltration) (9). Due to the potential complexity of drug dosing in the presence of renal replacement therapy, consulting a clinician who is readily familiar with the pharmacokinetic implications of intermittent or continuous renal replacement is advisable.
Hepatic Disease
Like renal insufficiency, hepatic disease can also significantly alter the pharmacokinetic profile of a drug and the ultimate effect on the patient. Unfortunately, data relating the degree of hepatic dysfunction in relation to drug metabolism and elimination is limited, and dosage adjustment guidelines for hepatic dysfunction are available for only some medications.
Because patients with severe liver failure often suffer from secondary gastrointestinal problems, the bioavailability of some orally administered drugs may be altered (18). Severe liver failure and cirrhosis have been associated with delayed gastric emptying and delayed absorption (18). For example, enteral furosemide has been shown to have delayed absorption in patients with mild and severe cirrhosis irrespective of the presence of ascites (19). In addition, patients with cirrhosis are at increased risk for severe upper gastrointestinal tract bleeding, which can limit enteral medication tolerance (20).
Liver failure also affects the volume of distribution of some drugs. Patients with significant ascites comprise a larger portion of their body weight as water. Therefore, the volume of distribution of hydrophilic drugs is increased (18). Most patients with significant liver dysfunction are also hypoalbuminemic, leading to a higher free fraction of highly protein-bound drugs.
Finally, while useful in evaluating long-term prognosis, liver cirrhosis severity scores, such as the Child-Pugh score, are less helpful in assessing the need for dosage adjustments of drugs (18). Many factors influence hepatic drug clearance; however, the most significant characteristics are hepatic blood flow and enzyme activity.
High extraction drugs are significantly removed (>60%) during the first passage across the liver, and the clearance of these drugs is significantly dependent on hepatic blood flow (18). In significant liver disease, such as cirrhosis, hepatic blood flow is decreased or totally shunted if portosystemic shunt is present (21). When a drug is administered enterally in these patients, drug enters the systemic circulation with minimal or no first-pass effect (22). Thus, the bioavailability of some orally administered drugs with extensive first-pass effect is greatly increased, and dosages should be appropriately reduced to prevent adverse side effects (23). Table 62.1 lists some of the high extraction drugs that may be used in the ICU.
|
Table 62.1 High Extraction Drugs Commonly Used in the Intensive Care Unit |
||||||||||||||||||||
|
Low extraction drugs are those that experience a relatively minimal first-pass effect (<30%), and initial bioavailability is not largely affected by liver disease (18). The clearance of hepatically metabolized, low extraction drugs may be influenced by the effects of liver disease on the enzyme system responsible for the metabolic process. Research has shown that a reduction in the activity of drug-metabolizing enzymes in livers from cirrhotic patients is associated with increasing disease severity (24,25). However, a large variation in the degree of enzyme activity impairment exists, probably due to the heterogeneity of the enzymes involved in drug metabolism processes. Numerous enzymes are affected differently by liver disease. For example, oxidation reactions tend to be more sensitive than glucuronidation (26).
Low extraction ratio drugs are less dependent on hepatic blood flow but are highly dependent on protein binding and the level of enzymatic capacity of the liver. These drugs are susceptible to changing clearance because of the induction or inhibition of liver enzymes and are sometimes referred to as enzyme limited or capacity limited (27). The hepatic clearance of some enzyme-limited drugs is termed binding sensitive or binding restrictive when only the circulating free drug is removed. Therefore, a decrease in drug binding causes an increase in the hepatic clearance of the drug. Drugs that are highly protein bound, such as phenytoin and warfarin, are most sensitive to this change in clearance.
Unfortunately, multiple factors determine the degree of impairment for the hepatic clearance of drugs, and no standard guidelines aid practitioners in the dosage adjustments of these medications. In addition, most available data that addresses the effect of liver disease on drug clearance are from studies evaluating patients with a single organ dysfunction (e.g., alcoholic cirrhosis); this is frequently not the case in critically ill patients. Therefore, any degree of other organ dysfunction, such as renal impairment or low cardiac output state, could further alter the pharmacokinetics and pharmacodynamics of the drug. The critical care team must initially evaluate the extent of organ dysfunction and determine whether a dosage reduction for a given drug is likely to be needed, and then monitor the patient carefully for early signs of toxicity. When possible, blood concentrations of the affected medications should be monitored.
Pregnancy
Because pregnant females represent a subset of critically ill patients with special needs and characteristics, considerations beyond the predicted physiologic changes to risk/benefit therapy assessment for both mother and fetus apply. Each subspecialty discipline must share their expertise with the intensivist team, and when possible, decisions should be made collaboratively with both the mother and fetus in mind. A fine balance must be obtained between avoiding undertreating the mother and presenting unnecessary risks to the fetus. Even in the otherwise healthy pregnant female, determining the optimal dosing regimen presents a considerable challenge due to the significant pharmacokinetic changes that occur during pregnancy.
Few gastrointestinal tract changes may affect drug absorption during pregnancy (28). With the exception of the active labor period, gastric emptying time appears to be unchanged (29). However, due to a decrease in gastrin secretion, pregnant women have a higher gastric pH and slower intestinal motility (30). The clinical significance of these changes in relation to drug absorption is not known. Some women experience significant nausea and vomiting, especially during the first trimester, which may affect their ability to tolerate some enteral medications. Noxious stimuli of any kind should be minimized.
Unlike absorption, the volume of distribution of certain drugs can be significantly changed by pregnancy. Most impressively, blood volume increases by approximately 40% to 50% during pregnancy (31). The exception is patients with severe pre-eclampsia or eclampsia; blood volume may expand very little in this population (28). Changes in plasma protein concentrations, such as a decrease in albumin concentration, can affect the free fraction of some drugs. Some examples of drugs that have a higher free fraction during pregnancy include diazepam, valproic acid, and phenytoin (32).
Pregnancy can also affect the metabolism and elimination of drugs as well. Interestingly, hepatic metabolism via the cytochrome P450 (CYP) system may be increased or decreased, depending on the specific enzyme. Specifically, pregnancy decreases the activity of CYP1A2 and CYP2C19. However, the activities of CYP3A4, CYP2D6, CYP2C9, and CYP2A6 are increased (30). The renal clearance of some drugs is also significantly increased during pregnancy due to an increase in glomerular filtration rate (GFR) of approximately 50% (30). For example, several β-lactam antibiotics, such as ampicillin, cefazolin, and piperacillin, have been shown to have increased clearance in pregnant women (33,34,35). Alterations in tubular secretion/reabsorption may also occur; however, few data are available in this area. Based on the available pharmacokinetic studies of renally excreted drugs, the effect of pregnancy on drug clearance varied widely from 20% to 65% above the clearance in nonpregnant females (30). For this reason, drugs that are primarily renally excreted unchanged may require a dosage increase of 20% to 65% to maintain prepregnancy blood concentrations (30).
In addition to pharmacokinetic changes that occur during pregnancy, the critical care practitioner also has to consider other physiologic changes that may affect the patient's need for drug therapy. For example, pregnant females are normally hypercoagulable and are at risk for venous thrombosis. The addition of other factors commonly seen in the ICU, such as immobility and endothelial injury, may put them at even higher risk. Unfractionated heparin and low-molecular-weight-heparins (LMWHs) are the drugs of choice for the prevention or treatment of thrombosis during pregnancy. Practitioners should be aware that many pregnant females require higher doses of heparin compared to nonpregnant females due to increased concentrations of heparin-binding proteins, increased volume of distribution, and increased clearance (36). In fact, the dose required for therapeutic anticoagulation may be as much as twice the typical weight-based dose (37). LMWHs are considered to be a safe alternative to unfractionated heparin during pregnancy, but their use in the acute setting for the treatment of venous thrombosis is controversial. Similar to unfractionated heparin, the clearance of LMWHs is increased during pregnancy, making dosing less straightforward than in the nonpregnant patient. Pregnant females have been shown to require higher than standard doses of LMWH to maintain goal anti–factor Xa concentrations (37,38). Therefore, frequent monitoring of anti–factor Xa concentrations is recommended, and multiple dosage adjustments may be required. With this increase in monitoring and related potential dosage adjustments, some argue that many of the advantages, both clinical and financial, of LMWHs are lost in this setting.
In conclusion, when the critical care practitioner is faced with determining the drug regimen for a pregnant patient, several steps should be taken. Most important, a multidisciplinary approach, including experts in the area of obstetrics and drug therapy, is invaluable. Next, the need to prescribe any drug must be carefully considered prior to the ordering process, and standard protocols should be modified as appropriate. Once the need for a drug is determined, the practitioner must consider the implications on fetal development and choose the safest drug available that will appropriately treat the mother. In considering the safety of a drug during pregnancy, the practitioner must go beyond simply identifying the “Pregnancy Risk Factor” that is included in the drug package labeling. A thorough search for information should be conducted, and the current trimester of the pregnancy should be considered. Some medications are safer for use during certain periods of the pregnancy. For example, nonsteroidal anti-inflammatory drugs (NSAIDs) are thought to carry the most risk during the first and third trimesters (39). In addition to primary literature, a medical reference that specifically addresses medication use in pregnancy should be consulted (39). In summary, once the treatment has been chosen, the practitioner must also consider the physiologic changes during pregnancy that may lead to a need for a dosage adjustment and monitor both the drug level, when possible, and the clinical picture.
Adverse drug Reactions
The term adverse drug reactions (ADRs) encompasses a broad range of drug effects, ranging from exaggerated but predictable pharmacologic actions of the drug to toxic effects unrelated to intended pharmacologic effects (40). As such, the true incidence rate of ADRs is difficult to determine, with estimates ranging from 5% to 47% (41,42,43). Only by maintaining a high index of clinical suspicion, keeping the drug therapy to the minimum necessary, and stopping the suspect drug, if possible, could one minimize the incidence of ADRs, thereby decreasing patient morbidity, perhaps even mortality, and also keeping the cost of therapy low.
A small group of widely used drugs account for a disproportionate number of ADRs; aspirin, anticoagulants, diuretics, digoxin, antimicrobials, steroids, and hypoglycemic agents account for 90% of reactions (40).
The most frequent ADRs result from the exaggerated but predicted pharmacologic actions of the drug, and, as such, are readily identifiable and often are preventable. Examples include hemorrhagic complications caused by anticoagulants or hypoglycemia caused by oral hypoglycemic agents or insulin. The most important determinant for such adverse effect is the abnormally high drug concentration at the receptor site. This can occur for various reasons, ranging from errors in calculating drug dosage or administration to an alteration in the pharmacokinetics (such as reduction in the volume of distribution, rate of metabolism, or rate of excretion), or because of a drug interaction resulting in increased concentration of free drug at the receptor site, leading to untoward effects.
Other ADRs result from toxic effects unrelated to the intended pharmacologic actions. Such events, therefore, often are unpredictable and frequently are severe. Various mechanisms are involved: genetic, immunologic and nonimmunologic, or idiosyncratic. A list of genetic susceptibilities leading to ADRs with certain drugs is presented in Table 62.2. The most relevant examples are succinylcholine-induced prolonged apnea (suxamethonium sensitivity) and drug-induced malignant hyperthermia (MH).
Succinylcholine is a depolarizing muscle relaxant that is rapidly metabolized by plasma pseudocholinesterase, with a duration of paralysis for 2 to 4 minutes. Approximately 1 in 3,200 patients is homozygous for a defective pseudocholinesterase, which has an autosomal recessive transmission and demonstrates markedly prolonged block with succinylcholine in the range of 3 to 8 hours (44).
MH is a clinical syndrome of muscle rigidity, tachycardia, tachypnea, rapidly increasing temperature, hypoxia, hypercapnia, hyperglycemia, hyperkalemia, hypercalcemia, lactic acidosis, and eventual cardiovascular collapse that occurs during or after general anesthesia (45). Several drugs have been implicated in triggering MH, particularly the halogenated inhalational agents (halothane, enflurane, and isoflurane), succinylcholine, and possibly an amino amide local anesthetic (lidocaine or bupivacaine) (46,47).
|
Table 62.2 Examples of Inherited Disorders Involving an Abnormal Response to Drugs |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
The cause of MH seems to be an inability of the sarcoplasmic reticulum of skeletal muscle to take up released myoplasmic calcium. When a MH episode is triggered, the myoplasmic levels of calcium rise tremendously, accelerating muscle metabolism and contraction, and leading to the clinical manifestations of the syndrome (45). The manifestation of MH may appear in the operating room or in the ICU shortly after the end of the surgical procedure.
Dantrolene is the cornerstone of therapy for MH (48). It reduces rigidity and restores muscle function by preventing calcium release from the sarcoplasmic reticulum and antagonizes its effects on muscle contraction (49). Treatment of MH is initiated by discontinuing the anesthetic and other possible triggering agents, ventilating the patient with 100% oxygen, providing hemodynamic support as needed, and administering dantrolene. The mortality rate from the fulminant syndrome was approximately 70% before the use of dantrolene (50). With early recognition and optimal therapy, reported mortality rates decreased from 10% to 7% (50).
An immunologically mediated ADR is epitomized by the classic anaphylactic reaction, which is IgE mediated, and a serum sickness–like condition. A similar reaction, but not immunologically mediated, known as anaphylactoid reaction, is sometimes seen as an indirect histamine release from the mast cells by morphine, or a complement activation leading to mediator release from the mast cell and basophil by aspirin and radiographic contrast media. There are a host of drug reactions for which the exact underlying mechanism of toxicity is not well understood, which are termed idiosyncratic reactions. Every organ system can potentially be adversely affected by drug exposures, or ADRs can present with effects on multiple organ systems. Of the multisystem manifestations of ADRs, drug fever, drug withdrawal reactions, and anaphylaxis are worth elaborating.
Drug-induced fever should always be considered in the workup of pyrexia in the intensive care unit, and more so when no other obvious source is apparent. Fever is thought to be relatively rare as a primary or sole manifestation of a drug reaction and is usually associated with other hypersensitivity type reactions like anaphylaxis, serum sickness, rash, or eosinophilia. Yet, sometimes fever may be the only manifestation of an ADR. Antibiotics, cardiovascular drugs, and central-acting agents are the largest categories of drugs causing fever (51). Patients with hypersensitivity-induced drug fever have been observed to have fevers as high as 40°C and yet generally appear well, which can be an important clue to the presence of a drug-related fever (52).
Medication withdrawal can trigger fever. Opiate and benzodiazepine tolerance and dependence can develop over a short period, and subsequent attempts at withdrawing these medications might be associated with hypermetabolism and “sympathetic overdrive” characterized by fever, hypertension, mental confusion, seizures, and cardiac arrhythmias, a clinical picture that may be confused with an underlying disease process (53). Management consists of the gradual withdrawal of the drug and substitution of longer-acting agents; clonidine, a centrally acting drug, has been used with some success in these conditions (54). The pathophysiology of anaphylactic shock from drug reactions has been described elsewhere in this text.
Mental confusion is a common problem in the ICU, and the cause is often multifactorial (55). Drugs that have been reported to alter mood and increase mental confusion, especially in the elderly, include corticosteroids, histamine-2 receptor antagonists, fluoroquinolones, theophylline, barbiturates, benzodiazepines, digoxin, antidepressants, penicillin, lidocaine, antihistamines, quinidine, opiates, and phenothiazines. Occasionally, confusion is associated with discontinuation of one of the aforementioned medications if used for a long period of time. Drugs like imipenem-cilastatin have been implicated as an inducer or promoter of seizure-like activity (56).
Some of the antiarrhythmic drugs, particularly procainamide, have a strong proarrhythmic effect, and drug levels should be closely monitored during use. Torsade de pointes, a form of polymorphic ventricular tachyarrhythmia caused by drugs that can prolong the QTc interval (like phenothiazines and tricyclic antidepressants), can be fatal. Treatment with intravenous magnesium and overdrive pacing has been used with some success to reverse cardiac arrest associated with this dysrhythmia.
Drug-induced renal injury remains a major cause of increased morbidity, and contributes to the overall mortality of the critically ill. The aminoglycoside antibiotics and radiographic contrast agents are the leading causes of acute nephrotoxicity in the ICU. Other risk factors that make patients prone to develop nephrotoxicity include advanced age, prior renal disease, intravascular volume depletion, simultaneous use of other nephrotoxic agents, and certain disease states such as congestive heart failure, liver cirrhosis, and diabetes mellitus (57).
Drugs can cause abnormalities in any of the formed elements of blood, which can lead to diagnostic confusion in separating these from primary disease processes. Drug-induced pancytopenia, hemolytic anemia, thrombocytopenia, and granulocytopenia all have been well described (57). Some of these conditions are more pertinent to ICU patients than others; heparin-induced thrombocytopenia (HIT) is an example. The mechanism is believed to be the induction of platelet-specific IgG antibody by heparin, with subsequent aggregation of platelets causing vascular thrombosis and fall in circulating platelets (58). Once the syndrome occurs, even trivial amounts of heparin can perpetuate the pathology. Heparin-containing flush solutions, indwelling catheters, and even heparin-coated pulmonary artery catheters have been reported to sustain the syndrome.
Generalized skin reactions of various forms occur as a part of a hypersensitivity reaction to various drugs. The drugs frequently involved are sulfonamides, the penicillins, and phenytoin. Of concern to the intensivists are the three major types of drug-induced skin diseases with the potential for life-threatening complications, including erythema multiforme (Stevens-Johnson syndrome), toxic epidermal necrolysis, and exfoliative erythroderma (40). The lesions range from typical target-shaped eruptions to extensive bullous eruptions and large areas of skin sloughing. These conditions are usually associated with hypovolemic shock, a hypercatabolic state, and multisystem organ dysfunction; the management is mainly supportive.
Drug Interactions
Conventionally, a drug interaction is regarded as the modification of the effect of one drug by prior or concomitant administration of another (59). Several textbooks (60,61,62,63,64) and reviews (65) have compiled extensive listings of potential interactions. Most drug interaction studies report on small numbers of noncritically ill patients or volunteers, and thus, extrapolation of these studies to the ICU setting is undesirable, possibly leading to an unnecessary restriction of useful medications. This section reviews clinically significant drug interactions encountered in the ICU.
Drug interactions can result from pharmacokinetic or pharmacodynamic causes. Pharmacokinetic interactions affect the process of drug absorption, distribution, metabolism, and excretion. Pharmacodynamic interactions alter the biochemical or physiologic effect of a drug.
Pharmacokinetic Interactions
Absorption
Many drug interactions affect the bioavailability of drugs through their effects on absorption. These include adsorption and formation of drug complexes, changes in gastric emptying time and pH, alteration in intestinal motility and mucosal function, and reduction in splanchnic perfusion. Phenytoin absorption has been found to vary with type of enteral feeding (66). Anion exchange resin (cholestyramine) aluminum-containing drugs such as sucralfate and antacids, kaolin pectin, activated charcoal, and iron-containing preparations impair the absorption of various drugs such as digoxin, warfarin, and levothyroxine by forming insoluble complexes. In response, the safest action is to not give any oral medication within 2 hours of administering these chelating agents.
Drug incompatibilities in intravenous preparations also can present as drug interaction or absorption problems. Precipitation or chemical alteration may occur before parenteral dosing. Knowledge of in vitro drug incompatibilities is, therefore, essential, and in-depth resources are available (66,67). The precipitation of phenytoin in glucose-containing solution is a relevant example (68).
|
Table 62.3 Drugs With High Plasma Protein Binding Affinity |
|||||||||||||||
|
|||||||||||||||
Distribution
Many drugs circulate in the plasma partly bound to plasma proteins. Because the free or unbound serum concentration of a drug determines its biologic activity, changes in protein binding induced by another agent can have an important effect on the drug's pharmacologic response (69,70,71,72). Displacement from a protein of one drug by another depends on the concentration and relative binding affinities of the drugs. A drug with a higher serum concentration and higher protein-binding affinity displaces a second drug more readily. For example, warfarin's displacement by another drug can result in clinical bleeding complications. The acute elevation of free drug concentration in serum often is accompanied by an increased distribution to other tissues or increased elimination by metabolism and excretion until a new steady state is reached. Thus, when a new steady state is reached, the total drug level in the blood will be lower because of less protein-bound drug, whereas the free drug level will be in the therapeutic range. Individualization of drug therapy should be based on the clinical response or the plasma concentration of unbound drug, if available. Highly protein-bound drugs are particularly susceptible to these interactions (Table 62.3).
Metabolism
The metabolism of most drugs occurs largely in the liver. Quantitatively, the cytochrome P450 microsomal enzyme system containing mixed-function oxidases present in smooth endoplasmic reticulum is most important for initial metabolic conversion. The activity of these enzymes can be profoundly influenced by genetic factors and the coadministration of many drugs. These enzymes can be induced or inhibited by various agents (73,74).
|
Table 62.4 Drugs Commonly Affecting Metabolizing Enzymes in Liver |
||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||
|
Table 62.5 Drugs Commonly Affected by Enzyme-Inducing or Enzyme-Inhibiting Agents |
||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||
Some common enzyme-inducing agents include the anticonvulsants, phenytoin, phenobarbital, and carbamazepine; ethanol; phenylbutazone; and rifampin (Table 62.4). Cigarette smoking has been shown to be an excellent inducer of isoenzymes responsible for theophylline metabolism. Enzyme induction increases the rate of elimination of various drugs, leading to lower plasma levels (Table 62.5).
Several compounds (Table 62.4) inhibit microsomal enzyme function, inhibiting the metabolism of many of the same drugs affected by enzyme induction (Table 62.5). For example, cimetidine is a potent inhibitor of oxidative metabolism of warfarin, quinidine, nifedipine, lidocaine, theophylline, and phenytoin. Erythromycin inhibits the metabolism of cyclosporine, warfarin, carbamazepine, and theophylline.
Excretion
The most important route of drug excretion is renal and involves the processes of filtration, secretion, and reabsorption (75,76). All of these aspects of renal handling of compounds can potentially lead to drug interactions.
Several drugs, such as the aminoglycoside antibiotics, are eliminated almost completely by glomerular filtration. Furosemide, a potent loop diuretic, by causing intravascular volume depletion, can decrease renal perfusion pressure and filtration rate, thereby reducing elimination of gentamicin.
Many drugs are actively secreted in the proximal tubule. The most important of these agents include the organic acids, captopril, cephalosporins, sulfonamides, sulfonylureas, penicillins, diuretics, probenecid, and nonsteroidal anti-inflammatory agents. These compounds can block each others' secretions, thus decreasing their urinary excretion. Also, quinidine decreases the tubular secretion of digoxin. Thus, the coadministration of quinidine and digoxin approximately doubles the serum digoxin concentration. Inhibition of the tubular cation transport system by cimetidine impedes the renal clearance of procainamide and its active metabolite, N-acetyl procainamide.
The reabsorption of filtered or secreted compounds occurs in the distal tubule or collecting duct as a function of drug concentration, urinary flow, and pH of the urine. Alteration in distal urine pH can cause ion trapping of certain weak acids or bases and reduce passive reabsorption. Thus, alkalinization of urine by sodium bicarbonate facilitates the excretion of acidic drugs such as phenobarbital, salicylates, and amphetamines (40).
Lithium is reabsorbed with sodium in the kidney by the same renal mechanism. In case of volume depletion from chronic diuretic use, renal increases in sodium and lithium reabsorption occur, leading to a potentially toxic lithium level (40).
Pharmacodynamic Interactions
Unlike pharmacokinetic interactions, pharmacodynamic interactions occur when a pharmacodynamic effect of one medication affects the actions or effects of a second medication. Because critically ill patients are frequently on many medications for multiple indications, they are at high risk for these types of interactions. In some cases, pharmacodynamic interactions may be purposely used to achieve a desired therapeutic end point. For example, opioids and benzodiazepines are frequently used in combination to take advantage of their synergistic effects, possibly allowing for lower doses of each to be used (77,78). More frequently in the ICU, however, the pharmacodynamic interactions experienced are not desired and may lead to suboptimal therapy and adverse events.
Because of the numerous drug combinations and disease states that are seen in critical care units, the number of potential pharmacodynamic drug interactions is infinite and continues to grow as more medications are developed. For example, the chronic use of some antiepileptic medications may cause resistance to nondepolarizing neuromuscular blocking agents due to both enzyme induction, a pharmacokinetic interaction, and an up-regulation of acetylcholine receptors—namely, a pharmacodynamic interaction (79). In addition, the fluoroquinolone gatifloxacin has been reported to cause severe hypoglycemia when used in combination with oral hypoglycemic medications (80). Perhaps one of the most well-recognized pharmacodynamic interactions is the increased risk of torsade de pointes and death when two drugs that are known to cause QT prolongation are used in combination (81).
The principles of pharmacokinetics and pharmacodynamics are key to optimizing drug therapy of all types. If practitioners are unfamiliar with a medication, they should investigate the pharmacokinetic and pharmacodynamic characteristics of that drug to avoid subtherapeutic or supratherapeutic dosing, maximize efficacy, and minimize adverse events. Practitioners must be particularly careful in the ICU because critically ill patients frequently have some degree of organ dysfunction and may be receiving numerous medications.
A Practical Approach to Pharmacological Management in ICU
Pharmacotherapy is a complex science in the management of the critically ill patient. However, knowledge of pharmacokinetic principles, drug–drug interactions, changes related to systemic diseases, and possible drug toxicities permits the critical care clinician to design an appropriate medication regimen. The considerations listed below are recommended:
1. Review the medication administration record daily.
2. Monitor therapeutic end points and toxic effects for each drug.
3. Be knowledgeable of the pharmacokinetics of each drug (e.g., first-order versus zero-order kinetics, serum elimination half-life).
4. Remember that systemic disease (e.g., renal, hepatic) may create the need to alter the dosage regimen: an increase in volume of distribution may increase the required loading dose; and a decrease in clearance may decrease the required maintenance dose.
5. Look for possible drug–drug interactions.
6. Minimize the number of drugs.
7. Substitute equally effective, less expensive medications when possible.
8. Plan an approach to monitoring therapeutic and toxic effects. Check serum levels as appropriate, allowing four to five half-lives for steady-state achievement.
The Clinical Pharmacist's Role in Critical Care
In the multidisciplinary approach to care of the critically ill patient, the pharmacist is an essential member of the healthcare team (77,78,79). The impact of a pharmacist on the delivery of cost-effective care has been established in multiple settings, including the spectrum of intensive care units (80,81,82,83,84,85,86,87,88,89,90,91). In addition to affecting economics, the critical care pharmacist directly influences clinical outcomes. Pharmacist involvement in critically ill patient care has been associated with optimal fluid and neuromuscular blockade management as well as significant reductions in adverse drug events, medication errors, unnecessary serum drug measurements, and ventilator-associated pneumonia rates (92,93,94,95,96,97,98,99,100,101,102,103,104).
As outlined by the Society of Critical Care Medicine and American College of Clinical Pharmacy Task Force on Critical Care Pharmacy Services position paper, fundamental critical care pharmacist activities include: prospective medication therapy evaluation; prevention, management, and reporting of adverse drug events; pharmacokinetic monitoring; drug information and education services; medication policy and procedure implementation and maintenance; cost-effectiveness analyses; and quality assurance measures and reviews (79). With departmental and service support, many critical care pharmacists participate in didactic and clinical teaching programs as well as clinical critical care pharmacotherapy research endeavors.
Summary
Pharmacotherapy in the critically ill patient is extremely complex. A good understanding of pharmacologic principles, special population considerations, impact of single or multiple organ dysfunction, drug–drug interactions, and adverse drug reactions is paramount to maximize good outcome and minimize iatrogenic complications. Although the exact role and activities may vary in any given institution and patient population, the inclusion of a critical care pharmacist in daily patient care is recommended given the demonstrated cost savings and morbidity and mortality reductions.
References
1. Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: the dynamics of drug absorption, distribution, and elimination. In: Hardin JG, Limbird LE, Gilman AG, eds. Goodman and Gilman's The Pharmacologic Basis of Therapeutics. New York, NY: McGraw-Hill; 1996:3.
2. Bauer LA. Individualization of drug therapy: clinical pharmacokinetics and pharmacodynamics. In: DiPiro JT, ed. Pharmacotherapy: A Pathophysiologic Approach. New York, NY: Elsevier; 1992:15.
3. Winter ME. Clinical pharmacokinetics. In: Applied Therapeutics: The Clinical Use of Drugs. Vancouver, WA: Applied Therapeutics; 1992.
4. Riegelman S, Loo JC, Rowland M. Shortcomings in pharmacokinetic analysis by conceiving the body to exhibit properties of a single compartment. J Pharm Sci. 1968;57(1):117–123.
5. Greenblatt DJ, Kock-Weser J. Drug therapy. Clinical Pharmacokinetics (first of two parts). N Engl J Med. 1975;293(14):702–705.
6. Winter ME. Basic Clinical Pharmacokinetics. 6th ed. Vancouver, WA: Applied Therapeutics; 1999.
7. Koch-Weser J. Bioavailability of drugs (first of two parts). N Engl J Med. 1974;291(5):233–237.
8. Ross EM, Kennakin TP. Pharmacodynamics mechanisms of drug action and the relationship between drug concentration and effect. In: Hardman JG, Limbird LE, Gilman AG, eds. Goodman and Gilman's The Pharmacological Basis of Therapeutics. New York, NY: McGraw-Hill; 2001:31–43.
9. Frye RF, Matzke GK. Drug therapy individualization for patients with renal insufficiency. In: DiPiro JT, et al., eds. Pharmacotherapy: A Pathophysiologic Approach. New York, NY: McGraw-Hill; 1999:939–952.
10. Vanholder R, Van Landschoot N, De Smet R, et al. Drug protein binding in chronic renal failure: evaluation of nine drugs. Kidney Int. 1988;33(5):996–1004.
11. Cheng JW, Charland SL, Shaw LM, et al. Is the volume of distribution of digoxin reduced in patients with renal dysfunction? Determining digoxin pharmacokinetics by fluorescence polarization immunoassay. Pharmacotherapy. 1997;17(3):584–590.
12. Taber SS, Mueller BA. Drug-associated renal dysfunction. Crit Care Clin. 2006;22(2):357–374.
13. National Kidney Disease Education Program. Creatinine standardization program: recommendations for pharmacists and authorized drug prescribers. July 2006. http://www.nkdep.nih.gov/labprofessionals/Pharm_Recommendations_508.pdf. Accessed August 2, 2007.
14. Lötsch J. Opioid metabolites. J Pain Symptom Manage. 2005;29(5 Suppl):S10–24.
15. Jacobi J, Fraser GL, Coursin DB, et al. Clinical practice guidelines for the sustained use of sedatives and analgesics in the critically ill adult. Crit Care Med. 2002;30(1):119–141.
16. De Schoenmakere G, De Waele J, Terryn W, et al. Phenytoin intoxication in critically ill patients. Am J Kidney Dis. 2005;45(1):189–192.
17. Veltri MA, Neu AM, Fivush BA, et al. Drug dosing during intermittent hemodialysis and continuous renal replacement therapy: special considerations in pediatric patients. Paediatr Drugs. 2004;6(1):45–65.
18. Delcò F, Tchambaz L, Schlienger R, et al. Dose adjustment in patients with liver disease. Drug Saf. 2005;28(6):529–545.
19. Fredrick MJ, Pound DC, Hall SD, et al. Furosemide absorption in patients with cirrhosis. Clin Pharmacol Ther. 1991;49(3):241–247.
20. Schemmer P, Decker F, Dei-Anane G, et al. The vital threat of an upper gastrointestinal bleeding: risk factor analysis of 121 consecutive patients. World J Gastroenterol 2006;12(22):3597–3601.
21. Vyas K, Gala B, Sawant P, et al. Assessment of portal hemodynamics by ultrasound color Doppler and laser Doppler velocimetry in liver cirrhosis. Indian J Gastroenterol. 2002;21(5):176–178.
22. Rodighiero V. Effects of liver disease on pharmacokinetics. An update. Clin Pharmacokinet. 1999;37(5):399–431.
23. Hoyumpa AM, Schenker S. Influence of liver disease on the disposition and elimination of drugs. In: Schiff L, Schiff ER, eds. Diseases of the Liver. Philadelphia, PA: JB Lippincott Co; 1993.
24. George J, Murray M, Byth K, et al. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21(1):120–128.
25. Iqbal S, Vickers C, Elias E. Drug metabolism in end-stage liver disease. In vitro activities of some phase I and phase II enzymes. J Hepatol. 1990;11(1):37–42.
26. McLean AJ, Morgan DJ. Clinical pharmacokinetics in patients with liver disease. Clin Pharmacokinet. 1991;21(1):42–69.
27. Blaschke TF. Protein binding and kinetics of drugs in liver diseases. Clin Pharmacokinet. 1977;2(1):32–44.
28. Yeomans ER, Gilstrap LC III. Physiologic changes in pregnancy and their impact on critical care. Crit Care Med. 2005;33(10 Suppl):S256–258.
29. Cunningham FG, MacDonald PC, Gant NF, eds. Williams Obstetrics. 18th ed. Norwalk, CO: Appleton & Lange; 1989.
30. Anderson GD. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet. 2005;44(10):989–1008.
31. Pritchard JA. Changes in the blood volume during pregnancy and delivery. Anesthesiology. 1965;26:393–399.
32. Loebstein R, Lalkin A, Koren G. Pharmacokinetic changes during pregnancy and their clinical relevance. Clin Pharmacokinet. 1997;33(5):328–343.
33. Philipson A. Pharmacokinetics of ampicillin during pregnancy. J Infect Dis. 1977;136(3):370–376.
34. Philipson A, Stiernstedt G, Ehrnebo M. Comparison of the pharmacokinetics of cephradine and cefazolin in pregnant and non-pregnant women. Clin Pharmacokinet. 1987;12(2):136–144.
35. Heikkilä A, Erkkola R. Pharmacokinetics of piperacillin during pregnancy. J Antimicrob Chemother. 1991;28(3):419–423.
36. Stone SE, Morris TA. Pulmonary embolism during and after pregnancy. Crit Care Med. 2005;33(10 Suppl):S294–300.
37. Barbour LA, Smith JM, Marlar RA, et al. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol. 1995;173(6):1869–1873.
38. Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG. 2003;110(2):139–144.
39. Briggs GG, Freeman RK, Yaffe SJ, eds. Drugs in Pregnancy and Lactation. 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
40. Oates JA, Wood AJ. Adverse reaction to drugs. In: Harrison's Principles of Internal Medicine. New York, NY: McGraw-Hill; 1991:373.
41. Gray TK, Adams LL, Fallon HJ. Short-term intense surveillance of adverse drug reactions. J Clin Pharmacol New Drugs, 1973;13(2):61–67.
42. Jick H. Adverse drug effects in relation to renal function. Am J Med. 1977;62(4):514–517.
43. Leape LL, Cullen DJ, Dempsey Clapp M, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit. JAMA. 1999;281(3):267–270.
44. Whittaker M. Plasma cholinesterase variants and the anaesthetist. Anaesthesia 1980;35(2):174–197.
45. Nelson TE, Flewellen EH. Current concepts. The malignant hyperthermia syndrome. N Engl J Med. 1983;309(7):416–418.
46. Gronert GA. Malignant hyperthermia. Anesthesiology. 1980;53(5):395–423.
47. Gronert GA. Malignant hyperthermia. In: Miller RD, ed. Anesthesia. New York, NY: Churchill Livingstone; 1986:1971.
48. Kolb ME, Horne ML, Martz R. Dantrolene in human malignant hyperthermia. Anesthesiology. 1982;56(4):254–262.
49. Morgan KG, Bryant SH. The mechanism of action of dantrolene sodium. J Pharmacol Exp Ther. 1977;201(1):138–147.
50. Gronert GA. Malignant hyperthermia. Semin Anesth. 1983;2:197.
51. Norwood S. An approach to the febrile patient. In: Civetta JM, Taylor RW, Kirby RR, eds. Critical Care. Philadelphia, PA: JB Lippincott Co; 1992:992.
52. Mackowiak PA, LeMaistre CF. Drug fever: a critical appraisal of conventional concepts. An analysis of 51 episodes in two Dallas hospitals and 97 episodes reported in the English literature. Ann Intern Med. 1987;106(5):728–733.
53. George CF, Robertson D. Clinical consequences of abrupt drug withdrawal. Med Toxicol Adverse Drug Exp, 1987;2(5):367–382.
54. Bohrer H, et al. Clonidine as a sedative adjunct in intensive care. Intensive Care Med. 1990;16(4):265–266.
55. Easton C, MacKenzie F. Sensory-perceptual alterations: delirium in the intensive care unit. Heart Lung. 1988;17(3):229–237.
56. Messing RO, Closson RG, Simon RP. Drug-induced seizures: a 10-year experience. Neurology. 1984;34(12):1582–1586.
57. Albertson TE, Foulke GE, Tharratt S. Pharmacokinetics and iatrogenic drug toxicity in the intensive care unit. In: Hall JB, Schmidt GA, Wood LH, eds. Principles of Critical Care. New York, NY: McGraw-Hill; 1992:2061.
58. Warkentin TE, Kelton JG. Heparin and platelets. Hematol Oncol Clin North Am. 1990;4(1):243–264.
59. McInnes GT, Brodie MJ. Drug interactions that matter. A critical reappraisal. Drugs. 1988;36(1):83–110.
60. Chernow B, ed. The Pharmacologic Approach to the Critically Ill Patient. Baltimore, MD: Williams & Wilkins; 1988.
61. Hardman JG, Limbird LE, Gilman AG, eds. The Pharmacologic Basis of Therapeutics. 10th ed. New York, NY: Mc-Graw-Hill; 2001.
62. Hansen PD, Horn JR. Drug Interaction. St. Louis, MO: Wolters Kluwer Health; 2006.
63. Morselli PL, Garattinni S, Cohen SN. Drug Interactions. New York, NY: Raven Press; 1974.
64. Stockley I. Drug Interactions. 3rd ed. Oxford, UK: Blackwell Science; 1995.
65. Prescott LF. Pharmacokinetic drug interactions. Lancet. 1969;2(7632):1239–1243.
66. Guidry JR, Eastwood TF, Curry SC. Phenytoin absorption in volunteers receiving selected enteral feedings. West J Med. 1989;150(6):659–661.
67. Trissel LA. Handbook of Injectable Drugs. 13th ed. Bethesda, MD: American Society of Health-System Pharmacists; 2005.
68. Cloyd JC, Bosch DE, Sawchuk RJ. Concentration-time profile of phenytoin after admixture with small volumes of intravenous fluids. Am J Hosp Pharm. 1978;35(1):45–48.
69. Koch-Weser J, Sellers EM. Binding of drugs to serum albumin (first of two parts). N Engl J Med. 1976;294(6):311–316.
70. MacKichan JJ. Pharmacokinetic consequences of drug displacement from blood and tissue proteins. Clin Pharmacokinet. 1984;9(Suppl 1):32–41.
71. McElnay JC, D'Arcy PF. Protein binding displacement interactions and their clinical importance. Drugs. 1983;25(5):495–513.
72. Wood M. Plasma drug binding: implications for anesthesiologists. Anesth Analg. 1986;65(7):786–804.
73. Burns JJ, Conney AH. Enzyme stimulation and inhibition in the metabolism of drugs. Proc R Soc Med. 1965;58(11 Pt 2):955–960.
74. Gelehrter TD. Enzyme induction (first of three parts). N Engl J Med. 1976;294(10):522–526.
75. Prescott LF. Mechanisms of renal excretion of drugs (with special reference to drugs used by anaesthetists). Br J Anaesth, 1972;44(3):246–251.
76. Weiner IM, Mudge GH. Renal tubular mechanisms for excretion of organic acids and bases. Am J Med. 1964;36:743–762.
77. Gilliland HE, Prasad BK, Mirakhur RK, et al. An investigation of the potential morphine sparing effect of midazolam. Anaesthesia. 1996;51(9):808–811.
78. Richman PS, Baram D, Varela M, et al. Sedation during mechanical ventilation: a trial of benzodiazepine and opiate in combination. Crit Care Med. 2006;34(5):1395–1401.
79. Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol. 2006;61(3):246–255.
80. LeBlanc M, Bélanger C, Cossette P. Severe and resistant hypoglycemia associated with concomitant gatifloxacin and glyburide therapy. Pharmacotherapy. 2004;24(7):926–931.
81. Allen LaPointe NM, Curtis LH, Chan KA, et al. Frequency of high-risk use of QT-prolonging medications. Pharmacoepidemiol Drug Saf. 2006;15(6):361–368.
82. Brilli RJ, Spevetz A, Branson RD, et al. Critical care delivery in the intensive care unit: defining clinical roles and the best practice model. Crit Care Med. 2001;29(10):2007–2019.
83. Haupt MT, Bekes CE, Brilli RJ, et al. Guidelines on critical care services and personnel: recommendations based on a system of categorization of three levels of care. Crit Care Med. 2003;31(11):2677–2683.
84. Rudis MI, Brandl KM. Position paper on critical care pharmacy services. Society of Critical Care Medicine and American College of Clinical Pharmacy Task Force on Critical Care Pharmacy Services. Crit Care Med. 2000;28(11):3746–3750.
85. Baldinger SL, Chow MS, Gannon RH, et al. Cost savings from having a clinical pharmacist work part-time in a medical intensive care unit. Am J Health Syst Pharm. 1997;54(24):2811–2814.
86. Gandhi PJ, Smith BS, Tataronis GR, et al. Impact of a pharmacist on drug costs in a coronary care unit. Am J Health Syst Pharm. 2001;58(6):497–503.
87. Krupicka MI, Bratton SL, Sonnenthal K, et al. Impact of a pediatric clinical pharmacist in the pediatric intensive care unit. Crit Care Med. 2002;30(4):919–921.
88. Patel NP, Brandt CP, Yowler CJ. A prospective study of the impact of a critical care pharmacist assigned as a member of the multidisciplinary burn care team. J Burn Care Res. 2006;27(3):310–313.
89. Hatoum HT, Hutchinson RA, White KW, et al. Evaluation of the contribution of clinical pharmacists: inpatient care and cost reduction. Drug Intell Clin Pharm. 1988;22(3):252–259.
90. Herfindal ET, Bernstein LR, Kishi DT. Impact of clinical pharmacy services on prescribing on a cardiothoracic/vascular surgical unit. Drug Intell Clin Pharm. 1985;19(6):440–444.
91. Katona BG, Ayd PR, Walters JK, et al. Effect of a pharmacist's and a nurse's interventions on cost of drug therapy in a medical intensive-care unit. Am J Hosp Pharm. 1989;46(6):1179–1182.
92. McMullin ST, Hennenfent JA, Ritchie DJ, et al. A prospective, randomized trial to assess the cost impact of pharmacist-initiated interventions. Arch Intern Med. 1999;159(19):2306–2309.
93. Miyagawa CI, Rivera JO. Effect of pharmacist interventions on drug therapy costs in a surgical intensive-care unit. Am J Hosp Pharm. 1986;43(12):3008–3013.
94. Montazeri M, Cook DJ. Impact of a clinical pharmacist in a multidisciplinary intensive care unit. Crit Care Med. 1994;22(6):1044–1048.
95. Schumock GT, Meek PD, Ploetz PA, et al. Economic evaluations of clinical pharmacy services—1988–1995. The Publications Committee of the American College of Clinical Pharmacy. Pharmacotherapy. 1996;16(6):1188–1208.
96. Smythe MA, Shah PP, Spiteri TL, et al. Pharmaceutical care in medical progressive care patients. Ann Pharmacother. 1998;32(3):294–299.
97. Broyles JE, Brown RO, Vehe KL, et al. Pharmacist interventions improve fluid balance in fluid-restricted patients requiring parenteral nutrition. DICP. 1991;25(2):119–122.
98. Calabrese AD, Erstad BL, Brandl K, et al. Medication administration errors in adult patients in the ICU. Intensive Care Med. 2001;27(10):1592–1598.
99. Devlin JW, Holbrook AM, Fuller HD. The effect of ICU sedation guidelines and pharmacist interventions on clinical outcomes and drug cost. Ann Pharmacother. 1997;31(6):689–695.
100. Kaye J, Ashline V, Erickson D, et al. Critical care bug team: a multidisciplinary team approach to reducing ventilator-associated pneumonia. Am J Infect Control. 2000;28(2):197–201.
101. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279(15):1200–1205.
102. Leape LL, Cullen DJ, Clapp MD, et al. Pharmacist participation on physician rounds and adverse drug events in the intensive care unit. JAMA. 1999;282(3):267–270.
103. Rudis MI, Sikora CA, Angus E, et al. A prospective, randomized, controlled evaluation of peripheral nerve stimulation versus standard clinical dosing of neuromuscular blocking agents in critically ill patients. Crit Care Med. 1997;25(4):575–583.
104. Dager WE, Albertson TE. Impact of therapeutic drug monitoring of intravenous theophylline regimens on serum theophylline concentrations in the medical intensive care unit. Ann Pharmacother. 1992;26(10):1287–1291.