Takeru Shimizu
Taro Mizutani
Human body temperature is maintained within tight limits by a balance between heat production and dissipation. Heat is normally generated by muscular activity and metabolic reactions, the latter mainly by the liver. It is dissipated by a combination of radiation, convection, conduction, and evaporation. This balance between heat generation and dissipation is the key to maintaining optimal body temperature. Under normal circumstances, body temperature is 37°C under the tongue, 38°C in the rectum, 32°C at the skin, and 38.5°C in the central liver. Significant deviation from normal body temperature is a critical condition that requires prompt diagnosis, treatment, and normalization of the temperature alteration. Herein, we discuss hypothermia, hyperthermia, and malignant hyperthermia and the neuroleptic malignant hyperthermia.
Hypothermia
Hypothermia is generally defined as a core temperature below 35°C (95° F) (1,2,3). In a more detailed manner, the literature classifies hypothermia as mild, moderate, or severe: mild hypothermia is that between 35°C and 32°C, moderate hypothermia between 32°C and 28°C, and severe hypothermia below 28°C (4). Table 89.1 outlines the classification of severity and clinical manifestations.
Hypothermia may be precipitated by various acute and chronic medical conditions, environmental exposure, or drugs. Hypothermia caused without exposure to the extreme temperatures is generally limited to mild to moderate in degree. Interestingly, however, with equivalent body temperature, patients found indoors were more severely affected and died more frequently than those found outdoors (5).
Hypothermia is considered to be an underrecognized condition, especially in the aged (2,3). Elderly patients who develop hypothermia are more likely to live alone, have other intercurrent diseases, to have their home heating turned off or inadequate home heating, and to wear inappropriate clothing for actual ambient temperatures (6).
Several confounding factors can further impair temperature control. Intoxicants, medications, extremes of age, and the general state of health—including intercurrent diseases—can modify the heat loss. Hypothermia occurs in various clinical settings; Table 89.2 outlines the clinical causes and disorders associated with this finding.
Accidental hypothermia is defined as a spontaneous decrease in core temperature. It is often caused by a cold environment and associated with an acute problem, but without any primary disorder of the temperature regulatory center. This is most commonly observed in neonates; the elderly; unconscious, immobile, or drugged persons; and workers in an extremely cold environment. Mortality rates for accidental hypothermia have been reported to range between 10% and 80%. A multicenter review of 428 cases of accidental hypothermia reported an overall mortality of 17% (7,8,9). Intercurrent diseases or infection seem to contribute to most deaths, as it was shown that patients with sepsis had a markedly worse mortality rate when they presented with hypothermia, as opposed to fever (10).
Temperature Regulation and Mechanism of Heat Loss
Hypothermia presents when heat generation cannot keep up with heat dissipation. Heat generation depends on the metabolic process at rest and on skeletal muscle metabolism during exercise. Humans have a high capacity to dissipate heat, with four primary means of heat dissipation or transmission. It is important to know these mechanisms to prevent hypothermia and to develop effective rewarming strategies.
1. Conduction: Conduction is the transfer of heat between two masses in contact with one another. The rate of heat transfer depends on the temperature gradient at the interface and the size of the contact area. It is also determined by the thermal conductivity of the materials. Metals and liquids are most conductive, and gases are most insulating. For example, water has a 25- to 30-fold larger conductivity than air. This means that contacting a wet surface is one of the fastest ways to dissipate body temperature.
2. Convection: Convection is the transfer of heat due to the flow of liquids or gases over a surface. Convective heat loss occurs when air around a patient is continuously swept away, and it is directly proportional to the body surface area, the temperature gradient, and the air velocity.
3. Radiation: Transfer of radiant energy is due to electromagnetic transmission.
4. Evaporation: Evaporation is the process whereby atoms or molecules in a liquid state gain sufficient energy to enter the gaseous state. Evaporation proceeds more quickly at higher temperature and/or at higher flow rates between the gaseous and liquid phase. Therefore, the heat loss by this mechanism is proportional to the change of the vapor pressure from the surface to ambient air and the velocity of air movement. Approximately 30% of evaporative heat loss occurs in the lung, and the rest is from the skin surface at usual room temperature. Evaporation accounts for 10% to 15% of total body heat loss. The heat of evaporation of water is 0.58 kcal/g H2O. Given that 30 g of water is lost during the breathing of dry room air per hour, about 18 kcal, which is nearly half of an anesthetized patient's hourly heat production, is lost.
|
Table 89.1 Classification of Hypothermia |
||||||||||||||||||||||||||||||||||||
|
Clinical Syndromes
Cardiovascular
A sympathetic response increases myocardial oxygen consumption and causes tachycardia and peripheral vasoconstriction—that is, diminished pulses, pallor, acrocyanosis, and cold extremities—in patients with mild hypothermia (core temperature of 32°C–35°C). Blood pressure and heart rate are initially increased, followed by bradycardia, which further deteriorates at 32°C, and consequently, cardiac output, myocardial contractility, and arterial pressure fall.
Electrocardiographic (ECG) findings include the Osborne J-wave after the QRS complex as hypothermia becomes more severe (Fig. 89.1) The Osborne J-wave is an important diagnostic feature, which can be observed in other pathologic conditions such as central nervous system lesions and sepsis, but it is frequently absent (3,11). Atrial and ventricular fibrillation are common, and electrical defibrillation during hypothermia is often ineffective. It is important to remember that the hypothermic myocardium is irritable, making placement of pulmonary artery or other central catheters dangerous.
|
Table 89.2 Causes of Hypothermia |
||||||||||||||||
|
Respiratory System
Respiratory rate falls. The patient becomes apneic or has an agonal respiratory pattern when the body temperature is less than 28°C.
Central Nervous System
The electroencephalogram (EEG) becomes flat at 19°C to 29°C (12). Cerebrovascular autoregulation remains intact until the core temperature falls to below 25°C, but mentation starts to drop at 30°C. Dysarthria and hyperreflexia occur below 35°C, and hyporeflexia occurs below 32°C.
Coagulation
Hypothermia produces coagulopathy via three major mechanisms (13,14). First, the enzymatic coagulation cascade is impaired; second, platelet dysfunction occurs; and third, plasma fibrinolytic activity is enhanced. Because coagulation tests, such as prothrombin time (PT) or partial thrombin time (PTT), are performed at 37°C in the laboratory, a major disparity between clinical coagulopathy and the reported values is frequently observed (15). A disseminated intravascular coagulation (DIC) type of syndrome is also reported (16). Clinically significant coagulopathies occur and are often associated with trauma (17,18).
Renal System
Exposure to cold induces a diuresis irrespective of the state of hydration. Centralization of the blood volume—due to the initial peripheral vasoconstriction—stimulates the diuresis. Hypothermia depresses renal blood flow by 50% at 27°C to 30°C, and the renal cellular basal metabolic rate decreases (2). As a result, renal tubular cell reabsorptive function decreases and the kidney excretes a large amount of dilute urine. This is termed cold diuresis, resulting in a decreased blood volume and progressive hemoconcentration (19).
|
|
|
Figure 89.1. The electrocardiogram (ECG) shows atrial fibrillation with a very slow ventricular response, prominent J (Osborne) waves (late, terminal upright deflection of QRS complex; best seen in leads V3–V6), and nonspecific QRS widening. (Adapted from O'Keefe J, Hammill S, Freed M, et al. The Complete Guide to ECGs. 2nd ed. Royal Oak, MI: Physicians' Press; 2002.) |
Glucose Metabolism
Blood glucose concentration commonly increases because pancreatic function, insulin activity, and/or response to insulin decrease, along with activated function of the autonomic nervous system in hypothermia. At the same time, hemoconcentration results in elevated serum glucose concentration.
Therapeutic Approach
While rewarming is the common goal in the clinical treatment of the hypothermic patient, it is both difficult and controversial to treat. At the same time as treatment is initiated, a search for and discovery of the mechanism of heat loss will play a key role in achieving a better outcome. Treatment includes that delivered by prehospital providers as well as inpatient care.
Prehospital Treatment
Hypothermia is often combined with mental and physical exhaustion. Even if a patient is found down, cold, stiff, and cyanotic, the patient is not necessarily dead and may make a recovery even when signs of life are initially absent. Thus, rescue efforts should not be given up while the patient is cold. Since ventricular fibrillation or asystole may be induced by any stimuli, such as tracheal intubation, comatose patients should be treated with extreme care.
The initial primary focus of prehospital treatment is to avoid further loss of heat. Removal of wet clothing and applying dry insulating covers such as blankets, pads, coats, and sleeping bags are effective in treating all the mechanisms of heat loss described above. During transportation, an aluminized space blanket may be used. To make the most of its effect, the patient should be carefully wrapped with additional blankets. The patient should be kept horizontal to minimize the circulatory and sympathetic change. Vigorous rubbing should be avoided because it induces vasodilation, which may be followed by hypotension or “rewarming shock.” Hot water bottles or hot packs may be used if available but should be used cautiously to avoid burn injury.
Resuscitation using the ABCs—airway, breathing, circulation—of basic life support are implemented if needed. Warmed (42°C–46°C), humidified oxygen during bag/mask ventilation (20) and warmed intravenous fluids should be given if possible. Death should not be declared below 32°C.
In-hospital Treatment
Indicated monitoring includes an ECG, Doppler evaluation of pulses, and temperature. General laboratory studies include electrolytes, complete blood count, coagulation studies (prothrombin time and partial thromboplastin time), blood urea nitrogen, creatinine, amylase, calcium, magnesium, and glucose concentrations. Radiologic examinations are indicated. Patients with hypothermia are usually dehydrated, which should be corrected with IV fluids warmed to 43°C (20). The use of glucose-containing solutions should be used with caution as hypothermic patients are usually hyperglycemic due to hypoactivity of insulin.
|
|
|
Table 89.3 Rewarming Methods |
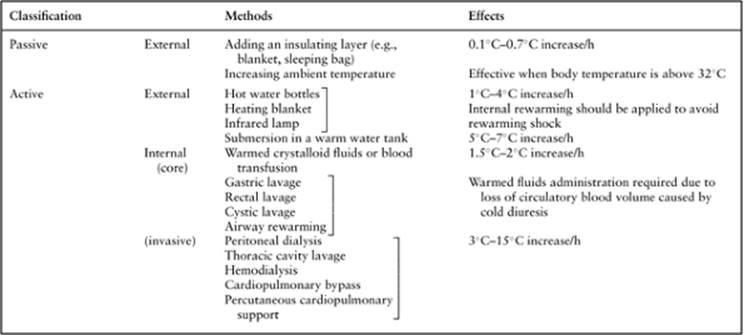
The various options for therapeutic approaches can be considered, and rewarming should be performed along with other supportive therapy. Techniques of rewarming include active and passive methods, and external and internal (core) methods (Table 89.3). Although no difference in survival and clinical or histopathologic morbidity has been demonstrated by some prospective studies comparing active and external rewarming versus core rewarming methods (21,22), percutaneous or open chest cardiopulmonary bypass is recommended for pulseless hypothermic patients if it is available (23,24,25,26,27,28,29,30,31).
Bronchopneumonia secondary to aspiration is a common complication. Oral intake of warm or hot drink should be avoided because obtunded, hypothermic patients may have suppressed airway protective mechanisms, including cough or gag reflexes. Prophylactic tracheal intubation may be considered if suppression of these reflexes is present. When the trachea is intubated, warmed (42°C–46°C), humidified oxygen should be administered (20).
Hyperthermia
There are many conditions that elevate the body temperature. Table 89.4 outlines the major causes, which may be classified as hyperthermia or fever. In this section, we discuss environmental hyperthermia as well as malignant hyperthermia and the neuroleptic malignant syndrome.
Environmental Hyperthermia—Heat Stroke
Heat stroke is a medical emergency, characterized by a high body temperature, altered mental status, and hot dry flushed skin (32). It may lead to multisystem organ dysfunction with hemorrhage and necrosis in the lungs, heart, liver, kidneys, brain, and intestines (33). Heat stroke is thought to be relatively uncommon in temperate climates. The documented body temperature with this disorder is 41.1°C or more. There has been no obvious decrease in mortality in the last 50 years, which is variably quoted as ranging between 10% and 50% (33).
There are several heat-related illnesses, which may take the form of heat syncope, heat cramps, heat exhaustion, and heat stroke. Heat stroke may be further classified as exertional and nonexertional (classic heatstroke). Table 89.5 outlines the syndromes.
· Heat syncope: Heat syncope is fainting due to peripheral vasodilation secondary to high ambient temperature.
· Heat cramp: Heat cramp refers to muscular cramping occurring during exercise in heat, which is related to electrolyte deficiency; it is usually benign.
· Heat exhaustion: This is often referred to as heat prostration. Heat exhaustion occurs when the individual becomes dehydrated and weak. The patient collapses from dehydration, salt depletion, and hypovolemia. Anorexia, nausea, and vomiting frequently occur. Excessive sweating leads to a loss of water and/or electrolytes. There are two mechanisms for this disorder: salt-depletion and water-depletion heat exhaustion. Salt-depletion heat exhaustion usually occurs when an unacclimatized person exercises and replaces only water. Water-depletion heat exhaustion is usually observed in an acclimatized person who has inadequate water intake during exposure to extreme heat. Serum sodium concentration may be normal or mildly elevated. The core temperature may or may not be raised (usually mild to moderate, less than 38°C) and tissue damage does not occur.
· Heat stroke: Heat stroke occurs when the core body temperature rises against a failing thermoregulatory system (32). The core temperature most often quoted is a rectal temperature exceeding 40.6°C (34). Heat stroke may be divided into exertional and nonexertional (classic) heat stroke (34). Exertional heat stroke occurs in previously healthy young people exercising in hot and humid climates without being acclimatized. Nonexertional heat stroke occurs during extreme heat waves, the elderly being particularly vulnerable (Table 89.6).
|
Table 89.4 Hyperthermia and Fever |
|||||||||
|
|||||||||
Temperature Regulation
Normal heat production is primarily due to metabolic activity in the liver and skeletal muscle, with the liver generating most body heat at rest and muscle being the major source with exercise or shivering. Skeletal muscle heat production ranges from 65 to 85 kcal/hour at basal level, but it may increase up to 900 kcal/hour (35). Heat elimination occurs by four major mechanisms as we have discussed in hypothermia. Convection and radiation are normally the most important mechanisms for heat elimination. Evaporation becomes a major mechanism for heat dissipation with incremental skeletal muscle metabolic activity. If the ambient temperature exceeds body temperature, heat loss may depend only on evaporation. However, sweating produces only 400 to 650 kcal/hour of heat dissipation. Therefore, blood flow regulation to skin and sweat gland activity are critical in maintaining thermal balance. There is a distinction made between exertional and nonexertional heat stroke at this point; failure of thermoregulation (lack of sweating) may be more important in nonexertional (classic) heat stroke and less so in exertional heat stroke (36).
|
Table 89.5 Heat Syndromes |
|||||||||||||||
|
The process of thermoregulation consists of three parts: (i) afferent thermal sensing, (ii) hypothalamic processing, and (iii) efferent responses through the sympathetic system. Heat stimuli are carried by C fibers from the skin to the spinal cord. Central temperature sensors in the abdominal and thoracic viscera, spinal cord, and brain may play a significant role in preventing hyperthermia, although peripheral sensors in the skin seem to be most important. The hypothalamus integrates all afferent temperature input to alter body temperature by regulating vasomotor tone to the skin and inducing sweat formation. Neural output to the cerebral cortex is also important in modifying behavior to compensate for changes in temperature. The efferent hypothalamic response to heat consists of cutaneous vasodilation, sweat formation, and inhibition of muscle tone. Vasomotor tonic changes result in cutaneous dilation and shunting of blood away from the liver and splanchnic circulation, facilitating heat transfer from the core to skin. Sweat formation is under cholinergic sympathetic control. Removing clothes, limiting physical activity, and moving to a cooler place are important behaviors.
|
Table 89.6 Exertional and Nonexertional Heat Stroke Syndromes |
||||||||||||||||||
|
Acclimatization is a physiologic process whereby an individual adapts to work in a hot environment (35). Acclimatization to sustained increases in body temperature is slow and requires 1 to 2 weeks for peak effect. Sweat volume increases from 1.5 L/hour up to 4 L/hour. The sweating threshold decreases over an extended period of time. Sweat sodium concentration decreases from 30 to 60 mEq/L to about 5 mEq/L. Plasma antidiuretic hormone, growth hormone, and aldosterone levels increase. Cardiovascular mechanisms include a 10% to 25% increase in plasma volume and an increased stroke volume and cardiac output with a slowing of heart rate.
Clinical Syndrome
Heat stroke is mostly defined as a core temperature above 40.6°C, but neurologic impairment may occur at lower temperatures in some cases; indeed, neurologic dysfunction is a cardinal feature of heat stroke (37). Neurologic manifestations include slurred speech, delirium, stupor, lethargy, coma, and seizures (38). Seizures occur more commonly at temperatures above 41°C. Ataxia, dysmetria, and dysarthria may also be observed.
The cardiovascular system is commonly compromised in the presence of heat stroke. Tachydysrhythmia and hypotension frequently occur (38). Hypotension may result from translocation of blood from the central circulation to the periphery to dissipate heat, or the increased production of nitric oxide may result in vasodilation (37,39). A study of Doppler and echocardiographic findings in patients with classic heat stroke and heat exhaustion reported a circulation that was hyperdynamic, with tachycardia, resulting in high cardiac output (34). It also reported that hypovolemia was more pronounced in heat stroke patients with signs of peripheral vasoconstriction. Heat exhaustion patients were more likely to demonstrate peripheral vasodilation.
Lactic acidosis may occur. Since patients in heat stroke are in shock, the mechanism by which lactate is cleared by the liver and converted to glucose is less effective, and restoration of the circulating volume may lead to worsening lactic acidosis as skeletal muscle is reperfused and the elevated lactate cleared. Patients typically hyperventilate to compensate for the acute acidosis with an acute respiratory alkalosis. This may lead to heat-induced tetany. After several hours, a mixed acid-base disorder may occur because of sustained tissue damage.
Significant dehydration is noted in most patients with exertional heat stroke and may be reflected as elevated blood urea nitrogen and creatinine levels or hemoconcentration. Sodium, potassium, phosphate, calcium, and magnesium serum concentrations are frequently low in the early period (35,40,41,42,43,44). Sodium, potassium, and magnesium are lost through increased sweating. Hypokalemia may be as a result of catecholamine release or may occur secondary to hyperventilation. Hypokalemia decreases sweat secretion and skeletal muscle blood flow, which may impair heat dissipation. Cellular death begins to occur throughout the body at temperatures above 42°C. Hyperkalemia may occur if significant skeletal muscle damage or cellular lysis develops. If significant rhabdomyolysis develops, injured cells release phosphate, which reacts with serum calcium and may lead to hypocalcemia.
Renal dysfunction is well documented in exertional heat stroke, with the incidence of acute renal failure approximately 25% (45). The cause is usually multifactorial, including direct thermal injury, the prerenal insults of volume depletion, and renal hypotension, rhabdomyolysis, and disseminated intravascular coagulation (DIC) (38).
Liver damage is very frequently seen and is probably related to splanchnic redistribution (46,47). Elevated liver enzymes are common.
Hemorrhagic complications may be observed. These may be petechial hemorrhages and ecchymoses, which may represent direct thermal injury or may be related to the development of DIC. Damatte et al. (38) reported that 45% of patients had laboratory evidence of DIC. This consumption coagulopathy may be further compounded by hepatocellular damage.
Complications
Cardiac complications include myocardial pump failure, tachydysrhythmia, high cardiac output, and myocardial infarction. ECG abnormalities are also observed (48). Sinus tachycardia and QT prolongation are followed by nonspecific ST-T wave changes, suggesting cardiac ischemia.
Neurologic complications include seizures, cerebral edema, and localized brain hemorrhages. Irreversible brain damage occurs above 42°C. Cerebellar impairment may persist after recovery.
Pulmonary edema may be caused by a limited cardiac function or may develop secondary to the acute respiratory distress syndrome (ARDS) (49). Pulmonary aspiration may be observed in obtunded patients.
Acute renal failure may be caused by direct heat damage, renal hypoperfusion, or rhabdomyolysis. The incidence of renal failure is about 35% with exertional heat stroke and about 5% in classic nonexertional heat stroke, with which rhabdomyolysis is less likely to coexist.
Liver damage and dysfunction occur in most patients with heat stroke. Cholestasis and centrilobular necrosis elevate bilirubin and liver enzymes, which may not be apparent until 48 to 72 hours after injury (44).
Hematologic complications include hemolysis, thrombocytopenia, and DIC (49). DIC is triggered by diffuse endothelial and organ damage, has an onset delay of 2 to 3 days after the initiating event, and is associated with high mortality.
Therapeutic Approach
Heat stroke requires prompt and effective treatments. Oxygen therapy, rapid cooling, and cautious hydration should be immediately instituted to avoid complications and achieve recovery. Tracheal intubation should be considered if the patient is obtunded or in respiratory distress. Oxygen delivery is often less than normal, and pulmonary shunt fraction is increased (50).
Rapid cooling is accomplished by external techniques. These include immersion in ice water or application of cooling blankets above and below the patient (conductive cooling technique), and wetting the skin with water or alcohol, followed by the use of fans to facilitate evaporation and heat dissipation (evaporative-convective cooling technique). Both techniques usually reduce core temperature below 40°C in 1 hour. A significant disadvantage of immersion is impairment of access to the patient and limited monitoring. Another disadvantage of immersion is that intense vasoconstriction can slow the rate of heat loss (51). Vasoconstriction may also have adverse cardiovascular effects in patients with limited cardiac function because it increases cardiac afterload. Vasoconstriction may be reduced by skin massage, which prevents dermal stasis of cooled blood. More aggressive cooling techniques include gastric lavage with iced saline, cold hemodialysis, and cardiopulmonary bypass. They are only rarely required, being used in cases of refractory temperature elevation or malignant hyperthermia, in which thermogenesis is ongoing. In addition, the efficacy of rapid infusion of large-volume ice-cold intravenous fluid (LVICF)—using either lactated Ringer solution or normal saline—has been implicated in clinical trials of induced hypothermia. Bernard et al. (52) showed that 30 mL/kg of LVICF (lactated Ringer solution at 4°C) over 30 minutes decreased core temperature by 1.7°C immediately after infusion, with improvements in acid-base and renal function.
Core body temperature should be monitored closely at the rectum, bladder, or tympanic membrane. Vital signs, neurologic functions, urine output, and laboratory measurements should also be monitored closely. Laboratory measurements include arterial blood gas and serum electrolyte concentrations, especially potassium, which may increase significantly and result in life-threatening hyperkalemia. Glucose–insulin therapy should be instituted emergently in patients with ECG changes.
Intravenous volume repletion should be individualized. Volume deficit is not a prominent feature in classic nonexertional heat stroke. Central venous catheter and pulmonary artery catheter placement may be invaluable to assess volume depletion, peripheral vascular vasodilation, or primary myocardial dysfunction, especially in patients with limited cardiac reserve. Hypotension usually responds to intravenous fluids, but if an inotropic drug is needed, dobutamine is the drug of choice for heat stroke.
Seizures occur commonly in heat stroke patients and should be treated with intravenous diazepam or other benzodiazepines. The efficacy or clinical rationale for the administration of dehydrating drugs is uncertain, but these drugs may be potentially beneficial for some patients at risk of acute renal failure secondary to rhabdomyolysis, as acute renal failure can be a major cause of patient morbidity. This may be prevented by prompt repletion of intravascular volume and restabilizing adequate renal perfusion pressure. Hemodialysis may be required if hyperkalemia or other metabolic disturbances exist.
DIC may be treated with continuous infusion heparin therapy. Although this therapy brings some benefit, its utility seems uncertain (40,42,44).
Malignant Hyperthermia and the Neuroleptic Malignant Syndrome
Malignant hyperthermia (MH) and the neuroleptic malignant syndrome (NMS) are disorders of rising body temperature related to an imbalance between heat production and heat dissipation. MH was not clearly described as a syndrome until 1960 (53,54). NMS was first described by Delay et al. (55) after the introduction of neuroleptics in 1960. These disorders are uncommon but life-threatening complications related to the administration of anesthetic or neuroleptic drugs. Their main features include hyperthermia, muscle rigidity, metabolic acidosis, and autonomic disturbances. Endogenous heat production resulting from impaired physiologic heat-dissipating mechanisms and hypothalamic temperature regulation is responsible for elevation of core body temperature in NMS. On the other hand, it usually appears intact in MH (56). Both of them are uniquely characterized by their association with various drugs, although they are distinctive from each other; associated drugs are listed in Table 89.7 (56,57,58,59,60,61,62,63,64,65). An additive in commercial succinylcholine, chlorocresol, has been reported as an additional trigger in MH (66). The in vitro halothane–caffeine contracture test on skeletal muscle helps to identify susceptible individuals and to establish with certainty the genetic nature of the disorder in most individuals (67).
|
Table 89.7 Drugs Associated with Malignant Hyperthermia and the Neuroleptic Malignant Syndrome |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
Incidence
A Danish survey indicates an incidence of fulminant MH of 1 in 250,000 patients. The overall incidence of MH is between 1 in 50,000 and 1 in 100,000 patients receiving general anesthesia (68,69,70). Suspected MH occurred in 1 in 16,000 anesthetics overall and 1 in 4,200 anesthetics involving potent volatile agents and succinylcholine. Incidence rates of MH reported vary by country. The mortality rate was initially 70%; earlier diagnosis and use of dantrolene have reduced it to less than 5% (71).
The incidence of NMS ranges from 0.07% to 2.2% among patients receiving neuroleptic agents (72,73,74). A decrease in mortality has been reported; NMS has had a 76% mortality before 1970, a 22% mortality from 1970 to 1980, and a 15% mortality since 1980 (75).
Temperature Regulation
Most patients with an episode of MH have a history of relatives with a similar episode or an abnormal response to the halothane–caffeine contracture test. Genetic inheritance patterns reflect the complexity of the responsible genes of MH. Genes on chromosomes 1, 3, 5, 7, 17, and 19 (1q32, 3q13, 5p, 7q21–24, 17q21–24) have been indicated (76,77,78). The exact mechanisms of MH are poorly understood. The initial focus was on an abnormal calcium channel receptor, ryanodine RYR1 receptor, in patients with MH, which is responsible for calcium release from the sarcoplasmic reticulum and plays a critical role in muscle depolarization. Further studies have shown that many patients with MH have a normal ryanodine receptor. However, mutations in RYR1 occur in at least 50% of susceptible subjects and almost all families. More than 30 missense mutations and one deletion have been associated with a positive contracture test result or clinical MH (79). Other than RYR1, only two other genes—α1s-subunit of DHPR, and CACNL1A3 in MHS3—are implicated, and they are responsible for less than 1% of the cases (80). For practical purposes, the RYR1 gene remains the target for genetic analysis.
Resultant from this mutation, free inbound ionized calcium can be released from the storage sites, which normally maintain skeletal muscle relaxation by sequestering calcium from the muscle contractile apparatus (56). The administration of anesthetics may unpredictably trigger rapid calcium release into the myoplasm, followed by the development of muscle contracture, rigidity, and increased muscle metabolic activity. This process can cause core body temperature to rise vigorously at a rate of 1°C every 5 minutes.
|
Table 89.8A Scoring Rules for the Malignant Hyperthermia (MH) Clinical Grading Scale |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
The administration of certain neuroleptic drugs may induce a similar elevation of body core temperature. A common pathophysiology of NMS and MH has been suggested (81,82). This suggestion is based mainly on three points: (i) NMS and MH have clinical features in common, such as hyperthermia, rigidity, an elevated creatine kinase concentration, and a mortality rate of 10% to 30%; (ii) sodium dantrolene has been used successfully in both syndromes; and (iii) abnormal findings have been observed in in vitro contractility tests in patients with either of the syndromes. Caroff et al. (83) suggested that patients with a genetic predisposition for MH might also be at risk for developing NMS. However, Adnet et al. (84) reported that abnormal sarcolemmal calcium permeability was not shared in the pathogenesis of these disorders. The mechanism of hyperthermia and muscle rigidity is not yet defined, but two major theories have been postulated, which are central dopamine receptor blockade and the direct toxic effect of skeletal muscle induced by neuroleptics. Hypothalamic thermoregulation involves noradrenergic, serotoninergic, cholinergic, and central dopaminergic pathways (85). Dopamine plays a role in central thermoregulation in mammals. A dopamine injection into the hypothalamus causes a reduction in core temperature (86). Since neuroleptics block dopamine receptors, the hyperthermia associated with NMS may result from a blockade of hypothalamic dopamine sites. In addition, the blockade of dopamine receptors in the corpus striatum is thought to cause muscular rigidity and heat generation. Muscle contracture has been induced in vitro by chlorpromazine (87), which is reported to influence calcium ion transport across the sarcoplasmic reticulum and the contractile system (88). However, other studies that do not support the mechanism have also been reported (83,84).
Clinical Syndrome
MH may occur shortly after induction of anesthesia, at any time during the administration of anesthetics, or postoperatively. Trismus is the initial event in 50% of patients, and other early signs are tachycardia and hypercapnia due to increased metabolism (89). These are followed by whole-body rigidity and a marked increase in core body temperature. Trismus may occur in up to 1% of normal patients, and it has been also reported that fewer than 50% of patients prove to be susceptible to MH by muscle testing (90). Tachypnea is obvious when muscle relaxants are not administered. Sympathetic system overactivity produces tachycardia, hypertension, and mottled cyanosis. These symptoms precede hyperthermia, hyperkalemia, hypercalcemia, and lactic acidosis. Capnography may provide an early warning, since carbon dioxide production is remarkably increased while MH is in progress (91). Core body temperature can rise at a rate of 1°C every 5 minutes when hyperthermia occurs. Hypertension may be rapidly followed by hypotension as cardiac depression occurs. Anesthesia should be aborted if these signs appear or if MH is suspected. Laboratory evaluation reveals increased serum myoglobin, creatine kinase (CK, greater than 20,000 U/L), lactate dehydrogenase, and aldolase levels. Dark urine reflects myoglobinemia and myoglobinuria. However, elevation of both myoglobin and CK levels can be observed in some normal patients after succinylcholine administration without MH. The recent most significant study on a clinical grading scale for the prediction of MH was reported and is summarized in Table 89.8A and B (89).
|
Table 89.8B Clinical Indicators for use in Determining the Malignant Hyperthermia (MH) Raw score |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
NMS should be suspected in patients given any neuroleptic drugs who subsequently develop signs of muscular rigidity, dystonia, or unexplained catatonic behavior, followed by hyperpyrexia. Other symptoms include unstable blood pressure, confusion, coma, and delirium. Although laboratory data may vary, a raised CK may be observed in patients who develop rhabdomyolysis. Some authors incline to make a diagnosis of NMS if certain signs are present. Levenson (92), for example, suggested that the presence of all three major signs, or two major and four minor signs (Table 89.9), indicates a high probability of NMS. These criteria are commonly used in clinical research studies (83,84).
|
Table 89.9 Criteria for Guidance in the Diagnosis of Neuroleptic Malignant Syndrome |
||||||
|
Complications
Complications arising from MH and NMS are in general parallel to those of heat stroke syndrome, but complications associated with MH may be more severe because of extreme elevation of temperature. Rhabdomyolysis and hepatic necrosis may be fulminant, and DIC is more common (56). Renal failure is seen almost exclusively in patients with severe rhabdomyolysis. Ventricular fibrillation can occur, and cerebral edema with seizures is uncommon but may be seen. Patients with NMS are at risk for aspiration pneumonia because of dystonia and the inability to handle secretions (93).
Therapeutic Approach
Successful treatment of MH and NMS depend on early clinical recognition and prompt withdrawal of the suspected drugs. In MH, discontinuation alone is effective if the syndrome is not well established (56). NMS may be similarly aborted with discontinuation of the drugs. It may take 5 to 7 days to return to the patient's baseline (89) because neuroleptics cannot be removed by dialysis and blood concentrations decline slowly. General symptomatic treatment, such as hydration, nutrition, and reduction of fever, is essential.
Dantrolene should be administered emergently to prevent further release of calcium from the sarcoplasmic reticulum. The dose is 2 mg/kg intravenously every 5 minutes to a total dose of 10 mg/kg until the episode terminates (94). Dantrolene also decreases temperature in NMS and thyroid storm.
Acidosis should be treated aggressively with intravenous administration of bicarbonate, 2 to 4 mEq/kg. Hyperkalemia should be treated with insulin and glucose infusion, and diuresis.
Fever should be controlled by iced fluids, surface cooling, and cooling of body cavities with sterile iced fluids. Cold dialysis and cardiopulmonary bypass may also be applicable if other measures fail.
Mannitol infusion—0.5 g/kg—with or without furosemide should be used to establish a diuresis and prevent the onset of acute renal failure from myoglobinuria.
Further therapy is guided by blood gases, electrolytes, temperature, arrhythmia, muscle tone, and urinary output. Blood chemical analyses include electrolytes, CK concentrations, liver enzymes, blood urea nitrogen, lactate, glucose, serum hemoglobin and myoglobin, and urine hemoglobin and myoglobin. Coagulation studies also should be done.
Summary
In general, when significant deviation from normal body temperature exists, prompt diagnosis, treatment, and normalization of the temperature alteration are required immediately, followed by careful review of each patient's condition. Therapeutic approaches vary from conservative to invasive methods, and thus risk–benefit balance always should be taken into consideration for a better outcome.
References
1. Moss J. Accidental severe hypothermia. Surg Gynecol Obstet. 1986;162:501.
2. Larach MG. Accidental hypothermia. Lancet. 1995;345:493.
3. Danzl DF, Pozos RS. Accidental hypothermia. N Engl J Med. 1994;331:1756.
4. Maclean D, Emslie-Smith D. Accidental hypothermia. Edinburgh, Scotland: Blackwell Scientific Publications; 1977.
5. Megarbane B, Axler O, Chary I, et al. Hypothermia with indoor occurrence is associated with a worse outcome. Intensive Care Med. 2000;26:1843–1849.
6. Woodhouse P, Keatinge WR, Coleshaw SR. Factors associated with hypothermia in patients admitted to a group of inner city hospitals. Lancet. 1989;2:1201–1205.
7. Fox RH, Brooke OG, Collins JC, et al. Measurement of deep body temperature from the urine. Clin Sci Mol Med. 1975;48:1–7.
8. Miller JW, Danzl DF, Thomas DM. Urban accidental hypothermia: 135 cases. Ann Emerg Med. 1980;9:456–461.
9. Koutsavlis AT, Kosatsky T. Environmental-temperature injury in a Canadian metropolis. J Environ Health. 2003;66:40–45.
10. Clemmer TP, Fisher CJ Jr, Bone RC, et al. Hypothermia in the sepsis syndrome and clinical outcome. The Methylprednisolone Severe Sepsis Study Group. Crit Care Med. 1992;20:1395–1401.
11. Solomon A, Barish RA, Browne B, et al. The electrocardiographic features of hypothermia. J Emerg Med. 1989;7:169–173.
12. Ehrmantraut WR, Fazekas JF, Ticktin HE. Cerebral hemodynamics and metabolism in accidental hypothermia. AMA Arch Intern Med. 1957;99:57–59.
13. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg. 1990;160:515–518.
14. Ferraro FJ Jr, Spillert CR, Swan KG, et al. Cold-induced hypercoagulability in vitro: a trauma connection? Am Surg. 1992;58:355–357.
15. Rohrer MJ, Natale AM. Effect of hypothermia on the coagulation cascade. Crit Care Med. 1992;20:1402–1405.
16. Patt A, McCroskey BL, Moore EE. Hypothermia-induced coagulopathies in trauma. Surg Clin North Am. 1988;68:775–785.
17. Kashuk JL, Moore EE, Millikan JS, et al. Major abdominal vascular trauma–a unified approach. J Trauma. 1982;22:672–679.
18. Cosgriff N, Moore EE, Sauaia A, et al. Predicting life-threatening coagulopathy in the massively transfused trauma patient: hypothermia and acidoses revisited. J Trauma. 1997;42:857–861.
19. Cupples WA, Fox GR, Hayward JS. Effect of cold water immersion and its combination with alcohol intoxication on urine flow rate of man. Can J Physiol Pharmacol. 1980;58:319–321.
20. American Heart Association. Hypothermia. 2005 American Heart Association (AHA) guidelines for cardiopulmonary resuscitation (CPR) and emergency cardiovascular care (ECC). Circulation. 2005;112:IV136–IV138.
21. Ledingham IM, Mone JG. Treatment of accidental hypothermia: a prospective clinical study. Br Med J. 1980;280:1102–1105.
22. Moss JF, Haklin M, Southwick HW, et al. A model for the treatment of accidental severe hypothermia. J Trauma. 1986;26:68–74.
23. Gregory JS, Bergstein JM, Aprahamian C. Comparison of three methods of rewarming from hypothermia: advantages of extracorporeal blood warming. J Trauma. 1991;31:1247–1251.
24. Wong PS, Pugsley WB. Partial cardiopulmonary bypass for the treatment of profound accidental hypothermic circulatory collapse. J R Soc Med. 1992;85:640.
25. Gentilello LM, Cobean RA, Offner PJ, et al. Continuous arteriovenous rewarming: rapid reversal of hypothermia in critically ill patients. J Trauma. 1992;32:316–325.
26. Vretenar DF, Urschel JD, Parrott JC, et al. Cardiopulmonary bypass resuscitation for accidental hypothermia. Ann Thorac Surg. 1994;58:895–898.
27. Antretter H, Dapunt OE, Mueller LC. Portable cardiopulmonary bypass: resuscitation from prolonged ice-water submersion and asystole. Ann Thorac Surg. 1994;58:1786–1787.
28. Mair P, Schwarz B, Komberger E, et al. Case 5-1997. Successful resuscitation of a patient with severe accidental hypothermia and prolonged cardiocirculatory arrest using cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 1997;11:901–904.
29. Dobson JA, Burgess JJ. Resuscitation of severe hypothermia by extracorporeal rewarming in a child. J Trauma. 1996;40:483–485.
30. Roeggla G, Wagner A, Roeggla M, et al. Immediate use of cardiopulmonary bypass in patients with severe accidental hypothermia in the emergency department. Eur J Emerg Med. 1994;1:155.
31. Mayor Pleines AF, Guyot E, Yersin B. Accidental hypothermia: an extreme case of successful resuscitation [in German]. Schweiz Rundsch Med Prax. 2006;95:1075–1079.
32. Duthie DJ. Heat-related illness. Lancet. 1998;352:1329–1330.
33. Bouchama A. Heatstroke: a new look at an ancient disease. Intensive Care Med. 1995;21:623–625.
34. Shahid MS, Hatle L, Mansour H, et al. Echocardiographic and Doppler study of patients with heatstroke and heat exhaustion. Int J Card Imaging. 1999;15:279–285.
35. Porter AM. Heat illness and soldiers. Mil Med. 1993;158:606–609.
36. Knochel JP. Exertional heat stroke—pathophysiology of heat stroke. In: Hopkins PM, Ellis FR, eds. Hyperthermic and Hypermetabolic Disorders. Cambridge, UK: Cambridge University Press; 1996:42–46.
37. Alzeer AH, Al-Arifi A, Warsy AS, et al. Nitric oxide production is enhanced in patients with heat stroke. Intensive Care Med. 1999;25:58–62.
38. Dematte JE, O'Mara K, Buescher J, et al. Near-fatal heat stroke during the 1995 heat wave in Chicago. Ann Intern Med. 1998;129:173–181.
39. Howorth PJN. The biochemistry of heat illness. J R Army Med Corps. 1995;141:40–41.
40. Knochel JP. Environmental heat illness. An eclectic review. Arch Intern Med. 1974;133:841–864.
41. Hart GR, Anderson RJ, Crumpler CP, et al. Epidemic classical heat stroke: clinical characteristics and course of 28 patients. Medicine (Baltimore). 1982;61:189–197.
42. O'Donnell TF. Acute heat stroke. Epidemiologic, biochemical, renal, and coagulation studies. JAMA. 1975;234:824–828.
43. Tucker LE, Stanford J, Graves B, et al. Classical heatstroke: clinical and laboratory assessment. South Med J. 1985;78:20–25.
44. Hassanein T, Razack A, Gavaler JS, et al. Heatstroke: its clinical and pathological presentation, with particular attention to the liver. Am J Gastroenterol. 1992;87:1382–1389.
45. Yu F, Lu K, Lin S, et al. Energy metabolism in exertional heat stroke with acute renal failure. Nephrol Dial Transplant. 1997;2087–2092.
46. Giercksky T, Boberg KM, Farstad IN, et al. Severe liver failure in exertional heat stroke. Scand J Gastroenterol. 1999;34:824–827.
47. Saissy JM. Liver transplantation in a case of fulminant liver failure after exertion. Intensive Care Med. 1996;22:831.
48. Akhtar MJ, Al-Nozha M, Al-Harthi S, et al. Electrocardiographic abnormalities in patients with heat stroke. Chest. 1993;104:411–414.
49. El Kassimi FA, Al-Mashhadani SA, Akhtar J. Adult respiratory distress syndrome and disseminated intravascular coagulation complicating heat stroke. Chest. 1986;90:571–574.
50. Dahmash NS, Al-Harthi SS, Akhtar J. Invasive evaluation of patients with heat stroke. Chest. 1993;103:1210–1214.
51. Gonzalez-Alonso J, Mora-Rodriguez R, Below PR, et al. Dehydration markedly impairs cardiovascular function in hyperthermic endurance athletes during exercise. J Appl Physiol. 1997;82:1229–1236.
52. Bernard S, Buist M, Monteiro O, et al. Induced hypothermia using large volume, ice-cold intravenous fluid in comatose survivors of out-of-hospital cardiac arrest: a preliminary report. Resuscitation. 2003;56:9–13.
53. Denborough MA, Lovell RR. Anaesthetic deaths in a family. Lancet. 1960;2:45.
54. Denborough MA, Forster JF, Lovell RR, et al. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395–396.
55. Delay J, Pichot P, Lemperiere T, et al. A non-phenothiazine and non-reserpine major neuroleptic, haloperidol, in the treatment of psychoses. Ann Med Psychol. 1960;118:145–152.
56. Urwyler A, Censier K, Kaufmann MA, et al. Genetic effects on the variability of the halothane and caffeine muscle contracture tests. Anesthesiology. 1994;80:1287–1295.
57. Gronert GA. Malignant hyperthermia. Anesthesiology. 1980;53:395–423.
58. Neuroleptic malignant syndrome. Lancet. 1984;1:545–546.
59. Smego RA, Durack DT. The neuroleptic malignant syndrome. Arch Intern Med. 1982;142:1183–1185.
60. Morris HH 3rd, McCormick WF, Reinarz JA. Neuroleptic malignant syndrome. Arch Neurol. 1980;37:462–463.
61. Guze BH, Baxter LR. Current concepts. Neuroleptic malignant syndrome. N Engl J Med. 1985;313:163–166.
62. Kemperman CJ. Zuclopenthixol-induced neuroleptic malignant syndrome at rechallenge and its extrapyramidal effects. Br J Psychiatry. 1989;154:562–563.
63. van Maldegem BT, Smit LM, Touw DJ, et al. Neuroleptic malignant syndrome in a 4-year-old girl associated with alimemazine. Euro J Pediatr. 2002;161:259–261.
64. Webster P, Wijeratne C. Risperidone-induced neuroleptic malignant syndrome. Lancet. 1994;344:1228–1229.
65. Heinemann F, Assion HJ, Hermes G, et al. Paroxetine-induced neuroleptic malignant syndrome [in German]. Nervenarzt. 1997;68:664–666.
66. Tegazzin V, Scutari E, Treves S, et al. Chlorocresol, an additive to commercial succinylcholine, induces contracture of human malignant hyperthermia-susceptible muscles via activation of the ryanodine receptor Ca2+ channel. Anesthesiology. 1996;84:1380–1385.
67. Rosenberg H, Reed S. In vitro contracture tests for susceptibility to malignant hyperthermia. Anesth Analg. 1983;62:415–420.
68. Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64:700–704.
69. Ording H. Investigation of malignant hyperthermia susceptibility in Denmark. Dan Med Bull. 1996;43:111–125.
70. Halliday NJ. Malignant hyperthermia. J Craniofac Surg. 2003;14:800–802.
71. Gronert GA, Pessah IN, Muldoon SM, et al. Malignant hyperthermia. In: Miller RD, ed. Miller's Anesthesia. 6th ed. Philadelphia, PA: 2005:1169.
72. Gelenberg AJ, Bellinghausen B, Wojcik JD, et al. A prospective survey of neuroleptic malignant syndrome in a short-term psychiatric hospital. Am J Psychiatry. 1988;145:517–518.
73. Hermesh H, Aizenberg D, Lapidot M, et al. Risk of malignant hyperthermia among patients with neuroleptic malignant syndrome and their families. Am J Psychiatry. 1988;145:1431–1434.
74. Hermesh H, Aizenberg D, Weizman A, et al. Risk for definite neuroleptic malignant syndrome. A prospective study in 223 consecutive in-patients. Br J Psychiatry. 1992;161:254–257.
75. Adnet P, Lestavel P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth. 2000;85:129–135.
76. Fletcher JE, Tripolitis L, Hubert M, et al. Genotype and phenotype relationships for mutations in the ryanodine receptor in patients referred for diagnosis of malignant hyperthermia. Br J Anaesth. 1995;75:307–310.
77. Wallace AJ, Wooldridge W, Kingston HM, et al. Malignant hyperthermia–a large kindred linked to the RYR1 gene. Anaesthesia. 1996;51:16–23.
78. Serfas KD, Bose D, Patel L, et al. Comparison of the segregation of the RYR1 C1840T mutation with segregation of the caffeine/halothane contracture test results for malignant hyperthermia susceptibility in a large Manitoba Mennonite family. Anesthesiology. 1996;84:322–329.
79. McWilliams S, Nelson T, Sudo RT, et al. Novel skeletal muscle ryanodine receptor mutation in a large Brazilian family with malignant hyperthermia. Clin Genet. 2002;62:80–83.
80. Robinson RL, Brooks C, Brown SL, et al. RYR1 mutations causing central core disease are associated with more severe malignant hyperthermia in vitro contracture test phenotypes. Hum Mutat. 2002;20:88–97.
81. Denborough MA, Collins SP, Hopkinson KC. Rhabdomyolysis and malignant hyperpyrexia. Br Med J. 1984;288:1878.
82. Tollefson G. A case of neuroleptic malignant syndrome: in vitro muscle comparison with malignant hyperthermia. J Clin Psychopharmacol. 1982;2:266–270.
83. Caroff SN, Rosenberg H, Fletcher JE, et al. Malignant hyperthermia susceptibility in neuroleptic malignant syndrome. Anesthesiology. 1987;67:20–25.
84. Adnet PJ, Krivosic-Horber RM, Adamantidis MM, et al. The association between the neuroleptic malignant syndrome and malignant hyperthermia. Acta Anaesthesiol Scand. 1989;33:676–680.
85. Bligh J, Cottle WH, Maskrey M. Influence of ambient temperature on the thermoregulatory responses to 5-hydroxytryptamine, noradrenaline and acetylcholine injected into the lateral cerebral ventricles of sheep, goats and rabbits. J Physiol. 1971;212:377–392.
86. Cox B, Kerwin R, Lee TF. Dopamine receptors in the central thermoregulatory pathways of the rat. J Physiol. 1978;282:471–483.
87. Kelkar VV, Doctor RB, Jindal MN. Chlorpromazine-induced contracture of frog rectus abdominis muscle. Pharmacology. 1974;12:32–38.
88. Takagi A. Chlorpromazine and skeletal muscle: a study of skinned single fibers of the guinea pig. Exp Neurol. 1981;73:477–486.
89. Larach MG, Localio AR, Allen GC, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80:771–779.
90. O'Flynn RP, Shutack JG, Rosenberg H, et al. Masseter muscle rigidity and malignant hyperthermia susceptibility in pediatric patients. An update on management and diagnosis. Anesthesiology. 1994;80:1228–1233.
91. Meier-Hellman A, Romer M, Hannemann L, et al. Early recognition of malignant hyperthermia using capnometry. Anaesthesist. 1990;39:41–43.
92. Levenson JL. Neuroleptic malignant syndrome. Am J Psychiatry. 1985;142:1137–1145.
93. Wedel DJ, Quinlan JG, Iaizzo PA. Clinical effects of intravenously administered dantrolene. Mayo Clin Proc. 1995;70:241–246.
94. Brandom BW, Larach MG, North American MH registry. Reassessment of the safety and efficacy of dantrolene. Anesthesiology. 2002;97:A1199.