Edith A. Nutescu and Stuart T. Haines
LEARNING OBJECTIVES
Upon completion of the chapter, the reader will be able to:
1. Identify risk factors and signs and symptoms of deep vein thrombosis (DVT) and pulmonary embolism (PE).
2. Describe the processes of hemostasis and thrombosis, including the role of the vascular endothelium, platelets, coagulation cascade, and thrombolytic proteins.
3. Determine a patient’s relative risk (low, moderate, or high) of developing venous thrombosis.
4. Formulate an appropriate prevention strategy for a patient at risk for DVT.
5. State at least two potential advantages of the low-molecular weight heparins (LMWHs) and fondaparinux over unfractionated heparin (UFH).
6. Select and interpret laboratory test(s) to monitor antithrombotic drugs.
7. Identify factors that place a patient at high-risk of bleeding while receiving antithrombotic drugs.
8. Identify warfarin drug-drug and drug-food interactions.
9. Manage a patient with an elevated International Normalized Ratio (INR) with or without bleeding.
10. Formulate an appropriate treatment plan for a patient who develops a DVT or PE, and develop a comprehensive education plan for a patient who is receiving an antithrombotic drug.
KEY CONCEPTS
![]() Antithrombotic therapies require meticulous and systematic monitoring, as well as ongoing patient education. Well-organized anticoagulation management services improve the quality of patient care and reduce the overall cost.
Antithrombotic therapies require meticulous and systematic monitoring, as well as ongoing patient education. Well-organized anticoagulation management services improve the quality of patient care and reduce the overall cost.
![]() The risk of venous thromboembolism (VTE) is related to several identifiable factors including age, prior history of VTE, major surgery (particularly orthopedic procedures of the lower extremities), trauma, malignancy, pregnancy, estrogen use, and hypercoagulable states. These risks are additive.
The risk of venous thromboembolism (VTE) is related to several identifiable factors including age, prior history of VTE, major surgery (particularly orthopedic procedures of the lower extremities), trauma, malignancy, pregnancy, estrogen use, and hypercoagulable states. These risks are additive.
![]() The diagnosis of VTE must be confirmed by objective testing.
The diagnosis of VTE must be confirmed by objective testing.
![]() At the time of hospital admission, all patients should be evaluated for their risk of VTE, and strategies to prevent VTE appropriate for each patient’s level of risk should be routinely employed. Prophylaxis should be continued throughout the period of risk.
At the time of hospital admission, all patients should be evaluated for their risk of VTE, and strategies to prevent VTE appropriate for each patient’s level of risk should be routinely employed. Prophylaxis should be continued throughout the period of risk.
![]() In the absence of contraindications, the treatment of VTE should initially include a rapid-acting anticoagulant (e.g., unfractional heparin [UFH], low-molecular weight heparin [LMWH], or fondaparinux) overlapped with warfarin for at least 5 days and until the patient’s International Normalized Ratio (INR) is greater than 2 and stable. Anticoagulation therapy should be continued for a minimum of 3 months. However, the duration of anticoagulation therapy should be based on the patient’s risk of VTE recurrence and major bleeding.
In the absence of contraindications, the treatment of VTE should initially include a rapid-acting anticoagulant (e.g., unfractional heparin [UFH], low-molecular weight heparin [LMWH], or fondaparinux) overlapped with warfarin for at least 5 days and until the patient’s International Normalized Ratio (INR) is greater than 2 and stable. Anticoagulation therapy should be continued for a minimum of 3 months. However, the duration of anticoagulation therapy should be based on the patient’s risk of VTE recurrence and major bleeding.
![]() Bleeding is the most common adverse effect associated with antithrombotic drugs. A patient’s risk of major hemorrhage is related to the intensity and stability of therapy, age, concurrent drug use, history of GI bleeding, risk of falls or trauma, and recent surgery.
Bleeding is the most common adverse effect associated with antithrombotic drugs. A patient’s risk of major hemorrhage is related to the intensity and stability of therapy, age, concurrent drug use, history of GI bleeding, risk of falls or trauma, and recent surgery.
![]() Most patients with an uncomplicated deep vein thrombosis (DVT) can be managed safely at home.
Most patients with an uncomplicated deep vein thrombosis (DVT) can be managed safely at home.
![]() Warfarin is prone to numerous clinically important drug-drug and drug-food interactions.
Warfarin is prone to numerous clinically important drug-drug and drug-food interactions.
INTRODUCTION
Venous thromboembolism (VTE) is one of the most common cardiovascular disorders in the United States. VTE is manifested as deep vein thrombosis (DVT) and pulmonary embolism (PE) resulting from thrombus formation in the venous circulation (Fig. 10–1).1 It is often provoked by prolonged immobility and vascular injury and is most frequently seen in patients who have been hospitalized for a serious medical illness, trauma, or major surgery. VTE can also occur with little or no provocation in patients who have an underlying hypercoagulable disorder.

FIGURE 10–1. Venous circulation. (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:332.)
While VTE may initially cause few or no symptoms, the first overt manifestation of the disease may be sudden death.2 Death from PE can occur within minutes, before effective treatment can be given. In addition to the symptoms produced by the acute event, the long-term sequelae of VTE such as the post-thrombotic syndrome (PTS; a complication of VTE occurring due to damage to the vein caused by a blood clot and that leads to development of symptomatic venous insufficiency such as chronic lower extremity swelling, pain, tenderness, skin discoloration, and ulceration) and recurrent thromboembolic events cause long-term pain and suffering.
The treatment of VTE is fraught with substantial risks.3 ![]() Antithrombotic drugs require precise dosing and meticulous monitoring, as well as ongoing patient education.4,5 Well-organized anticoagulation management services improve the quality of patient care and reduce the overall cost. A systematic approach to drug therapy management substantially reduces these risks, but bleeding remains a common and serious complication.5 Therefore, preventing VTE is paramount to improving outcomes. When VTE is suspected, a rapid and accurate diagnosis is critical to making appropriate treatment decisions. The optimal use of antithrombotic drugs requires not only an in-depth knowledge of their pharmacology and pharmacokinetic properties, but also a comprehensive approach to patient management.6
Antithrombotic drugs require precise dosing and meticulous monitoring, as well as ongoing patient education.4,5 Well-organized anticoagulation management services improve the quality of patient care and reduce the overall cost. A systematic approach to drug therapy management substantially reduces these risks, but bleeding remains a common and serious complication.5 Therefore, preventing VTE is paramount to improving outcomes. When VTE is suspected, a rapid and accurate diagnosis is critical to making appropriate treatment decisions. The optimal use of antithrombotic drugs requires not only an in-depth knowledge of their pharmacology and pharmacokinetic properties, but also a comprehensive approach to patient management.6
EPIDEMIOLOGY AND ETIOLOGY
The true incidence of VTE in the general population is unknown because many patients, perhaps more than 50%, have no overt symptoms or go undiagnosed.7 An estimated 2 million people in the United States develop VTE each year; 600,000 are hospitalized and 60,000 die. The estimated annual direct medical costs of managing the disease are well over $1 billion. The incidence of VTE nearly doubles in each decade of life over the age of 50 and is slightly higher in men. As the population ages, the total number of cases of DVT and PE continues to rise.2
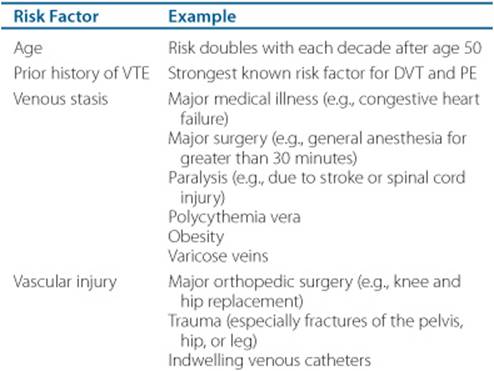
![]() The risk of VTE is related to several factors including age, prior history of VTE, major surgery (particularly orthopedic procedures of the lower extremities), trauma, malignancy, pregnancy, estrogen use, and hypercoagulable states (Table 10–1).2 VTE risk factors can be categorized in one of the three elements ofVirchow’s triad: stasis in blood flow, vascular endothelial injury, and inherited or acquired changes in blood constituents that cause hypercoagulation states. These risk factors are additive and some can be easily identified in clinical practice. A prior history of venous thrombosis is perhaps the strongest risk factor for recurrent VTE, presumably because of the destruction of venous valves and obstruction of blood flow caused by the initial event. Rapid blood flow has an inhibitory effect on thrombus formation, but a slow rate of flow reduces the clearance of activated clotting factors in the zone of injury and slows the influx of regulatory substances. Stasis tips the delicate balance of procoagulation and anticoagulation in favor of thrombogenesis. The rate of blood flow in the venous circulation, particularly in the deep veins of the lower extremities, is relatively slow. Valves in the deep veins of the legs, as well as contraction of the calf and thigh muscles, facilitate the flow of blood back to the heart and lungs. Damage to the venous valves and periods of prolonged immobility result in venous stasis. Vessel obstruction, either from a thrombus or external compression, promotes clot propagation. Numerous medical conditions and surgical procedures are associated with reduced venous blood flow and increase the risk of VTE (Table 10–1). Greater than normal blood viscosity, seen in myeloproliferative disorders like polycythemia vera, for example, may also contribute to slowed blood flow and thrombus formation.
The risk of VTE is related to several factors including age, prior history of VTE, major surgery (particularly orthopedic procedures of the lower extremities), trauma, malignancy, pregnancy, estrogen use, and hypercoagulable states (Table 10–1).2 VTE risk factors can be categorized in one of the three elements ofVirchow’s triad: stasis in blood flow, vascular endothelial injury, and inherited or acquired changes in blood constituents that cause hypercoagulation states. These risk factors are additive and some can be easily identified in clinical practice. A prior history of venous thrombosis is perhaps the strongest risk factor for recurrent VTE, presumably because of the destruction of venous valves and obstruction of blood flow caused by the initial event. Rapid blood flow has an inhibitory effect on thrombus formation, but a slow rate of flow reduces the clearance of activated clotting factors in the zone of injury and slows the influx of regulatory substances. Stasis tips the delicate balance of procoagulation and anticoagulation in favor of thrombogenesis. The rate of blood flow in the venous circulation, particularly in the deep veins of the lower extremities, is relatively slow. Valves in the deep veins of the legs, as well as contraction of the calf and thigh muscles, facilitate the flow of blood back to the heart and lungs. Damage to the venous valves and periods of prolonged immobility result in venous stasis. Vessel obstruction, either from a thrombus or external compression, promotes clot propagation. Numerous medical conditions and surgical procedures are associated with reduced venous blood flow and increase the risk of VTE (Table 10–1). Greater than normal blood viscosity, seen in myeloproliferative disorders like polycythemia vera, for example, may also contribute to slowed blood flow and thrombus formation.
Table 10–1 Risk Factors for VTE

A growing list of hereditary deficiencies, gene mutations, and acquired diseases have been linked to hypercoagulability (Table 10–1).8 Activated protein C resistance is the most common genetic disorder of hypercoagulability, found in nearly 5% of individuals of northern European descent and in as many as 40% of those who suffer an idiopathic DVT. Although these patients have normal plasma concentrations of protein C, they have a mutation on factor V that renders it resistant to degradation by activated protein C. This mutation is known as factor V Leiden, named after the city of Leiden, Holland, where the defect was initially reported. The prothrombin gene 20210A mutation is also a relatively common defect, occurring in as many as 3% of healthy individuals of southern European descent and 16% of those with an idiopathic DVT. Although less common, inherited deficiencies of the natural anticoagulants protein C, protein S, and antithrombin (AT) place patients at a high lifetime risk for VTE. Conversely, high concentrations of factors VIII, IX, and XI also increase the risk of VTE. Some patients have multiple genetic defects.
Acquired disorders of hypercoagulability include malignancy, antiphospholipid antibodies, estrogen use, and pregnancy.2 The strong link between cancer and thrombosis has been recognized since the late 1800s.9 Tumor cells secrete a number of procoagulant substances that activate the clotting cascade. Furthermore, patients with cancer often have suppressed levels of protein C, protein S, and AT. Antiphospholipid antibodies, commonly found in patients with autoimmune disorders such as systemic lupus erythematosus and inflammatory bowel disease, can cause venous and arterial thrombosis.8 The antiphospholipid antibody syndrome is associated with repeated pregnancy loss. The precise mechanism by which these antibodies provoke thrombosis is unclear, but they activate the coagulation cascade and platelets, as well as inhibit the anticoagulant activity of proteins C and S. Estrogen-containing contraceptives, estrogen replacement therapy, and many of the selective estrogen receptor modulators (SERMs) increase the risk of venous thrombosis.2,10,11 While the mechanisms are not clearly understood, estrogens increase serum clotting factor concentrations and induce activated protein C resistance. Increased serum estrogen concentrations may explain, in part, the increased risk of VTE during pregnancy and the postpartum period.11
PATHOPHYSIOLOGY
Hemostasis, the arrest of bleeding following vascular injury, is essential to life.12 Within the vascular system, blood remains in a fluid state, transporting oxygen, nutrients, plasma proteins, and waste. When a vessel is injured, a dynamic interplay between thrombogenic (activating) and antithrombotic (inhibiting) forces result in the local formation of a hemostatic plug that seals the vessel wall and prevents further blood loss (Figs. 10-2, 10-3, and 10-4). A disruption of this delicate system of checks and balances may lead to inappropriate clot formation within the blood vessel that can obstruct blood flow or embolize to a distant vascular bed.
Under normal circumstances, the endothelial cells that line the inside of blood vessels maintain blood flow by producing a number of substances that inhibit platelet adherence, prevent the activation of the coagulation cascade, and facilitate fibrinolysis.12 Vascular injury exposes the subendothelium (Fig. 10–3). Platelets readily adhere to the subendothelium, using glycoprotein (GP) lb receptors found on their surfaces and facilitated by von Willebrand’s factor (vWF). This causes platelets to become activated, releasing a number of procoagulant substances that stimulate circulating platelets to expose GP lib–IIIa receptors and allow platelets to adhere to one another, resulting in platelet aggregation. The damaged vascular tissue releases tissue factor which activates the extrinsic pathway of the coagulation cascade (Fig. 10–4).
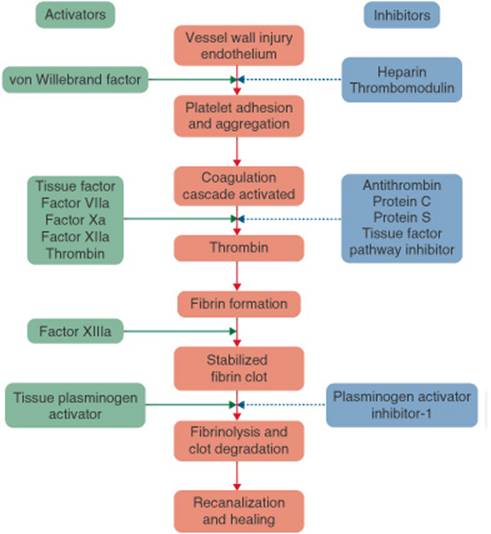
FIGURE 10–2. Hemostasis and thrombosis. (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:334.)
The clotting cascade is a stepwise series of enzymatic reactions that result in the formation of a fibrin mesh.12 Clotting factors circulate in the blood in inactive forms. Once a precursor is activated by specific stimuli, it activates the next precursor in the sequence. The final steps in the cascade are the conversion of prothrombin to thrombin and fibrinogen to fibrin. Thrombin plays a key role in the coagulation cascade; it is responsible not only for the production of fibrin, but also for the conversion of factors V and VIII, creating a positive feedback loop that greatly accelerates the entire cascade. Thrombin also enhances platelet aggregation. Traditionally, the coagulation cascade has been divided into three distinct parts: the intrinsic, the extrinsic, and the common pathways (Fig. 10–4). This artificial division is misleading because there are numerous interactions between the three pathways.
A number of tempering mechanisms control coagulation (Fig. 10–2).12 Without effective self-regulation, the coagulation cascade would proceed unabated until all the clotting factors and platelets are consumed. AT and heparin cofactor II (HCII) are circulating proteins that inhibit thrombin and factor Xa. The intact endothelium adjacent to the damaged tissue actively secretes several antithrombotic substances including heparan sulfate and thrombomodulin. Heparan sulfate exponentially accelerates AT and HCII activity. Protein C and its cofactor, protein S, are vitamin K-dependent anticoagulant proteins made in the liver. Activation of the clotting cascade activates protein C that, in turn, inhibits factor Va and Villa activity. Tissue factor pathway inhibitor (TFPI) inhibits the extrinsic coagulation pathway. When these self-regulatory mechanisms are intact, the formation of the fibrin clot is limited to the zone of tissue injury. However, disruptions in the system often result in inappropriate clot formation.
The fibrinolytic protein plasmin degrades the fibrin mesh into soluble end products collectively known as fibrin split products or fibrin degradation products.13 The fibrinolytic system is also under the control of a series of stimulatory and inhibitory substances. Tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA) convert plasminogen to plasmin. Plasminogen activator inhibitor-1 (PAI-1) inhibits the plasminogen activators and α2-antiplasmin inhibits plasmin activity. Aberrations in the fibrinolytic system have also been linked to hypercoagulability.
CLINICAL PRESENTATION AND DIAGNOSIS
Although a thrombus can form in any part of the venous circulation, the majority begin in the lower extremities. Once formed, a venous thrombus may behave in a combination of ways including: (a) remain asymptomatic, (b) spontaneously lyse, (c) obstruct the venous circulation, (d) propagate into more proximal veins, (e) embolize, and/or (f) slowly incorporate into the endothelial layer of the vessel.14 The majority of patients with VTE never develop symptoms.2 However, even those who initially experience no symptoms may suffer long-term consequences, such as PTS and recurrent VTE. ![]() The symptoms of DVT or PE are nonspecific, and it is extremely difficult to distinguish VTE from other disorders on clinical signs alone.15 Therefore, objective tests are required to confirm or exclude the diagnosis. Patients with DVT frequently present with unilateral leg pain and swelling. Similarly, the PTS, a long-term complication of DVT caused by damage to the venous valves, produces chronic lower extremity swelling, pain, and tenderness that lead to skin discoloration and ulceration. The D-dimer test, a quantitative measure of fibrin breakdown in the serum, is a marker of acute thrombotic activity and may help to distinguish between an acute DVT and the PTS. Symptomatic PE usually produces shortness of breath, tachypnea, and tachycardia.16 Hemoptysisoccurs in less than one-third of patients. The physical exam may reveal diminished breath sounds, crackles, wheezes, or a pleural friction rub during auscultation of the lungs. Cardiovascular collapse, characterized by cyanosis, shock, and oliguria, is an ominous sign.
The symptoms of DVT or PE are nonspecific, and it is extremely difficult to distinguish VTE from other disorders on clinical signs alone.15 Therefore, objective tests are required to confirm or exclude the diagnosis. Patients with DVT frequently present with unilateral leg pain and swelling. Similarly, the PTS, a long-term complication of DVT caused by damage to the venous valves, produces chronic lower extremity swelling, pain, and tenderness that lead to skin discoloration and ulceration. The D-dimer test, a quantitative measure of fibrin breakdown in the serum, is a marker of acute thrombotic activity and may help to distinguish between an acute DVT and the PTS. Symptomatic PE usually produces shortness of breath, tachypnea, and tachycardia.16 Hemoptysisoccurs in less than one-third of patients. The physical exam may reveal diminished breath sounds, crackles, wheezes, or a pleural friction rub during auscultation of the lungs. Cardiovascular collapse, characterized by cyanosis, shock, and oliguria, is an ominous sign.
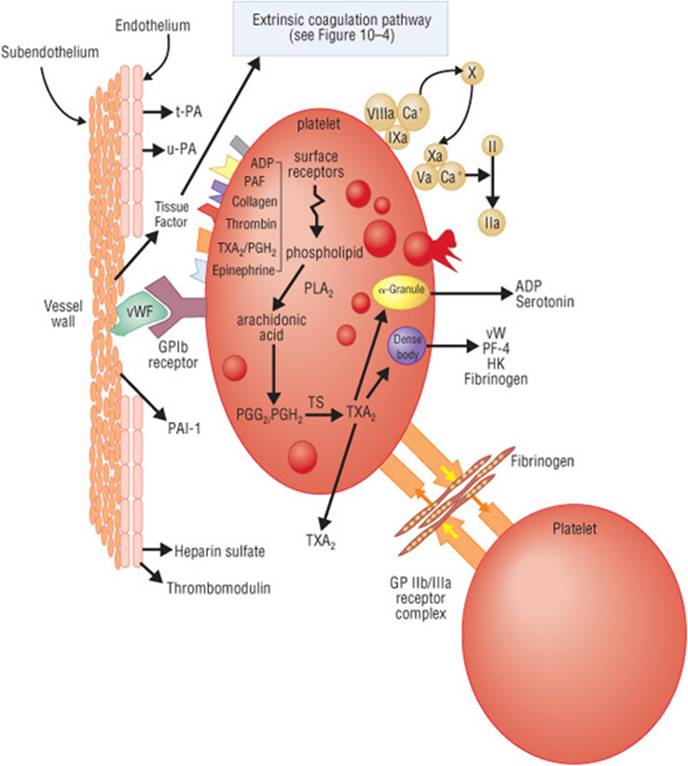
FIGURE 10–3. Vascular injury and thrombosis. (ADR, adenosine diphosphate; GP lb, glycoprotein lb; GP llb/lla, glycoprotein llb/lla; HK, high-molecular weight kininogen; PAF, platelet activating factor-1; PAI-1, plasminogen activator inhibitor; PF-4, platelet factor-4; PGG/PGH, prostaglandins; PLA, phospholipase A; TS, thromboxane synthetase; TXA2, thromboxane A2; t-PA, tissue plasminogen activator; u-PA, urokinase plasminogen activator; vWF, von Willebrand’s factor.) (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, etal., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:335.)
Given that VTE can be debilitating or fatal, it is important to treat it quickly and aggressively.17 On the other hand, because major bleeding induced by antithrombotic drugs can be equally harmful, it is important to avoid treatment when the diagnosis is not reasonably certain. Assessment of the patient’s status should focus on the search for risk factors in the patient’s medical history (Table 10—1).15,16Venous thrombosis is uncommon in the absence of risk factors, and the effects of these risks are additive. Indeed, if a patient has multiple risk factors, VTE should be strongly suspected even when the symptoms are very subtle.
Because radiographic contrast studies are the most accurate and reliable methods for the diagnosis of VTE, they are considered the gold standards in clinical trials.18,19 Contrast venography allows visualization of the entire venous system in the lower extremities and abdomen. Pulmonary angiography allows the visualization of the pulmonary arteries. The diagnosis of VTE can be made if there is a persistent intraluminal filling defect observed on multiple x-ray films. Contrast studies are expensive, invasive procedures that are technically difficult to perform and evaluate. Severely ill patients often are unable to tolerate the procedure, and many develop hypotension and cardiac arrhythmias. Furthermore, the contrast medium is irritating to vessel walls and toxic to the kidneys. For these reasons, noninvasive tests, such as ultrasonography, CT scans, MRI, and ventilation/perfusion (V/Q) scans are frequently used in clinical practice for the initial evaluation of patients with suspected VTE. See the Clinical Presentation and Diagnosis of PE, and Clinical Presentation and Diagnosis of DVT textboxes.
Clinical Presentation and Diagnosis of DVT
General
VTE most commonly develops in patients with identifiable risk factors (Table 10–1) during or following a hospitalization. Many, perhaps the majority of patients, have asymptomatic disease. Patients may die suddenly of PE.
Symptoms
• The patient may complain of leg swelling, pain, warmth, skin discoloration. Symptoms are nonspecific and objective testing must be performed to establish the diagnosis.
Signs
• The patient’s superficial veins may be dilated and a “palpable cord” may be felt in the affected leg.
• The patient may experience unilateral leg edema with measurable difference in leg circumference, erythema, increase in warmth, tenderness with palpation of calf muscles.
• The patient may experience pain in back of the knee in the affected leg when the examiner dorsiflexes the foot while the knee is slightly bent (Homan’s sign).
NOTE: The physical exam signs can be unreliable. Homan’s sign (an increased resistance to foot dorsiflexion without pain or discomfort as criteria for the positive test) can also be unreliable.
Laboratory Tests
• The initial lab evaluation should include CBC with differential, coagulation studies, serum chemistries with renal and liver function, and urinalysis.
• Serum concentrations of D-dimer, a by-product of thrombin generation, will be elevated in an acute event. The patient may have an elevated erythrocyte sedimentation rate (ESR) and WBC.
• The D-dimer test cannot be used alone to prove the presence of a DVT because it can be elevated in many other conditions. A D-dimer level of less than 500 ng/mL (less than 500 mcg/L) with a low clinical probability of DVT is helpful and cost effective in excluding DVT without the need for ultrasound testing.
Diagnostic Tests
• Duplex ultrasonography is the most commonly used test to diagnose DVT. It is a noninvasive test that can measure the rate and direction of blood flow and visualize clot formation in proximal veins of the legs. It cannot reliably detect small blood clots in distal veins. Coupled with a careful clinical assessment, it can rule in or out (include or exclude) the diagnosis in the majority of cases.
• Venography (also known as phlebography) is the gold standard for the diagnosis of DVT. However, it is an invasive test that involves injection of radiopaque contrast dye into a foot vein. It is expensive and can cause anaphylaxis and nephrotoxicity.
PREVENTION
Given that VTE is often clinically silent and potentially fatal, prevention strategies have the greatest potential to improve patient outcomes.2 To rely on the early diagnosis and treatment of VTE is unacceptable because many patients will die before treatment can be initiated. Furthermore, even clinically silent disease is associated with long-term morbidity from the PTS and predisposes the patient to future thromboembolic events. Despite an immense body of literature that overwhelmingly supports the widespread use of pharmacologic and nonpharmacologic strategies to prevent VTE, prophylaxis is underutilized in most hospitals. Even when prophylaxis is given, many patients receive prophylaxis that is less than optimal. Educational programs and computerized clinical decision support systems have been shown to improve the appropriate use of VTE prevention methods.2
The goal of an effective VTE prophylaxis program is to identify all patients at risk, determine each patient’s level of risk, and select and implement regimens that provide sufficient protection for the level of risk.20 ![]() At the time of hospital admission, all patients should be evaluated for their risk of VTE, and strategies to prevent VTE appropriate for each patient’s level of risk should be routinely employed. Prophylaxis should be continued throughout the period of risk. The risk classification criteria and recommended prophylaxis strategies published by the American College of Chest Physicians (ACCP) Conference on Antithrombotic Therapy are widely used in North America (Table 10–2).2 Several pharmacologic and nonpharmacologic methods are effective for preventing VTE, and these can be used alone or in combination. Nonpharmacologic methods improve venous blood flow by mechanical means, while drug therapy prevents thrombus formation by inhibiting the coagulation cascade.
At the time of hospital admission, all patients should be evaluated for their risk of VTE, and strategies to prevent VTE appropriate for each patient’s level of risk should be routinely employed. Prophylaxis should be continued throughout the period of risk. The risk classification criteria and recommended prophylaxis strategies published by the American College of Chest Physicians (ACCP) Conference on Antithrombotic Therapy are widely used in North America (Table 10–2).2 Several pharmacologic and nonpharmacologic methods are effective for preventing VTE, and these can be used alone or in combination. Nonpharmacologic methods improve venous blood flow by mechanical means, while drug therapy prevents thrombus formation by inhibiting the coagulation cascade.

FIGURE 10–4. Coagulation cascade. (AT, antithrombin; HCII, heparin cofactor II; TFPI, tissue factor pathway inhibitor.) (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: Di Piro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th eel. New York: McGraw-Hill; 2008:336.)
Nonpharmacologic Therapy
Ambulation as soon as possible following surgery lowers the incidence of VTE in low-risk patients.2 Walking increases venous blood flow and promotes the flow of natural antithrombotic factors into the lower extremities. Graduated compression stockings (GCS) reduce the incidence of VTE by approximately 60% following general surgery, neurosurgery, and stroke. Compared with anticoagulant drugs, GCS are relatively inexpensive and safe; however, in higher risk patients they are less effective then pharmacologic agents. They are a good choice in low-to moderate-risk patients when pharmacologic interventions are contraindicated. When combined with pharmacologic interventions, GCS have an additive effect. However, some patients are unable to wear compression stockings because of the size or shape of their legs.
Similar to GCS, intermittent pneumatic compression (IPC) devices increase the velocity of blood flow in the lower extremities.2 These devices sequentially inflate a series of cuffs wrapped around the patient’s legs from the ankles to the thighs and then deflate in 1-to 2-minute cycles. IPC has been shown to reduce the risk of VTE by more than 60% following general surgery, neurosurgery, and orthopedic surgery. Although IPC is well tolerated and safe to use in patients who have contraindications to pharmacologic therapies, it does have a few drawbacks: It is more expensive than the use of GCS, it is a relatively cumbersome technique, and some patients may have difficulty sleeping while using it. To be effective, IPC needs to be used throughout the day. In practice, this has been difficult to achieve and special efforts should be made to ensure that the devices are worn and operational for the majority of the day.
Clinical Presentation and Diagnosis of PE
General
PE most commonly develops in patients with risk factors for VTE (Table 10–1) during or following a hospitalization. While many patients will have symptoms of DVT prior to developing a PE, many do not. Patients may die suddenly before effective treatment can be initiated.
Symptoms
• The patient may complain of cough, chest pain, chest tightness, shortness of breath, wheezing, or palpitations.
• The patient may present with hemoptysis (spit or cough up blood).
• The patient may complain of dizziness or lightheadedness.
• Symptoms may be confused for a myocardial infarction or pneumonia, and objective testing must be performed to establish the diagnosis.
Signs
• The patient may have tachypnea (increased respiratory rate) and tachycardia (increased heart rate).
• The patient may appear diaphoretic (sweaty).
• The patient’s neck veins may be distended reflecting increased jugular venous pressure.
• The examiner may hear diminished breath sounds, crackles, wheezes, or pleural friction rub, right ventricular S3, or parasternal lift during auscultation of the lungs.
• In massive PE, the patient may appear cyanotic and hypotensive. In such cases, oxygen saturation by pulse oximetry or arterial blood gas will likely indicate that the patient is hypoxic.
• In the worst cases, the patient may go into circulatory shock and die within minutes.
Laboratory Tests
• Serum concentrations of D-dimer, a by-product of thrombin generation, will be elevated. The patient may have an elevated ESR and WBC count.
• The patient may have elevated serum LDH or AST (SGOT) with normal bilirubin. Serum troponin I and troponin T can be elevated in a large PE.
Diagnostic Tests
• A CT scan is the most commonly used test to diagnose PE but some institutions still use a ventilation/perfusion (V/Q) scan. Spiral CT scans can detect emboli in the pulmonary arteries. A V/Q scan measures the distribution of blood and air flow in the lungs. When there is a large mismatch between blood and air flow in one area of the lung, there is a high probability that the patient has a PE.
• Pulmonary angiography is the gold standard for the diagnosis of PE. However, it is an invasive test that involves injection of radiopaque contrast dye into the pulmonary artery. The test is expensive and associated with a significant risk of mortality.
Table 10–2 Risk Classification and Consensus Guidelines for VTE Prevention

Inferior vena cava (IVC) filters, also known as Greenfield filters, provide short-term protection against PE in very-high-risk patients by preventing the embolization of a thrombus formed in the lower extremities into the pulmonary circulation.2 Insertion of a filter into the IVC is a minimally invasive procedure. Despite the widespread use of IVC filters, there are very limited data regarding their effectiveness and long-term safety. The evidence suggests that IVC filters, particularly in the absence of effective antithrombotic therapy, increase the long-term risk of recurrent DVT. In the only randomized clinical trial examining the short-and long-term effectiveness of the filters in patients with a documented proximal DVT, treatment with IVC filters reduced the risk of PE by more than 75% during the first 12 days following insertion.21 However, this benefit was not sustained during 2 years of follow-up and the long-term risk of recurrent DVT was nearly two-fold higher in those who received a filter. Although IVC filters can reduce the short-term risk of PE in patients at highest risk, they should be reserved for patients in whom other prophylactic strategies cannot be used. To further reduce the long-term risk of VTE in association with IVC filters, pharmacologic prophylaxis is necessary and warfarin therapy should begin as soon as the patient is able to tolerate it.2
Pharmacologic Therapy
Numerous randomized clinical trials have extensively evaluated pharmacologic strategies for VTE prophylaxis.2 Appropriately selected drug therapies can dramatically reduce the incidence of VTE following hip replacement, knee replacement, general surgery, myocardial infarction, and ischemic stroke (Table 10–2). The choice of medication and dose to use for VTE prevention must be based on the patient’s level of risk for thrombosis and bleeding complications, as well as the cost and availability of an adequate drug therapy monitoring system.
The ACCP Conference on Antithrombotic Therapy recommends against the use of aspirin as the primary method of VTE prophylaxis.2 Antiplatelet drugs clearly reduce the risk of coronary artery and cerebrovascular events in patients with arterial disease, but aspirin produces a very modest reduction in VTE following orthopedic surgeries of the lower extremities. The relative contribution of venous stasis in the pathogenesis of venous thrombosis compared with that of platelets in arterial thrombosis likely explains the reason for this difference.
The most extensively studied drugs for the prevention of VTE are unfractionated heparin (UFH), the low-molecular weight heparins (LMWHs; dalteparin, enoxaparin, and tinzaparin), fondaparinux, and warfarin.2 The LMWHs and fondaparinux provide superior protection against VTE when compared to low-dose UFH after hip and knee replacement surgery and in other high-risk populations. Even so, UFH remains an effective, cost-conscious choice for moderate-risk patient populations, provided that it is given in the appropriate dose (Table 10–2). Low-dose UFH (5,000 units every 12 or 8 hours) given subcutaneously (SC) has been shown to significantly reduce the risk of VTE in patients undergoing a wide range of general surgical procedures as well as following a myocardial infarction or stroke. For patients who are hospitalized with an acute medical illness, the available evidence supports the use of UFH (5,000 units every 12 or 8 hours), enoxaparin 40 mg SC daily, dalteparin 5,000 units SC daily, or fondaparinux 2.5 mg SC daily.
For the prevention of VTE following hip and knee replacement surgery, the effectiveness of low-dose UFH is considerably lower. Adjusted-dose UFH therapy provided SC, which requires dose adjustments to maintain the activated partial thromboplastin time (aPTT) at the high end of the normal range, may be used in the highest-risk patient populations. However, adjusted-dose UFH has been studied in only a few, relatively small clinical trials and requires frequent laboratory monitoring. The LMWHs and fondaparinux appear to provide a high degree of protection against VTE in most high-risk populations. The appropriate prophylactic dose for each LMWH product is indication-specific (Table 10–2). There is no evidence that one LMWH is superior to another for the prevention of VTE. Fondaparinux was significantly more effective than enoxaparin in several clinical trials that enrolled patients undergoing high-risk orthopedic procedures, but has not been shown to reduce the incidence of symptomatic PE or mortality, and heightened the risk of bleeding.22 To provide optimal protection, some experts believe that the LMWHs should be initiated prior to surgery.2
Warfarin is another commonly used option for the prevention of VTE following orthopedic surgeries of the lower extremities.2 Warfarin appears to be as effective as the LMWHs for the prevention of symptomatic VTE events in the highest-risk populations. When used to prevent VTE, the dose of warfarin must be adjusted to maintain an International Normalized Ratio (INR) between 2 and 3. Oral administration and low drug cost give warfarin some advantages over the LMWHs and fondaparinux. However, warfarin does not achieve its full antithrombotic effect for several days and requires frequent monitoring and periodic dosage adjustments, making therapy cumbersome. Warfarin should only be used when a systematic patient monitoring system is available.
The optimal duration for VTE prophylaxis is not well established.2 Prophylaxis should be given throughout the period of risk. For general surgical procedures and medical conditions, once the patient is able to ambulate regularly and other risk factors are no longer present, prophylaxis can be discontinued. The risk of VTE in the first month following hospital discharge among patients who have undergone total knee replacement, total hip replacement or hip fracture repair is very high. Therefore, extended prophylaxis for 21 to 35 days following hospital discharge with an LMWH, fondaparinux, or warfarin is recommended.
Patient Encounter 1, Part 1
KK is a 69-year-old, obese female who fell on her way to church and fractured her right hip. She is hospitalized and will undergo surgery to repair her fractured right hip.
PMH: Hypertension × 12 years; dyslipidemia × 10 years; obesity × 20 years; degenerative joint disease × 5 years; recurrent urinary tract infections
FH: Nonsignificant
SH: Smoked half a pack per day for 25 years; occasional alcohol use. The patient has Medicare, but due to her fixed income, has difficulty paying for medications, leading to occasional periods of noncompliance
Current Meds: Metoprolol 100 mg by mouth twice daily; hydrochlorothiazide 25 mg by mouth daily; simvastatin 40 mg by mouth daily; salsalate 750 mg by mouth twice daily; trimethoprim-sulfamethoxazole SS tablets by mouth twice daily for 7 days (last treatment was 1 month ago); shark cartilage three tablets by mouth daily; enteric-coated aspirin 81 mg by mouth daily; ginseng two tablets by mouth daily
Allergies: NKDA
PE:
VS: BP 145/90 mm Hg, HR 72, RR 16, T 37.4°C (99.3°F), wt 127 kg (280 lb), BMI 40 kg/m2
Labs: Within normal limits; estimated glomerular filtration rate (GFR) = 74ml_/min
Which risk factor(s) predispose KK to VTE?
What is KK’s estimated risk for developing VTE?
Given KK’s presentation and history, create an appropriate VTE prophylaxis plan including the pharmacologic agent, dose, route and frequency of administration, duration of therapy, monitoring parameters, and patient education.
TREATMENT
Desired Therapeutic Outcomes
The goal of VTE treatment is to prevent short-and long-term complications of the disease. In the short term (i.e., the first few days to 6 months), the aim of therapy is to prevent propagation or local extension of the clot, embolization, and death. In the long term (i.e., more than 6 months after the first event), the aim of therapy is to prevent complications, such as PTS, pulmonary hypertension, and recurrent VTE.17,23
General Treatment Principles
Anticoagulant drugs are considered the mainstay of therapy for patients with VTE, and the therapeutic strategies for DVT and PE are essentially identical.17,23 ![]() In the absence of contraindications, the treatment of VTE should initially include a rapid-acting anticoagulant (e.g., UFH, LMWH, or fondaparinux) overlapped with warfarin for at least 5 days and until the patient’s INR is greater than 2. Anticoagulation therapy should be continued for a minimum of 3 months. However, the duration of anticoagulation therapy should be based on the patient’s risk of VTE recurrence and major bleeding. The treatment of VTE can be divided into acute, subacute, and chronic phases (Fig. 10–5).23,24 The acute treatment phase of VTE is typically accomplished by administering a fast-acting parenteral anticoagulant (Table 10–3). The subacute and chronic phase treatments of VTE are usually accomplished using oral anticoagulant agents, such as warfarin.17,23 In certain populations, such as patients with cancer and women who are pregnant, the LMWHs are the preferred agents during subacute and chronic treatment phases.17 In the last decade, several novel anticoagulants, such as direct thrombin inhibitors (DTIs) and factor Xa inhibitors have emerged as potential alternatives for the acute, subacute, and chronic phases of treatment. As data from clinical trials using these new agents in VTE treatment continue to emerge, their role in clinical practice will be better understood.17,23
In the absence of contraindications, the treatment of VTE should initially include a rapid-acting anticoagulant (e.g., UFH, LMWH, or fondaparinux) overlapped with warfarin for at least 5 days and until the patient’s INR is greater than 2. Anticoagulation therapy should be continued for a minimum of 3 months. However, the duration of anticoagulation therapy should be based on the patient’s risk of VTE recurrence and major bleeding. The treatment of VTE can be divided into acute, subacute, and chronic phases (Fig. 10–5).23,24 The acute treatment phase of VTE is typically accomplished by administering a fast-acting parenteral anticoagulant (Table 10–3). The subacute and chronic phase treatments of VTE are usually accomplished using oral anticoagulant agents, such as warfarin.17,23 In certain populations, such as patients with cancer and women who are pregnant, the LMWHs are the preferred agents during subacute and chronic treatment phases.17 In the last decade, several novel anticoagulants, such as direct thrombin inhibitors (DTIs) and factor Xa inhibitors have emerged as potential alternatives for the acute, subacute, and chronic phases of treatment. As data from clinical trials using these new agents in VTE treatment continue to emerge, their role in clinical practice will be better understood.17,23
Treatment Options
Pharmacologic Therapy
Thrombolytics
The role of thrombolysis in the treatment of VTE is controversial. Thrombolytic agents are proteolytic enzymes that have the ability to dissolve, or lyse, the fibrin clot (Table 10–4). Thrombolytics are administered systemically or directly into the thrombus using a catheter-directed infusion.17 Compared to anticoagulants, thrombolytics restore venous patency more quickly; however, the bleeding risk associated with their use is significantly higher.25 In patients with DVT, thrombolytics decrease short-term pain and swelling and prevent destruction of the venous valves. It is not clear if thrombolytics decrease the incidence and severity of PTS. Clinical trials have failed to show any long-term benefits from the routine use of thrombolytics; therefore, their use in the majority of patients is not recommended.17,25 In a select group of high-risk patients with massive iliofemoral DVT who are at risk of limb gangrene, thrombolysis may be considered.17

FIGURE 10–5. Treatment approach for patients with VTE. (INR, International Normalized Ratio; IV, intravenous; LMWH, low-molecular weight heparin; PO, oral; SC, subcutaneous; UFH, unfractionated heparin; VTE, venous thromboembolism.) (From Ref. 24.)
Table 10–3 Pharmacologic Options for the Initial Treatment of Acute VTE
|
UFH |
|
IV administration:a use weight-based dosing nomogram (Table 10–5) |
|
or |
|
SC administration: 17,500 units (250 units/kg) given every 12 hours (an initial 5,000 unit IV bolus dose is recommended to obtain rapid anticoagulation) |
|
Adjust subsequent doses to attain a goal aPTT based on the institution-specific therapeutic range |
|
or |
|
SC administration: 333 units/kg followed by 250 units/kg given every 12 hours (fixed-dose unmonitored dosing regimen) |
|
LMWHs |
|
Dalteparin: 200 units/kg SC once daily or 100 units/kg SC twice dailyb |
|
Enoxaparin: 1.5 mg/kg SC once daily or 1 mg/kg SC twice daily; if CrCl is less than 30 mL/min: 1 mg/kg SC once daily |
|
Tinzaparin: 175 units/kg SC once daily |
|
Factor Xa Inhibitor |
|
Fondaparinux: |
|
For body weight less than 50 kg (110 lb), use 5 mg SC once daily |
|
For body weight 50–100 kg (110-220 lb), use 7.5 mg SC once daily |
|
For body weight greater than 100 kg (220 lb), use 10 mg SC once daily |
aPTT, activated partial thromboplastin time; CrCl, creatinine clearance; SC, subcutaneous; VTE, venous thromboembolism; UFH, unfractionated heparin; LMWHs, low-molecular weight heparins.
aIV administration preferred due to improved dosing precision.
bNot FDA-approved for treatment of VTE in noncancer patients.
From Ref. 17.
In patients with acute PE, the use of thrombolytics provides short-term benefits such as restoring pulmonary artery patency and hemodynamic stability.17,26 A recent meta-analysis of nine small randomized clinical trials showed a slightly lower risk of death or recurrent PE in patients treated with thrombolytics when compared to those treated with heparin alone. However, this small benefit was offset by a higher risk of major bleeding.27 Streptokinase, urokinase, and tissue plasminogen activator (t-PA) have all been studied and are FDA-approved in the treatment of PE. All three agents have comparable thrombolytic capacity but t-PA has the potential advantage of a shorter infusion time. Reteplase is not currently FDA-approved for the treatment of PE, but it has also been studied. Reteplase is administered as two 10 unit IV boluses given 30 minutes apart.17,28 Given the relative lack of data to support their routine use, thrombolytics should be reserved for select high-risk circumstances (Table 10–4). Candidates for thrombolytic therapy are patients with acute massive embolism who are hemodynamically unstable (systolic blood pressure [SBP] less than 90 mm Hg) and at low risk for bleeding.17 The use of thrombolytics in hemodynamically stable patients with right ventricular dysfunction is controversial but some experts support their use.
Table 10–4 Thrombolysis for the Treatment of VTE
|
• Thrombolytic therapy should be reserved for patients who present with shock, hypotension, right ventricular strain, or massive DVT with limb gangrene |
|
• Diagnosis must be objectively confirmed before initiating thrombolytic therapy |
|
• Thrombolytic therapy is most effective when administered as soon as possible after PE diagnosis, but benefit may extend up to 14 days after symptom onset |
|
• Approved PE thrombolytic regimens: |
|
• Streptokinase 250,000 units IV over 30 minutes followed by 100,000 units/h for 24 hoursa |
|
• Urokinase 4,400 units/kg IV over 10 minutes followed by 4,400 units/kg/h for 12–24 hoursa |
|
• Alteplase 100 mg IV over 2 hours |
|
• Factors that increase the risk of bleeding must be evaluated before thrombolytic therapy is initiated (i.e., recent surgery, trauma or internal bleeding, uncontrolled hypertension, recent stroke, or ICH) |
|
• Baseline labs should include CBC and blood typing in case transfusion is needed |
|
• UFH should not be used during thrombolytic therapy. Neither the aPTT nor any other anticoagulation parameter should be monitored during the thrombolytic infusion |
|
• aPTT should be measured following the completion of thrombolytic therapy: |
|
• If aPTT less than 2.5 times the control value, UFH infusion should be started and adjusted to maintain aPTT in therapeutic range |
|
• If aPTT greater than 2.5 times the control value, remeasure every 2–4 hours and start UFH infusion when aPTT is less than 2.5 |
|
• Avoid phlebotomy, arterial puncture, and other invasive procedures during thrombolytic therapy to minimize the risk of bleeding |
aPTT, activated partial thromboplastin time; DVT, deep vein thrombosis; PE, pulmonary embolism; UFH, unfractionated heparin; VTE, venous thromboembolism.
aTwo-hour infusions of streptokinase and urokinase are as effective and safe as alteplase.
Unfractionated Heparin
UFH has traditionally been the drug of choice for indications requiring a rapid anticoagula-tion including the acute treatment of VTE. Commercially available UFH preparations are derived from porcine intestinal mucosa or bovine lung. UFH is composed of a heterogeneous mixture of glycosaminoglycans with variable length, molecular weight, and pharmacologic properties. Unlike thrombolytics, UFH and other anticoagulants will not dissolve a formed clot but prevent its propagation and growth.4,29 Heparin exerts its anticoagulant effect by augmenting the natural anticoagulant, AT. A specific pentasaccharide sequence on the heparin molecule binds to AT and causes a conformational change that greatly accelerates its activity (Fig. 10–6). This complex inhibits thrombin (factor IIa), as well as factors Xa, IXa, XIa, and Xlla. Thrombin and factor Xa are most sensitive to this inhibition and are inactivated in an equal 1:1 ratio. In order to inactivate thrombin, the UFH molecule needs to form a ternary complex by binding to both AT and thrombin. Only UFH molecules that are at least 18 saccharide units long are able to form this bridge between AT and thrombin. In contrast, inhibition of factor Xa does not require the formation of a ternary complex. UFH molecules as short as five saccharide units can catalyze the inactivation of factor Xa. Due to its nonspecific binding to cellular proteins, UFH has several limitations, including poor bioavail-ability when given SC and significant intra-and interpatient variability in anticoagulant response.4,29
UFH can be administered via the IV or SC route.4 When rapid anticoagulation is required, UFH should be administered IV and an initial bolus dose should be given. For the treatment of VTE, UFH is generally given as a continuous IV infusion. Intermittent IV bolus dosing is associated with a higher risk of bleeding and is therefore not recommended. When given SC, the bio availability of UFH ranges from 30% to 70%, depending on the dose given. Therefore, higher doses of UFH must be given if the SC route of administration is used. Onset of anticoagulation is delayed by 1 to 2 hours after the SC injection. Due to the risk of hematomas and erratic absorption, intramuscular (IM) administration is not recommended. The half-life of UFH is dose dependent and ranges from 30 to 90 minutes, but maybe significantly longer, up to 150 minutes, with high doses. UFH is eliminated by two mechanisms: (a) enzymatic degradation via a saturable zero-order process, and (b) renally via a first-order process. Lower UFH doses are primarily cleared via enzymatic processes, while higher doses are primarily renally eliminated. Clearance of UFH can be impaired in patients with renal and hepatic dysfunction. Patients with active thrombosis may require higher UFH doses due to a more rapid elimination or variations in the plasma concentrations of heparin-binding proteins. AT deficiency and elevated factor VIII levels are common in pregnant patients. AT deficiency has been linked to higher UFH dose requirements. The requirement of these higher UFH doses is termed “heparin resistance.” Factor VIII elevations can result in altered aPTT response to UFH and monitoring with antifactor Xa levels is recommended.4,29
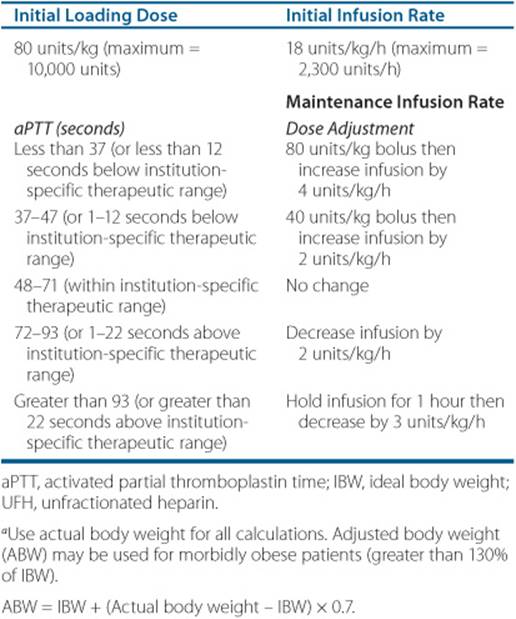
The dose of UFH required to achieve a therapeutic anticoagulant response is correlated to the patient’s weight.4 Weight-based dosing regimens should be utilized in order to exceed the therapeutic threshold in the first 24 hours after initiating treatment.30 Achieving a therapeutic aPTT in the first 24 hours after initiating UFH is critical because this has been shown to lower the risk of recurrent VTE.4,30 For nonobese patients, the actual body weight should be used to calculate the initial UFH dose (Table 10–5). For obese patients, using the actual body weight to calculate the initial dose is also generally recommended; however, data are more limited in morbidly obese patients; that is, weight greater than 150 kg (330 lb). Some experts recommend using an adjusted body weight (ABW) in these patients instead. The infusion rate is then adjusted based on laboratory monitoring of the patient’s response. UFH can also be administered via the SC route; however, IV infusion is preferred by most clinicians because it can be dosed more precisely. If the SC route is selected, an initial 5,000 unit IV bolus should be given followed by 17,500 units given SC every 12 hours. Subsequent doses of SC-UFH need to be adjusted based on the patient’s response.17,23,31 Alternatively, in patients with DVT, a fixed-dose, weight-based dose of unmonitored UFH regimen can also be used with an initial dose of 333 units/kg followed by 250 units/kg given SC every 12 hours.17,32
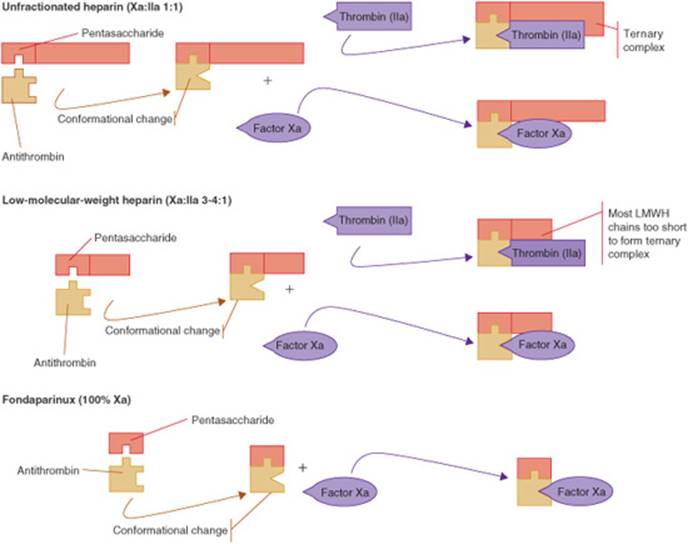
FIGURE 10–6. Mechanism of action of unfractionated heparin, low-molecular weight heparin (LMWH), and fondaparinux. (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:338.)
![]() Due to significant variability in interpatient response and changes in patient response over time, UFH requires close monitoring and periodic dose adjustment. The response to UFH can be monitored using a variety of laboratory tests including the aPTT, the whole blood clotting time, activated clotting time (ACT), antifactor Xa activity, and the plasma heparin concentration.4,33 Although it has several limitations, the aPTT is the most widely used test in clinical practice to monitor UFH. Traditionally, therapeutic aPTT range is defined as 1.5 to 2.5 times the control aPTT value. However, due to variations in the reagents and instruments used to measure the aPTT in different laboratories, each institution should establish a therapeutic range for UFH. The institution-specific therapy range should correlate with a plasma heparin concentration of 0.2 to 0.4 units/mL by protamine titration or 0.3 to 0.7 units/mL by an amidolytic antifactor Xa assay.4,33 An aPTT should be obtained at baseline, 6 hours after initiating the heparin infusion, and 6 hours after each dose change, as this is the time required to reach steady state. The UFH dose is then adjusted based on the aPTT measurement and the institutional-specific therapeutic range (Table 10–5). In patients with “heparin resistance,” antifactor Xa concentrations may be a more accurate method of monitoring the patient’s response.4,33
Due to significant variability in interpatient response and changes in patient response over time, UFH requires close monitoring and periodic dose adjustment. The response to UFH can be monitored using a variety of laboratory tests including the aPTT, the whole blood clotting time, activated clotting time (ACT), antifactor Xa activity, and the plasma heparin concentration.4,33 Although it has several limitations, the aPTT is the most widely used test in clinical practice to monitor UFH. Traditionally, therapeutic aPTT range is defined as 1.5 to 2.5 times the control aPTT value. However, due to variations in the reagents and instruments used to measure the aPTT in different laboratories, each institution should establish a therapeutic range for UFH. The institution-specific therapy range should correlate with a plasma heparin concentration of 0.2 to 0.4 units/mL by protamine titration or 0.3 to 0.7 units/mL by an amidolytic antifactor Xa assay.4,33 An aPTT should be obtained at baseline, 6 hours after initiating the heparin infusion, and 6 hours after each dose change, as this is the time required to reach steady state. The UFH dose is then adjusted based on the aPTT measurement and the institutional-specific therapeutic range (Table 10–5). In patients with “heparin resistance,” antifactor Xa concentrations may be a more accurate method of monitoring the patient’s response.4,33
Table 10–5 Weight-Baseda Dosing for UFH Administered by Continuous IV Infusion for VTE
Side effects associated with UFH include bleeding, thrombocytopenia, hypersensitivity reactions, and with prolonged use, alopecia, hyperkalemia, and osteoporosis.4,29 ![]() Bleeding is the most common adverse effect associated with antithrombotic drugs, including UFH therapy. A patient’s risk of major hemorrhage is related to the intensity and stability of therapy, age, concurrent drug use, history of GI bleeding, risk of falls or trauma, and recent surgery.Several risk factors can increase the risk of UFH-induced bleeding (Table 10–6). The risk of bleeding is related to the intensity of anticoagulation. Higher aPTT values are associated with an increased risk of bleeding. The risk of major bleeding is 1% to 5% during the first few days of treatment.3 In addition to the aPTT, hemoglobin, hematocrit, and blood pressure should also be monitored. Concurrent use of UFH with other antithrombotic agents, such as thrombolytics and antiplatelet agents, also increases the risk of bleeding. Patients receiving UFH therapy should be closely monitored for signs and symptoms of bleeding, including epistaxis, hemoptysis, hematuria, hematochezia, melena, severe headache, and joint pain. If major bleeding occurs, UFH should be stopped immediately and the source of bleeding treated.3,4 If necessary, use protamine sulfate to reverse the effects of UFH. The usual dose is 1 mg protamine sulfate per 100 units of UFH, up to a maximum of 50 mg, given as a slow IV infusion over 10 minutes. The effects of UFH are neutralized in 5 minutes, and the effects of protamine persist for 2 hours. If the bleeding is not controlled or the anticoagulant effect rebounds, repeated doses of protamine may be administered.4
Bleeding is the most common adverse effect associated with antithrombotic drugs, including UFH therapy. A patient’s risk of major hemorrhage is related to the intensity and stability of therapy, age, concurrent drug use, history of GI bleeding, risk of falls or trauma, and recent surgery.Several risk factors can increase the risk of UFH-induced bleeding (Table 10–6). The risk of bleeding is related to the intensity of anticoagulation. Higher aPTT values are associated with an increased risk of bleeding. The risk of major bleeding is 1% to 5% during the first few days of treatment.3 In addition to the aPTT, hemoglobin, hematocrit, and blood pressure should also be monitored. Concurrent use of UFH with other antithrombotic agents, such as thrombolytics and antiplatelet agents, also increases the risk of bleeding. Patients receiving UFH therapy should be closely monitored for signs and symptoms of bleeding, including epistaxis, hemoptysis, hematuria, hematochezia, melena, severe headache, and joint pain. If major bleeding occurs, UFH should be stopped immediately and the source of bleeding treated.3,4 If necessary, use protamine sulfate to reverse the effects of UFH. The usual dose is 1 mg protamine sulfate per 100 units of UFH, up to a maximum of 50 mg, given as a slow IV infusion over 10 minutes. The effects of UFH are neutralized in 5 minutes, and the effects of protamine persist for 2 hours. If the bleeding is not controlled or the anticoagulant effect rebounds, repeated doses of protamine may be administered.4
Table 10–6 Risk Factors for Major Bleeding While Taking Anticoagulation Therapy
|
Anticoagulation intensity (e.g., INR greater than 5, aPTT greater than 120 seconds) |
|
Initiation of therapy (first few days and weeks) |
|
Unstable anticoagulation response |
|
Age greater than 65 years |
|
Concurrent antiplatelet drug use |
|
Concurrent nonsteroidal anti-inflammatory drug or aspirin use |
|
History of GI bleeding |
|
Recent surgery or trauma |
|
High risk for fall/trauma |
|
Heavy alcohol use |
|
Renal failure |
|
Cerebrovascular disease |
|
Malignancy |
aPTT, activated partial thromboplastin time; INR, International Normalized Ratio.
Heparin-induced thrombocytopenia (HIT) is a very serious adverse effect associated with UFH use. Platelet counts should be monitored every 2 to 3 days during the course of UFH therapy.4 HIT should be suspected if the platelet count drops by more than 50% from baseline or to below 120 × 103/mm3 (120 × 109/L). HIT should also be suspected if thrombosis occurs despite UFH use. Immediate discontinuation of all heparin-containing products, including the use of LMWHs is in order. Alternative anticoagulation should be initiated. In patients with contraindications to anticoagulation therapy, UFH should not be administered (Table 10–7).
UFH is an FDA pregnancy category C drug and may be used to treat VTE during pregnancy. UFH should be used with caution in the peripartum period due to the risk of maternal hemorrhage. UFH is not secreted into the breast milk and is safe for use by women who wish to breast-feed.11 For the treatment of VTE in children, the UFH dose is 50 units/kg bolus followed by an infusion of 20,000 units/m2 per 24 hours. Alternatively, a loading dose of 75 units/kg followed by an infusion of 28 units/kg/h if less than 12 months old and 20 units/kg/h if greater than 1 year old may be considered.34
Low-Molecular Weight Heparins
The LMWHs are smaller heparin fragments obtained by chemical or enzymatic depolymerization of UFH. LMWHs are heterogeneous mixtures of glycosaminoglycans, and each product has slightly different molecular weight distributions and pharmacologic properties.4 Compared with UFH, LMWHs have improved pharmacodynamic and pharmacokinetic properties. They exhibit less binding to plasma and cellular proteins, resulting in a more predictable anticoagulant response. Consequently, routine monitoring of anticoagulation activity and dose adjustments are not required in the majority of patients. LMWHs have longer plasma half-lives, allowing once-or twice-daily administration, improved SC bio availability, and dose-independent renal clearance. In addition, LMWHs have a more favorable side effect profile than UFH. They are also associated with a lower incidence of HIT and osteopenia. Three LMWHs are currently available in the United States: dalteparin, enoxaparin, and tinzaparin.4,29
Like UFH, LMWHs prevent the propagation and growth of formed thrombi. The anticoagulant effect is mediated through a specific pentasaccharide sequence that binds to AT. The primary difference in the pharmacologic activity of UFH and LMWH is their relative inhibition of thrombin (factor IIa) and factor Xa. Smaller heparin fragments cannot bind AT and thrombin simultaneously (Fig. 10–6). Due to their smaller chain length, LMWHs have relatively greater activity against factor Xa and inhibit thrombin to a lesser degree. The antifactor Xa:IIa activity ratio for the LMWHs ranges from 2:1 to 4:1. The SC bioavailability of the LMWHs is greater than 90%. The peak anticoagulant effect of the LMWHs is reached 3 to 5 hours after a SC dose. The elimination half-life is 3 to 6 hours and is agent-specific. In patients with renal impairment, the half-life of LMWHs is prolonged.4,29
Table 10–7 Contraindications to Anticoagulation Therapy
|
General |
|
Active bleeding |
|
Hemophilia or other hemorrhagic tendencies |
|
Severe liver disease with elevated baseline PT |
|
Severe thrombocytopenia (platelet count less than 20 × 103/mm3[20x109/L]) |
|
Malignant hypertension |
|
Inability to meticulously supervise and monitor treatment |
|
Product-Specific Contraindications |
|
UFH |
|
Hypersensitivity to UFH History of HIT |
|
LMWHs |
|
Hypersensitivity to LMWH, UFH, pork products, methylparaben, or propylparaben |
|
History of HIT or suspected HIT |
|
Fondaparinux |
|
Hypersensitivity to fondaparinux |
|
Severe renal insufficiency (CrCI less than 30 mL/min) |
|
Body weight less than 50 kg (110 lb) |
|
Bacterial endocarditis |
|
Thrombocytopenia with a positive in vitro test for antiplatelet antibodies in the presence of fondaparinux |
|
Lepirudin |
|
Hypersensitivity to hirudins |
|
Argatroban |
|
Hypersensitivity to argatroban |
|
Warfarin |
|
Hypersensitivity to warfarin |
|
Pregnancy |
|
History of warfarin-induced skin necrosis |
|
Inability to obtain follow-up PT/INR measurements |
|
Inappropriate medication use or lifestyle behaviors |
CrCI, creatinine clearance; HIT, heparin-induced thrombocytopenia; INR, International Normalized Ratio; LMWHs, low-molecular weight heparins; PT, prothrombin time; UFH, unfractionated heparin.
The dose of LMWHs for the treatment of VTE is determined based on the patient’s weight and is administered SC once or twice daily (Table 10–3). Once-daily dosing of enoxaparin appears to be as effective as twice-daily dosing; however, some data suggest that twice-daily dosing may be more effective in patients who are obese or have cancer.17,23,35 The dose of enoxaparin is expressed in milligrams, whereas the doses of dalteparin and tinzaparin are expressed in units of antifactor Xa activity. Due to their predictable anticoagulant effect, routine monitoring is not necessary in the majority of patients.4 LMWHs have been evaluated in a large number of randomized trials and have been shown to be at least as safe and effective as UFH for the treatment of VTE.17,23 Indeed, the rate of mortality was lower in patients treated with a LMWH in clinical trials. This mortality benefit was primarily seen in patients with cancer.31,36
Prior to initiating treatment with a LMWH, baseline laboratory tests should include prothrombin time (PT)/INR, aPTT, CBC, and serum creatinine. Monitor the CBC with platelet count every 2 to 3 days during the first 2 weeks of therapy, and every 2 to 4 weeks with extended use.4 Use LMWHs cautiously in patients with renal impairment due to the potential of drug accumulation and risk of bleeding. Specific dosing recommendations for patients with a creatinine clearance (CrCl) less than 30 mL/min are currently available for enoxaparin but are lacking for other agents of the class (Table 10–3). Higher mortality rate has been reported in elderly patients with renal dysfunction who were treated with the LMWH tinzaparin. Current guidelines recommend the use of UFH over LMWH in patients with severe renal dysfunction (CrCl less than 30 mL/min).17,37
![]() Most patients with an uncomplicated DVT can be managed safely at home. LMWHs can be easily administered in the outpatient setting, thus enabling the treatment of VTE at home. Several large clinical trials have demonstrated the efficacy and safety of LMWHs for outpatient treatment of DVT17,20,31 Acceptance of this treatment approach has increased tremendously over the last several years among clinicians. Patients with DVT with normal vital signs, low-bleeding risk, no other comorbid conditions requiring hospitalization, and who are stable, may have anticoagulant initiated at home. Although the treatment of patients with PE in the outpatient setting is controversial, patients with submassive PE who are hemodynamically stable can be safely treated in the outpatient setting as well.17,26 Patients considered for outpatient therapy must be reliable or have adequate caregiver support and must be able to strictly adhere to the prescribed treatment regimen and recommended follow-up visits. Close patient follow-up is critical to the success of any outpatient DVT treatment program. Home DVT treatment results in cost savings and improved patient satisfaction and quality of life.20,37,38
Most patients with an uncomplicated DVT can be managed safely at home. LMWHs can be easily administered in the outpatient setting, thus enabling the treatment of VTE at home. Several large clinical trials have demonstrated the efficacy and safety of LMWHs for outpatient treatment of DVT17,20,31 Acceptance of this treatment approach has increased tremendously over the last several years among clinicians. Patients with DVT with normal vital signs, low-bleeding risk, no other comorbid conditions requiring hospitalization, and who are stable, may have anticoagulant initiated at home. Although the treatment of patients with PE in the outpatient setting is controversial, patients with submassive PE who are hemodynamically stable can be safely treated in the outpatient setting as well.17,26 Patients considered for outpatient therapy must be reliable or have adequate caregiver support and must be able to strictly adhere to the prescribed treatment regimen and recommended follow-up visits. Close patient follow-up is critical to the success of any outpatient DVT treatment program. Home DVT treatment results in cost savings and improved patient satisfaction and quality of life.20,37,38
Laboratory methods of measuring a patient’s response to LMWH maybe warranted in certain situations.4,39 Although controversial, measurement of antifactor Xa activity has been the most widely used method in clinical practice and is recommended by the College of American Pathologists.39 Monitoring of antifactor Xa activity may be considered in adult patients who are morbidly obese (weight greater than 150 kg [330 lb] or BMI greater than 50 kg/m2), weigh less than 50 kg (110 lb), or have significant renal impairment (CrCl less than 30 mL/min). Laboratory monitoring may also be useful in children and pregnant women. When monitoring antifactor Xa activity, the sample should be obtained approximately 4 hours after the SC dose is administered, when peak concentration is anticipated. The therapeutic range for antifactor Xa activity has not been clearly defined, and there is a limited correlation between antifactor Xa activity and safety or efficacy.39 For the treatment of VTE, an acceptable antifactor Xa activity range is 0.5 to 1 unit/mL with twice-daily dosing. In patients treated with once-daily LMWH regimens, a target level between 1 and 2 units/mL is recommended by some experts.4,17,35,39
Similar to UFH, bleeding is the major complication associated with LMWHs. The frequency of major bleeding appears to be numerically lower with LMWHs than with UFH.31 The incidence of major bleeding reported in clinical trials is less than 3%.17 Minor bleeding, especially bruising at the injection site, occurs frequently. Protamine sulfate will partially reverse the anticoagulant effects of the LMWHs and should be administered in the event of major bleeding. Due to its limited binding to LMWH chains, protamine only neutralizes 60% of their antithrombotic activity. If the LMWH was administered within the previous 8 hours, give 1 mg protamine sulfate per 1 mg of enoxaparin or 100 antifactor Xa units of dalteparin or tinzaparin. If the bleeding is not controlled, give 0.5 mg of protamine sulfate for every antifactor Xa 100 units of LMWH. Give smaller protamine doses if more than 8 hours have lapsed since the last LMWH dose.4
The incidence of HIT is lower with LMWHs than with UFH. However, LMWHs cross-react with heparin antibodies in vitro and should not be given as an alternative anticoagulant in patients with a diagnosis or history of HIT. Monitor platelet counts every few days during the first 2 weeks and periodically thereafter.4
In patients undergoing spinal and epidural anesthesia or spinal puncture, spinal and epidural hematomas have been linked to the use of LMWHs. In patients with in-dwelling epidural catheters, concurrent use of LMWHs and all other agents that impact hemostasis should be avoided. When inserting and removing the in-dwelling epidural catheters, the timing of LMWH administration around catheter manipulation should be carefully coordinated. Catheter manipulation should only occur at minimal or trough anticoagulant levels.4
LMWHs are an excellent alternative to UFH for the treatment of VTE in pregnant women.11 The LMWHs do not cross the placenta, and they are FDA pregnancy category B. Because the pharmacokinetics of LMWHs may change during pregnancy, monitor antifactor Xa activity every 4 to 6 weeks to make dose adjustments.11,40 LMWHs have also been used to treat VTE in pediatric patients. Children less than 1 year old require higher doses (e.g., enoxaparin 1.5 mg/kg SC every 12 hours). Monitor antifactor Xa activity to guide dosing in children.34
Factor Xa Inhibitors
Fondaparinux, the first agent in this class, is an indirect inhibitor of factor Xa, and exerts its anticoagulant activity by accelerating AT. Fondaparinux contains the specific five-saccharide sequence found in UFH that is responsible for its pharmacologic activity. Due to its small size, fondaparinux exerts inhibitory activity specifically against factor Xa and has no effect on thrombin (factor IIa; Fig. 10–6).29,41Fondaparinux is currently the only agent of the class that is commercially available in the United States. Idraparinux, rivaroxaban, and apixaban are antifactor Xa inhibitors currently undergoing Phase III clinical trials. Fondaparinux and idraparinux are administered SC; rivaroxaban and apixaban are administered orally. After SC administration, fondaparinux is completely absorbed, and peak plasma concentrations are reached within 2 to 3 hours.41–42
As synthetic drugs (unlike UFH and the LMWHs), factor Xa inhibitors cannot transmit animal pathogens, are consistent from batch-to-batch, and are available in an unlimited supply. Other favorable attributes of factor Xa inhibitors include a predictable and linear dose-response relationship, rapid onset of activity, and long half-life.29,41,42 Factor Xa inhibitors do not require routine coagulation monitoring or dose adjustments. Fondaparinux has a half-life of 17 to 21 hours, permitting once-daily administration, but the anticoagulant effects of fondaparinux will persist for 2 to 4 days after stopping the drug. In patients with renal impairment, the anticoagulant effect persists even longer. Idraparinux has a significantly longer duration of activity and is being developed for once-weekly injection. The oral agents, rivaroxaban and apixaban, have half-lives ranging from 5 to 14 hours allowing once-or twice-daily administration.41,42 Neither fondaparinux nor idraparinux are metabolized in the liver and therefore have few drug interactions. However, concurrent use with other antithrombotic agents increases the risk of bleeding. Unlike the heparins, factor Xa inhibitors do not affect platelet function and do not react with the heparin platelet factor-4 (PF-4) antibodies seen in patients with HIT. Some centers use fondaparinux in patients with subacute HIT or a history of HIT who require anticoagulation therapy.4,29,41
Fondaparinux has been evaluated for the treatment of DVT and PE in two large phase 3 trials and is approved by the FDA for these indications. Fondaparinux is as safe and effective as IV UFH for the treatment of PE and SC LMWH for DVT treatment.43,44 The recommended dose for fondaparinux in the treatment of VTE is based on the patient’s weight (Table 10–3). Fondaparinux is renally eliminated and accumulation can occur in patients with renal dysfunction. Due to the lack of specific dosing guidelines, fondaparinux is contraindicated in patients with severe renal impairment (CrCl less than 30 mL/min). Baseline renal function should be measured and monitored closely during the course of therapy. Based on limited available data at this time, monitoring antifactor Xa activity to guide fondaparinux dosing is not recommended.4,17,29
As with other anticoagulants, the major side effect associated with fondaparinux is bleeding. Fondaparinux should be used with caution in elderly patients because their risk of bleeding is higher. Patients receiving fondaparinux should be carefully monitored for signs and symptoms of bleeding. A CBC should be obtained at baseline and monitored periodically to detect the possibility of occult bleeding. In the event of major bleeding, fresh frozen plasma and factor concentrates should be given. In the case of a life-threatening bleed, recombinant factor VIIa may be considered, but this is a very costly option and it can also increase the risk of thrombosis. Fondaparinux is not reversed by protamine.4,17,29,41
Fondaparinux is pregnancy category B, but there are very limited data regarding its use during pregnancy. Use in pediatric patients has not been studied.4,29,41
DTIs
Given that thrombin is the central mediator of coagulation and amplifies its own production, it is a natural target for pharmacologic intervention. DTIs bind thrombin and prevent interactions with its substrates (Fig. 10–7). Several injectable DTIs are approved for use in the United States including lepirudin, bivalirudin, argatroban, and desirudin. Several oral DTIs are currently in development. These agents differ in terms of their chemical structure, molecular weight, and binding to the thrombin molecule. Potential advantages of DTIs include a targeted specificity for thrombin, the ability to inactivate clot-bound thrombin, and an absence of platelet interactions that can lead to HIT. Unlike heparins, DTIs do not require AT as a cofactor and do not bind to plasma proteins. Therefore they produce a more predictable anticoagulant effect. DTIs are considered the drugs of choice for the treatment of VTE in patients with a diagnosis or history of HIT.29,41,45
The prototype of this class is hirudin, which was originally isolated from the salivary glands of the medicinal leech, Hirudo medicinalis. Hirudin itself is not commercially available, but recombinant technology has permitted production of hirudin derivatives, namely lepirudin and desirudin.29,41,45 Lepirudin has a short half-life of approximately 40 minutes after IV administration and 120 minutes when given SC. Elimination of lepirudin is primarily renal; therefore, doses must be adjusted based on the patient’s renal function. The dose should be monitored and adjusted to achieve an aPTT ratio of 1.5 to 2.5 times the baseline measurement. Lepirudin is currently approved for use in patients with HIT and related thrombosis. Up to 40% of patients treated with lepirudin will develop antibodies to the drug.29,41,45

FIGURE 10–7. Mechanism of action of direct thrombin inhibitors. (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:345.)
Bivalirudin, a smaller-molecular-weight DTI, is given by IV infusion. Bivalirudin has a shorter elimination half-life (approximately 25 minutes) than lepirudin and is only partially eliminated renally. Unlike lepirudin, bivalirudin is a reversible inhibitor of thrombin and provides transient antithrombotic activity. Patients with moderate or severe renal impairment (CrCl less than 60 mL/min) may require dose adjustment because clearance of bivalirudin is reduced by approximately 20% in these patients. Bivalirudin is approved for use in patients with unstable angina undergoing percutaneous transluminal coronary angioplasty. The ACT is used to monitor the anticoagulant effect of bivalirudin during percutaneous coronary intervention (PCI).29,41,45
Desirudin is a SC administered DTI approved for VTE prevention after hip replacement surgery but is not yet commercially available in the United States. Desirudin has an elimination half-life of 2 to 3 hours and is typically dosed every 12 hours. It is primarily eliminated through the kidneys, so dose reduction is needed in patients with renal impairment. The aPTT should be used to measure desirudin’s anticoagulant activity.29,41,45
Argatroban is a small synthetic molecule that binds reversibly to the active site ofthrombin (Fig. 10–7). Argatroban is IV administered and has a 40-to 50-minute elimination half-life. The aPTT must be monitored to assess its anticoagulant activity. Argatroban is hepatically metabolized; therefore, dose reductions and careful monitoring are recommended in patients with hepatic dysfunction. Renal impairment has no influence on the elimination half-life or dosing of argatroban. Argatroban is approved for prevention and treatment of thrombosis in patients with HIT and in patients with HIT undergoing PCI.29,41,45
Small-molecule DTIs have been structurally modified for oral administration. Several oral DTIs are in development. Dabigatran is one of the oral DTI agents that is in the most advanced phases of clinical development. Dabigatran is undergoing phase 3 clinical trials for the treatment and prevention of VTE and for stroke prevention in patients with atrial fibrillation. The potential advantages of oral DTIs include the ability to give them in fixed once-or twice-daily doses, and without the need for routine coagulation monitoring. In addition, there appear to be a significantly lower number of drug and food interactions associated with oral DTIs as compared to some of the traditional oral anticoagulants such as warfarin.41,45
Contraindications to the use of DTIs and risk factors for bleeding are similar to those of other antithrombotic agents (Tables 10–6 and 10–7). Bleeding is the most common side effect reported with the use of DTIs. Concurrent use of DTIs with thrombolytics significantly increases bleeding complications. Currently, there are no known antidotes to reverse the effects of the DTIs. Fresh frozen plasma, factor concentrates, or recombinant factor VIIa should be given in the event of a major bleed. DTIs can increase the PT/INR and can interfere with the accuracy of monitoring and dosing of warfarin therapy. Data on the use of DTIs in pregnancy and pediatric patients are very limited.29,41,45
Warfarin
Warfarin has been the primary oral anticoagulant used in the United States for the past 60 years. Warfarin is the anticoagulant of choice when long-term or extended anticoagulation is required. Warfarin is FDA-approved for the prevention and treatment of VTE, as well as the prevention of thromboembolic complications in patients with myocardial infarction, atrial fibrillation, and heart valve replacement. While very effective, warfarin has a narrow therapeutic index, requiring frequent dose adjustments and careful patient monitoring.5,29
Warfarin exerts its anticoagulant effect by inhibiting the production of the vitamin K-dependent coagulation factors II (prothrombin), VII, IX, and X, as well as the anticoagulant proteins C and S (Fig. 10–8). Warfarin has no effect on circulating coagulation factors that have been previously formed, and its full antithrombotic activity is delayed for 5 to 7 days, and potentially longer in slower metabolizers. This delay is related to half-lives of the clotting factors: 60 to 100 hours for factor II (prothrombin), 6 to 8 hours for factor VII, 20 to 30 hours for factor IX, and 24 to 40 hours for factor X. Proteins C and S, the natural anticoagulants, are inhibited more rapidly due to their shorter half-lives, 8 to 10 hours and 40 to 60 hours, respectively. Reductions in the concentration of natural anticoagulants before the clotting factors are depleted can lead to a paradoxical hypercoagulable state during the first few days of warfarin therapy. It is for this reason that patients with acute thrombosis should receive a fast-acting anticoagulant (heparin, LMWH, or fondaparinux) while transitioning to warfarin therapy.5,20,29
Warfarin is a racemic mixture of two isomers, the S and the R forms. The S-isomer is two to five times more potent than the R-isomer. Both isomers are extensively bound to albumin. The two isomers are metabolized in the liver via several isoenzymes including cytochrome P-450 (CYP) 1A2, 2C9, 2C19, 2C18, and 3A4 (Fig. 10–8). Hepatic metabolism of warfarin varies greatly among patients, leading to very large interpatient differences in dose requirements and genetic variations in these isoenzymes; specifically, polymorphisms in the CYP2C9*2, CYP2C9*3 genotype result in significantly lower warfarin dose requirement to achieve a therapeutic response, and VKORC1 haplotype mutations can result in hereditary warfarin resistance and increased warfarin requirements.5 Several algorithms that incorporate CYP2C9 genotype and the VKORC1 haplotype with other patient characteristics to predict warfarin maintenance dosing requirements have been developed and are being tested in large populations. Whether pharmacogenomic-based dosing will improve clinical outcomes has yet to be determined and is not recommended at this time, but it does hold promise for a more personalized approach to warfarin dosing.5,46
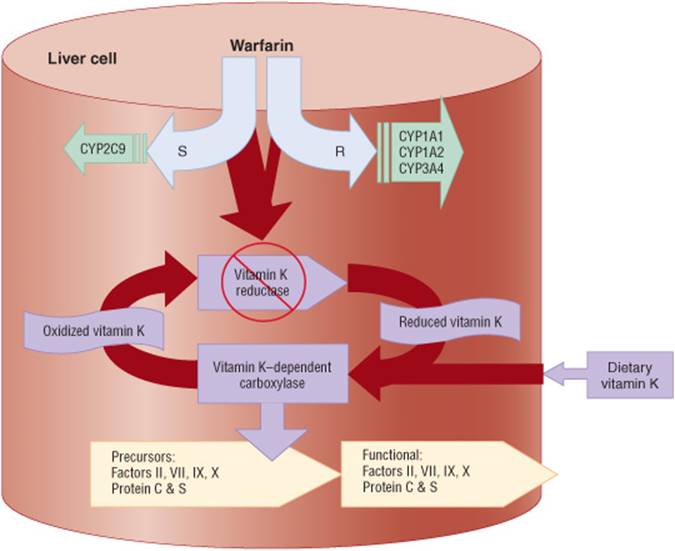
FIGURE 10–8. Pharmacologic activity and metabolism of warfarin. (CYP, cytochrome P-450 isoenzyme.) (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:347.)
Warfarin does not follow linear kinetics. Small dose adjustments can lead to large changes in anticoagulant response. The dose of warfarin is determined by each patient’s individual response to therapy and the desired intensity of anticoagulation. In addition to hepatic metabolism, warfarin dose requirements are influenced by diet, drug-drug interactions, and health status. Therefore, the dose of warfarin must be determined by frequent clinical and laboratory monitoring.5,20,29 While there are conflicting data regarding the optimal warfarin induction regimen, most patients can start with 5 mg daily, and subsequent doses are determined based on INR response (Fig. 10–9). When initiating therapy, it is difficult to predict the precise warfarin maintenance dose that a patient will require. Patients who are younger (less than 55 years of age) and otherwise healthy can safely use higher warfarin “initiation” doses (e.g., 7.5 or 10 mg). A more conservative “initiation” dose (e.g., 5 mg or less) should be given to elderly patients (greater than 75 years of age), patients with heart failure, liver disease, or poor nutritional status, and patients who are taking interacting medications or are at high risk of bleeding.5,23 Loading doses of warfarin (e.g., 15 to 20 mg) are not recommended. These large doses can lead to the false impression that a therapeutic INR has been achieved in 2 to 3 days and lead to potential future overdosing.5 Before initiating therapy, screen the patient for any contraindications to anticoagulation therapy and risk factors for major bleeding (Tables 10–6 and 10–7). In addition, conduct a thorough medication history including the use of prescription and over-the-counter drugs, and any herbal supplements to detect interactions that may affect warfarin dosing requirements. In patients with acute VTE, a rapid-acting anticoagulant (UFH, LMWH, or fondaparinux) should be overlapped with warfarin for a minimum of 5 days and until the INR is greater than 2 and stable. This is important because the full antithrombotic effect will not be reached until 5 to 7 days or even longer after initiating warfarin therapy.5,17 The typical maintenance dose of warfarin for most patients will be between 25 and 55 mg per week, although some patients require higher or lower doses. Adjustments in the maintenance warfarin dose should be determined based on the total weekly dose and by reducing or increasing the weekly dose by increments of 5% to 25%. When adjusting the maintenance dose, wait at least 7 days to ensure that a steady state has been attained on the new dose before checking the INR again. Checking the INR too soon can lead to inappropriate dose adjustments and unstable anticoagulation status.5
![]() Warfarin requires frequent laboratory monitoring to ensure optimal outcomes and minimize complications. The PT is the most frequently used test to monitor warfarin’s anticoagulant effect. The PT measures the biological activity of factors II, VII, and X. Due to wide variation in reagent sensitivity, different thromboplastins will result in different PT results, potentially leading to inappropriate dosing decisions.5 In order to standardize result reporting, the World Health Organization (WHO) developed a reference thromboplastin and recommended the INR to monitor warfarin therapy. The INR corrects for the differences in thromboplastin reagents and uses the following formula: INR =(PTPatient/PTControl)ISI. The International Sensitivity Index (IST) is a measure of the thromboplastin’s responsiveness compared to the WHO reference.5 The goal or target INR for each patient is based on the indication for warfarin therapy. For the treatment and prevention of VTE, the INR target is 2.5 with an acceptable range of 2 to 3. In certain high-risk patients (e.g., certain mechanical heart valves), a higher target INR of 3 with a range of 2.5 to 3.5 is recommended.5 Before initiating warfarin therapy, a baseline PT/INR and CBC should be obtained. After initiating warfarin therapy, the INR should be monitored at least every 2 to 3 days during the first week of therapy. Once a stable response to therapy is achieved, INR monitoring is performed less frequently, weekly for the first 1 to 2 weeks, then every 2 weeks, and monthly thereafter.5,47 At each encounter, the patient should be carefully questioned regarding any factors that may influence the INR result. These factors include adherence to therapy, the use of interacting medications, consumption of vitamin K-rich foods, alcohol use, and general health status. Patients should also be questioned about symptoms related to bleeding and thromboembolic events. Warfarin dose adjustments should take into account not only the INR result, but also patient-related factors that influence the result. Structured anticoagulation therapy management services (anticoagulation clinics) have been demonstrated to improve the efficacy and safety of warfarin therapy when compared to “usual” medical care.5,47Some patients engage in self-testing and self-management by using a point of care PT/INR device approved for home use. Highly motivated and well-trained patients are good candidates for self-testing or self-management.5,47
Warfarin requires frequent laboratory monitoring to ensure optimal outcomes and minimize complications. The PT is the most frequently used test to monitor warfarin’s anticoagulant effect. The PT measures the biological activity of factors II, VII, and X. Due to wide variation in reagent sensitivity, different thromboplastins will result in different PT results, potentially leading to inappropriate dosing decisions.5 In order to standardize result reporting, the World Health Organization (WHO) developed a reference thromboplastin and recommended the INR to monitor warfarin therapy. The INR corrects for the differences in thromboplastin reagents and uses the following formula: INR =(PTPatient/PTControl)ISI. The International Sensitivity Index (IST) is a measure of the thromboplastin’s responsiveness compared to the WHO reference.5 The goal or target INR for each patient is based on the indication for warfarin therapy. For the treatment and prevention of VTE, the INR target is 2.5 with an acceptable range of 2 to 3. In certain high-risk patients (e.g., certain mechanical heart valves), a higher target INR of 3 with a range of 2.5 to 3.5 is recommended.5 Before initiating warfarin therapy, a baseline PT/INR and CBC should be obtained. After initiating warfarin therapy, the INR should be monitored at least every 2 to 3 days during the first week of therapy. Once a stable response to therapy is achieved, INR monitoring is performed less frequently, weekly for the first 1 to 2 weeks, then every 2 weeks, and monthly thereafter.5,47 At each encounter, the patient should be carefully questioned regarding any factors that may influence the INR result. These factors include adherence to therapy, the use of interacting medications, consumption of vitamin K-rich foods, alcohol use, and general health status. Patients should also be questioned about symptoms related to bleeding and thromboembolic events. Warfarin dose adjustments should take into account not only the INR result, but also patient-related factors that influence the result. Structured anticoagulation therapy management services (anticoagulation clinics) have been demonstrated to improve the efficacy and safety of warfarin therapy when compared to “usual” medical care.5,47Some patients engage in self-testing and self-management by using a point of care PT/INR device approved for home use. Highly motivated and well-trained patients are good candidates for self-testing or self-management.5,47
FIGURE 10–9. Initiation of warfarin therapy. (INR, International Normalized Ratio; PT, prothrombin time.) (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:350.)
![]() Similar to other anticoagulants, warfarin’s primary side effect is bleeding. Warfarin can “unmask” an existing lesion. The incidence of warfarin-related bleeding appears to be highest during the first few weeks of therapy. The annual incidence of major bleeding ranges from 1% to 10% depending on the quality of warfarin therapy management. Bleeding in the GI tract is most common. Intracranial hemorrhage (ICH) is one of the most serious complications, as it often causes severe disability and death. The intensity of anticoagulation therapy is related to bleeding risk. Higher INRs result in higher bleeding risk, and the risk of ICH increases when the INR exceeds 4.3,5Instability and wide fluctuations in the INR are also associated with higher bleeding risk. In cases of warfarin overdose or over-anticoagulation, vitamin K may be used to reverse warfarin’s effect (Fig. 10–10).5 Vitamin K can be given by the IV or oral route; the SC route is not recommended. When given SC, vitamin K is erratically absorbed and frequently ineffective. The IV route is reserved for cases of severe warfarin overdose (e.g., INR greater than 20) or major bleeding. Anaphylactoid reactions have been reported with rapid IV administration, therefore slow infusion is recommended. An oral dose of vitamin K will reduce the INR within 24 hours. If the INR is still elevated after 24 hours, another dose of oral vitamin K can be given. The dose of vitamin K should be based on the INR elevation. A dose of 1 to 2.5 mg is sufficient when the INR is between 5 and 9, but 5 mg may be required for INRs greater than 9. Higher doses (e.g., 10 mg) can lead to prolonged warfarin resistance. In cases of life-threatening bleeding, fresh frozen plasma or clotting factor concentrates should be administered, in addition to IV vitamin K. In patients in whom the INR is less than 9 and there is no active bleeding or imminent risk of bleeding, simply withholding warfarin until the INR decreases to within therapeutic range and reducing the weekly dose with more frequent monitoring is appropriate.5
Similar to other anticoagulants, warfarin’s primary side effect is bleeding. Warfarin can “unmask” an existing lesion. The incidence of warfarin-related bleeding appears to be highest during the first few weeks of therapy. The annual incidence of major bleeding ranges from 1% to 10% depending on the quality of warfarin therapy management. Bleeding in the GI tract is most common. Intracranial hemorrhage (ICH) is one of the most serious complications, as it often causes severe disability and death. The intensity of anticoagulation therapy is related to bleeding risk. Higher INRs result in higher bleeding risk, and the risk of ICH increases when the INR exceeds 4.3,5Instability and wide fluctuations in the INR are also associated with higher bleeding risk. In cases of warfarin overdose or over-anticoagulation, vitamin K may be used to reverse warfarin’s effect (Fig. 10–10).5 Vitamin K can be given by the IV or oral route; the SC route is not recommended. When given SC, vitamin K is erratically absorbed and frequently ineffective. The IV route is reserved for cases of severe warfarin overdose (e.g., INR greater than 20) or major bleeding. Anaphylactoid reactions have been reported with rapid IV administration, therefore slow infusion is recommended. An oral dose of vitamin K will reduce the INR within 24 hours. If the INR is still elevated after 24 hours, another dose of oral vitamin K can be given. The dose of vitamin K should be based on the INR elevation. A dose of 1 to 2.5 mg is sufficient when the INR is between 5 and 9, but 5 mg may be required for INRs greater than 9. Higher doses (e.g., 10 mg) can lead to prolonged warfarin resistance. In cases of life-threatening bleeding, fresh frozen plasma or clotting factor concentrates should be administered, in addition to IV vitamin K. In patients in whom the INR is less than 9 and there is no active bleeding or imminent risk of bleeding, simply withholding warfarin until the INR decreases to within therapeutic range and reducing the weekly dose with more frequent monitoring is appropriate.5
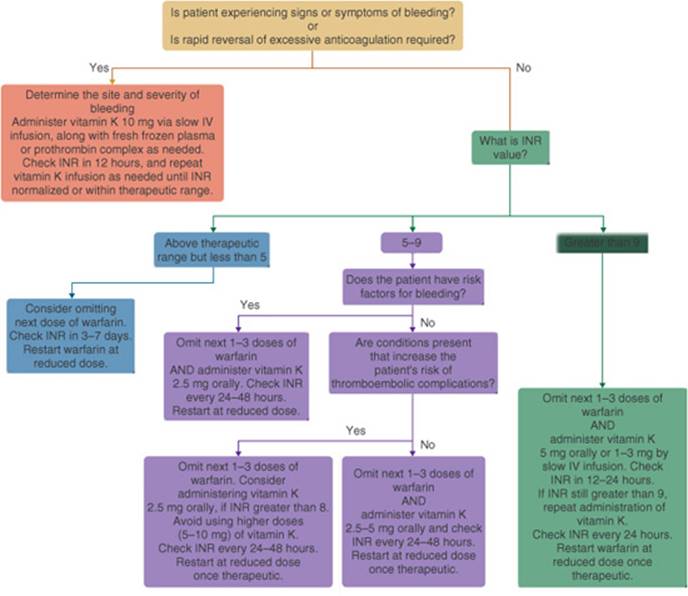
FIGURE 10–10. Management of an elevated INR in patients taking warfarin. Dose reductions should be made by determining the weekly warfarin dose and reducing the weekly dose by 10% to 25% based on the degree of INR elevation. Conditions that increase the risk of thromboembolic complications include history of hypercoagulability disorders (e.g., protein C or S deficiency, presence of antiphospholipid antibodies, antithrombin deficiency, or activated protein C resistance), arterial or venous thrombosis within the previous month, thromboembolism associated with malignancy, mechanical mitral valve in conjunction with atrial fibrillation, previous stroke, poor ventricular function, or coexisting mechanical aortic valve. (INR, International Normalized Ratio.) (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:353.)
Nonhemorrhagic side effects related to warfarin are rare but can be severe when they occur. Warfarin-induced skin necrosis presents as an eggplant-colored skin lesion or a maculopapular rash that can progress to necrotic gangrene. It usually manifests in fatty areas such as the abdomen, buttocks, and breasts. The incidence is less than 0.1%, and it generally appears during the first week of therapy. Patients with protein C or S deficiency or those who receive large loading doses of warfarin are at greatest risk.5,11 The mechanism is thought to be due to imbalances between procoagulant and anticoagulant proteins early in the course of warfarin therapy. Warfarin-induced purple toe syndrome is another rare side effect; patients present with a purplish discoloration of their toes. If these side effects are suspected, warfarin therapy should be discontinued immediately and an alternative anticoagulant given. There is a theoretical risk that warfarin may cause accelerated bone loss with long-term use, but to date there is no evidence to support this concern. Warfarin is teratogenic and is FDA pregnancy category X. It should be avoided during pregnancy, and women of child-bearing potential should be instructed to use an effective form of contraception. UFH and LMWH are the agents of choice for the treatment of VTE during pregnancy.5,11
![]() Warfarin is prone to numerous clinically significant drug-drug and drug-food interactions (Tables 10-8, 10-9, and 10-10). Patients on warfarin should be questioned at every encounter to assess for any potential interactions with foods, drugs, herbal products, and nutritional supplements. When an interacting drug is initiated or discontinued, more frequent monitoring should be instituted. In addition, the dose of warfarin can be modified (increased or decreased) in anticipation of the expected impact on the INR.5,48 Warfarin-related drug interactions can generally be divided into two major categories: pharmacokinetic and pharmacodynamic. Pharmacokinetic interactions are most commonly due to changes in hepatic metabolism or vitamins that can interact with warfarin.49 Patients on warfarin may experience changes in the INR due to fluctuating intake of dietary vitamin K. Patients should be instructed to maintain a consistent diet and avoid large fluctuations in vitamin K intake rather than strictly avoiding vitamin K-rich foods.49
Warfarin is prone to numerous clinically significant drug-drug and drug-food interactions (Tables 10-8, 10-9, and 10-10). Patients on warfarin should be questioned at every encounter to assess for any potential interactions with foods, drugs, herbal products, and nutritional supplements. When an interacting drug is initiated or discontinued, more frequent monitoring should be instituted. In addition, the dose of warfarin can be modified (increased or decreased) in anticipation of the expected impact on the INR.5,48 Warfarin-related drug interactions can generally be divided into two major categories: pharmacokinetic and pharmacodynamic. Pharmacokinetic interactions are most commonly due to changes in hepatic metabolism or vitamins that can interact with warfarin.49 Patients on warfarin may experience changes in the INR due to fluctuating intake of dietary vitamin K. Patients should be instructed to maintain a consistent diet and avoid large fluctuations in vitamin K intake rather than strictly avoiding vitamin K-rich foods.49
Table 10–8 Clinically Significant Warfarin Drug Interactions
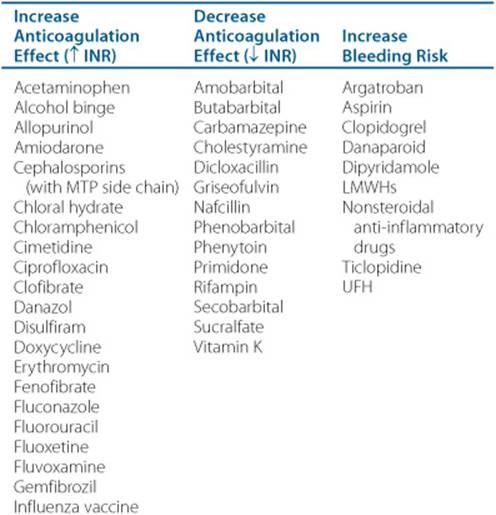

Table 10–9 Potential Warfarin Interactions With Herbal and Nutritional Products

Nonpharmacologic
Thrombectomy
Most cases of VTE can be successfully treated with anticoagulation. In some cases, removal of the occluding thrombus by surgical intervention may be warranted. Surgical or mechanical thrombectomy can be considered in patients with massive iliofemoral DVT when there is a risk of limb gangrene due to venous occlusion. The procedure can be complicated by recurrence of thrombus formation. In patients who present with massive PE, pulmonary embolectomy can be performed in emergency cases when conservative measures have failed. Patients who are hemodynamically unstable and have a contraindication to thrombolysis are candidates for pulmonary embolectomy. Administer heparin by IV infusion to achieve a therapeutic aPTT during the operation and postoperatively. Thereafter, give warfarin for the usual recommended duration.17,23
Vena Cava Interruption
IVC interruption is indicated in patients with PE who have a contraindication to anti-coagulation therapy and in patients who have recurrent VTE while taking anticoagulation therapy.17,23 IVC interruption is accomplished by inserting a filter through the internal jugular vein or femoral vein and advancing it into the IVC using ultrasound or fluoroscopic guidance. IVC filters reduce the short-term risk of PE, but this benefit is not sustained in the long term. The incidence of DVT at 1 year after IVC filter insertion is higher when compared to patients without filters. Survival after a PE is no different in patients with filters versus patients without filters.21 Therefore, anticoagulation therapy should be resumed as soon as possible after filter insertion and continued as long as the filter is in place due to the high risk of DVT.17Temporary or removable filters are now increasingly used and are replacing the use of permanent filters.17
Table 10–10 Vitamin K Content of Select Foodsa
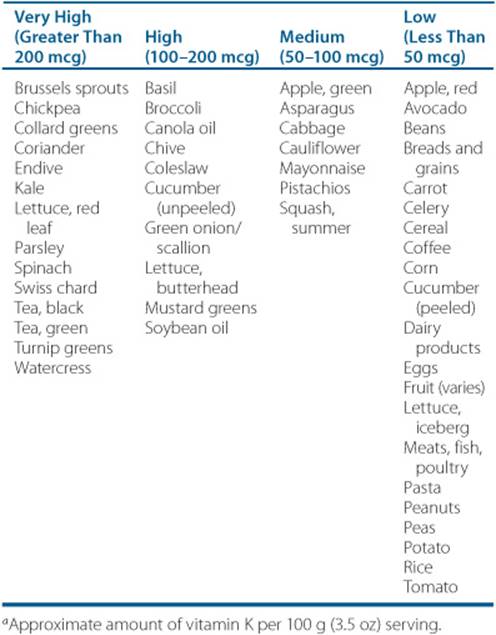
Patient Encounter 1, Part 2
KK has been hospitalized for right hip fracture repair. Two weeks after her discharge from the hospital, she presents to the emergency department with complaints of swelling, redness, and pain in her right lower extremity. KK states her symptoms started 3 days ago and have gotten progressively worse. During your interview, the patient states that she was sent home with a prescription for fondaparinux 2.5 mg SC daily. A duplex ultrasound shows a proximal DVT in her right lower extremity. All of her other laboratory values are within normal limits.
Which ofKK’s symptoms are consistent with an acute DVT?
Design an appropriate treatment plan forKK. Your plan should include acute and chronic therapy—specify the drug(s), dose(s), route, frequency of administration, and duration of each therapy, as well monitoring parameters, patient education, and follow-up plan.
Assuming KK continues to take the prescription and over-the-counter medications listed in her medication history obtained during her hospitalization, should any of these medications be discontinued or changed? If changed, what alternative therapy would you recommend?
Is KKa candidate for outpatient treatment of her DVT?
Compression Stockings
PTS occurs in 20% to 50% of patients within 8 years after a DVT. Wearing GCS after a DVT reduces the risk of PTS by as much as 50%. Current guidelines recommend the use of GCS with an ankle pressure of 30 to 40 mm Hg for 2 years after a DVT. To be effective, GCS must fit properly. Traditionally, strict bed rest has been recommended after a DVT, but this approach has now been refuted and patients should be encouraged to ambulate as tolerated.17,20
APPROACH TO TREATING PATIENTS WITH VTE
![]() Once the diagnosis of VTE has been confirmed with an objective test, promptly start anticoagulation therapy in full therapeutic doses. If there is high clinical suspicion of VTE, anticoagulation therapy can be initiated while waiting for the results of diagnostic tests.17 Initiate therapy with a quick-acting anticoagulant such as UFH (given IV or SC), an LMWH (given SC), or fondaparinux (given SC; Figs. 10-5and 10-11). In patients with adequate renal function, the LMWHs are preferred over UFH.17 Recent evidence also supports the use of SC fondaparinux as an alternative option to UFH or LMWH for the initial treatment of VTE.17 For the long-term treatment phase, warfarin is the preferred approach except for patients with cancer, in whom an LMWH is recommended due to better efficacy in preventing recurrent thromboembolic events. Initiate warfarin on the first day of therapy after the first dose of UFH, LMWH, or fondaparinux is given. Overlap the injectable agent with warfarin therapy for a minimum of 5 days. Warfarin should be dosed to achieve a goal INR range of 2 to 3. Once the INR is stable and above 2, the injectable anticoagulant should be discontinued. Anticoagulation therapy is continued for a minimum of 3 months but should be given longer depending on the underlying etiology of the VTE and the patient’s risk factors (Table 10–11).17,20,21 Use a thrombolytic only if the patient has a massive iliofemoral DVT and is at risk of limb gangrene. In patients with PE, use a thrombolytic if the patient is hemodynamically unstable (i.e., SBP less than 90 mm Hg). If there is a contraindication to anticoagulation therapy or the patient has failed therapy with an anticoagulant, a vena cava filter should be inserted. Encourage early ambulation as tolerated by the patient during the initial treatment phase.17,20,21
Once the diagnosis of VTE has been confirmed with an objective test, promptly start anticoagulation therapy in full therapeutic doses. If there is high clinical suspicion of VTE, anticoagulation therapy can be initiated while waiting for the results of diagnostic tests.17 Initiate therapy with a quick-acting anticoagulant such as UFH (given IV or SC), an LMWH (given SC), or fondaparinux (given SC; Figs. 10-5and 10-11). In patients with adequate renal function, the LMWHs are preferred over UFH.17 Recent evidence also supports the use of SC fondaparinux as an alternative option to UFH or LMWH for the initial treatment of VTE.17 For the long-term treatment phase, warfarin is the preferred approach except for patients with cancer, in whom an LMWH is recommended due to better efficacy in preventing recurrent thromboembolic events. Initiate warfarin on the first day of therapy after the first dose of UFH, LMWH, or fondaparinux is given. Overlap the injectable agent with warfarin therapy for a minimum of 5 days. Warfarin should be dosed to achieve a goal INR range of 2 to 3. Once the INR is stable and above 2, the injectable anticoagulant should be discontinued. Anticoagulation therapy is continued for a minimum of 3 months but should be given longer depending on the underlying etiology of the VTE and the patient’s risk factors (Table 10–11).17,20,21 Use a thrombolytic only if the patient has a massive iliofemoral DVT and is at risk of limb gangrene. In patients with PE, use a thrombolytic if the patient is hemodynamically unstable (i.e., SBP less than 90 mm Hg). If there is a contraindication to anticoagulation therapy or the patient has failed therapy with an anticoagulant, a vena cava filter should be inserted. Encourage early ambulation as tolerated by the patient during the initial treatment phase.17,20,21
Patient Encounter 2, Part 1
BA is a 38-year-old female who presents to the emergency department complaining of chest pain, shortness of breath, and lightheadedness. The patient states that her symptoms started with some mild left calf pain approximately 5 days ago. She started feeling short of breath and experiencing chest pain last evening. She could not sleep and her shortness of breath has gotten progressively worse in the last several hours. BA was hospitalized because she was suspected to have a PE.
PMH: Obesity × 12 years; asthma
FH: Mother died of a stroke; paternal grandmother had clots in her legs
SH: The patient is a school teacher
Current Meds: Albuterol (salbutamol) metered-dose inhaler as needed; ortho-Tri-Cyclen Lo by mouth daily; echinacea one to two tablets by mouth daily as needed; multivitamin one tablet by mouth daily
Allergies: Shellfish, NKDA
PE:
VS: BP 104/64 mm Hg, HR 102, RR 20, T 38°C (100.4°F), wt96kg (211 lb), ht 65 in. (165 cm)
Labs: Within normal limits; estimated GFR 101 mL/min
Procedures/Tests
ECG: Normal sinus rhythm
CXR: Slightly enlarged heart
V/Q scan: High probability of PE
What signs and symptoms are consistent with the diagnosis ofPE in BA’s case? What are the most likely etiologies forPE in this case?
What are appropriate initial and chronic treatment options for BA?
If UFH is chosen as the initial anticoagulation treatment option, what is the goal aPTT?
What is BA’s goal INR for warfarin therapy?
How long should BA remain on anticoagulation therapy?
Given the list of medications BA took prior to hospitalization, should any of these medications be discontinued or changed? If changed, what alternative therapy would you recommend?
OUTCOME EVALUATION
• Achieve optimal outcomes by: (a) preventing the occurrence of VTE in patients who are at risk, (b) administering effective treatments in a timely manner to patients who develop VTE, (c) preventing treatment-related complications, and (d) reducing the likelihood of long-term complications including recurrent events.

FIGURE 10–11. Treatment of VTE. (LMWH, low-molecular weight heparin; PE, pulmonary embolism; SBP, systolic blood pressure; UFH, unfractionated heparin; VTE, venous thromboembolism.) (From Haines ST, Witt DM, Nutescu EA. Venous thromboembolism. In: DiPiro JT, Talbert RL, Yee GC, et al., eds. Pharmacotherapy: A Pathophysiologic Approach, 7th ed. New York: McGraw-Hill; 2008:357.)
• Given that VTE is often clinically silent and potentially fatal, strategies to increase the widespread use of prophylaxis have the greatest potential to improve patient outcomes. Thus, relying on the early diagnosis and treatment of VTE is unacceptable because many patients will die before treatment can be initiated.
• Effective VTE prophylaxis programs screen and identify all patients at risk, determine each patient’s level of risk, and select and implement regimens that provide sufficient protection for the level of risk.
Table 10–11 Duration of Anticoagulation Therapy for the Treatment of VTE
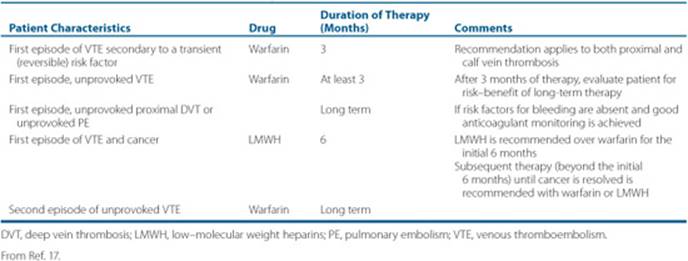
Patient Encounter 2, Part 2
BA is discharged home on warfarin therapy. She was referred to a local area antithrombosis center for monitoring of her oral anticoagulation therapy and has been maintained on warfarin 6 mg daily for the last 3 months. The patient presents today for a routine visit for anticoagulation monitoring and her INR is 8.3. She reports that 6 days ago she was started on metronidazole 500 mg by mouth twice daily, which was prescribed by her primary care physician for a vaginal infection. In addition, the primary care physician told the patient that her thyroid gland was enlarged and ordered some lab tests to determine if she has a thyroid problem. The patient has not heard what the results are. She also reports that her intake of vitamin K-rich foods (spinach, broccoli, and cabbage) has increased significantly over the last month because she is trying to lose weight. BA has no other complaints today and denies any signs or symptoms of bleeding.
What is the most likely explanation for elevated INR in BA’s case?
Should BA be given vitamin K? If yes, discuss the dose, route of administration, and an appropriate patient monitoring plan.
How will you manage BA’s warfarin therapy? Outline a plan including specific dose changes, timing of monitoring, and patient education.
• Periodically evaluate patients who receive prophylaxis during the course of treatment for signs and symptoms of VTE, such as swelling, pain, warmth, and redness of lower extremities, and for DVT, as well as chest pain, shortness of breath, palpitations, and hemoptysis.
• Providing effective treatment in a timely manner is the primary goal for patients who develop VTE. Treat DVT and PE quickly and aggressively with effective doses of anticoagulant drugs.
• The short-term aim of therapy is to prevent propagation or local extension of the clot, embolization, and death.
• Regularly monitor patients for the development of new symptoms or worsening of existing symptoms.
• Anticoagulant drugs require precise dosing and meticulous monitoring. Closely monitor patients receiving anticoagulant therapy for signs and symptoms of bleeding including epistaxis, hemoptysis, hematuria, bright red blood per rectum, tarry stools, severe headache, and joint pain. If major bleeding occurs, stop therapy immediately and treat the source of bleeding. In addition, closely monitor patients for potential drug-drug and drug-food interactions and adherence with the prescribed regimen.
• The long-term (more than 3 months after the first event) goals of therapy are to prevent complications such as the PTS, pulmonary hypertension, and recurrent VTE.
• Encourage all patients who have had DVT to wear GCS.
• Continue warfarin therapy for an appropriate duration based on the etiology of the initial clot and the presence of ongoing risk factors.
Patient Care and Monitoring
Day 1
1. It is critical to first confirm diagnosis of VTE
• Clinical assessment: look for risk factors for VTE
• If DVT symptoms are present, obtain a venous ultrasound
• If PE is suspected, obtain a V/Q or CT scan
• D-dimer: this test may be a helpful adjunct to either a venous ultrasound or V/Q scan
2. Obtain baseline laboratory tests. These tests must be obtained prior to initiating anticoagulation therapy:
• PT and calculated INR
• aPTT
• Serum creatinine
• CBC with platelets
3. Medications:
• Screen the patient’s pharmacy profile for potential drug-drug interactions with anticoagulation therapy
• Initiate UFH or LMWH or fondaparinux by injection (see Table 10–3 for dosing guidelines)
• Start warfarin sodium orally every evening (see Fig. 10–9 for dosing guidelines)
• Start pain medication if necessary (avoid nonsteroidal anti-inflammatory drugs)
4. Patient education:
• Educate the patient regarding the purpose of therapy and importance of proper monitoring of anticoagulant drugs. Assist the patient to determine an appropriate provider for long-term monitoring of anticoagulation therapy.
• If LMWH or fondaparinux is selected, teach the patient how to self-administer (if the patient or a family member is unwilling or unable to self-administer, visiting nurse services should be arranged). Initial injection should be administered in the medical office or hospital.
• Inform patient about the effects of vitamin K-rich foods on warfarin therapy. Moderate intake (less than 500-1,000 mcg) of vitamin K is acceptable. Provide patient with written material regarding vitamin K content of foods.
• Inform the patient about the potential drug-drug interactions with warfarin, including over-the-counter medications and dietary supplements (Tables 10–8, 10–9, and 10–10). Instruct the patient to call the health care practitioner responsible for monitoring warfarin therapy before starting any new medications or dietary supplements.
• Instruct the patient regarding nonpharmacologic strategies including elevation of the affected extremity and antiembolic exercises such as flexion/extension of the ankle (for lower extremity VTE) or hand squeezing/relaxation (for upper extremity VTE).
5. Next steps:
• If the patient is to be treated at home, dispense to the patient a 5-to 7-day supply of prefilled LMWH or fondaparinux syringes in patient-specific dose.
• If the patient is to be treated with UFH, measure aPTT (or antifactor Xa activity) 6 hours after initiating the IV infusion. Adjust dose if necessary (Table 10–5) and measure aPTT (or antifactor Xa activity) every 6 hours after each dose change until therapeutic. Measure aPTT (or antifactor Xa activity) daily thereafter.
• Arrange for follow-up and long-term anticoagulation therapy management. Communicate with the patient’s primary care physician and/or refer to a local antithrombosis service, if available. If the patient is to be treated primarily in the hospital, these arrangements can be made 1 to 2 days prior to hospital discharge.
6. Document all activities in medical record.
Day 2
1. If the patient is being treated with UFH, remeasure the aPTT, and adjust dose if necessary. If patient is being treated with LMWH or fondaparinux, continue therapy.
2. Interview the patient to determine if there is worsening or new symptoms related to VTE. Ask the patient about overt bruising or bleeding, particularly at the injection site, as well as changes in stool or urine color.
3. Advise the patient to limit physical activity if pain persists and to elevate the extremity; increase activity as tolerated.
4. Document activities in medical record.
Days 3 to 5
1. Measure PT/INR every 1 to 2 days.
2. Interview the patient to determine if there is worsening or new symptoms related to VTE. Inquire about and evaluate patient adherence to therapy. Ask the patient about overt bruising or bleeding, particularly at the injection site, as well as changes in stool or urine color. Advise the patient to limit physical activity if pain persists and to elevate the extremity; increase activity as tolerated. Reinforce previous patient education regarding vitamin K intake and potential drug-drug interactions with warfarin.
3. Hold or adjust warfarin dose as necessary. If the patient is being treated with UFH, measure aPTT daily, and adjust dose if necessary. If the patient is being treated with an LMWH or fondaparinux, continue therapy.
4. Document activities in medical record.
Days 6 to 8
1. Measure PT/INR every 2 to 3 days. Obtain CBC or platelet count.
2. Interview the patient to determine if there is worsening or new symptoms related to VTE. Inquire about and evaluate patient adherence to therapy. Ask the patient about overt bruising or bleeding, particularly at the injection site, as well as changes in stool or urine color. Advise the patient to limit physical activity if pain persists and to elevate the extremity; increase activity as tolerated. Reinforce previous patient education regarding vitamin K intake and potential drug-drug interactions with warfarin.
3. Hold or adjust warfarin dose as necessary. Discontinue UFH, LMWH, or fondaparinux if INR is greater than 2 on two consecutive occasions. If the patient requires continued treatment with UFH, measure aPTT, and adjust dose if necessary.
4. If the patient is treated with UFH or LMWH and platelet count has dropped by greater than 50% from baseline or is less than 120 × 103/μL, evaluate the patient for HIT.
5. Document activities in medical record.
Days 9 to 14
1. Measure PT/INR every 3 to 5 days.
2. Interview the patient to determine if there is worsening or new symptoms related to VTE. Inquire about and evaluate patient adherence to therapy. Ask the patient about overt bruising or bleeding, particularly at the injection site, as well as changes in stool or urine color. Advise the patient to elevate the extremity and increase activity as tolerated. Reinforce previous patient education regarding vitamin K intake and potential drug-drug interactions with warfarin.
3. Hold or adjust warfarin dose as necessary. Discontinue UFH, LMWH, or fondaparinux if INR is greater than 2 on two consecutive occasions. If the patient requires continued treatment with UFH, remeasure aPTT, and adjust dose if necessary.
4. Obtain CBC or platelet count. If the patient is treated with UFH or LMWH and platelet count has dropped by more than 50% from baseline or is less than 120 × 103/mm3 (120 × 109L), evaluate the patient for HIT.
5. Document activities in medical record.
Days 15 to 90
1. Measure PT/INR every 1 to 4 weeks based on the stability of the INR and patient’s health status.
2. Interview the patient to determine if there is worsening or new symptoms related to VTE. Inquire about and evaluate patient adherence to therapy. Ask the patient about overt bruising or bleeding as well as changes in stool or urine color. Encourage the patient to increase activity as tolerated. Reinforce previous patient education regarding vitamin K intake and potential drug-drug interactions with warfarin.
3. Adjust warfarin dose as necessary. Consider restarting LMWH or fondaparinux if INR drops below 1.5.
4. Document activities in medical record.
Three Months and Beyond
1. Measure PT/INR every 1 to 4 weeks based on the stability of the INR and patient’s health status.
2. Interview the patient to determine if there is worsening or new symptoms related to VTE. Inquire about and evaluate patient adherence to therapy. Ask the patient about overt bruising or bleeding as well as changes in stool or urine color. Reinforce previous patient education regarding vitamin K intake and potential drug-drug interactions with warfarin.
3. Reevaluate the risks and benefits of continuing warfarin therapy.
4. Document activities in medical record.
Abbreviations Introduced in This Chapter


 Self-assessment questions and answers are available at http://www.mhpharmacotherapy.com/pp.html.
Self-assessment questions and answers are available at http://www.mhpharmacotherapy.com/pp.html.
REFERENCES
1. Turpie AGG, Chin BSP, Lip GYH. Venous thromboembolism: Pathophysiology, clinical features, and prevention. BMJ 2002;325: 887-890.
2. Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:381S-453S.
3. Schulman S, Beyth RJ, Kearon C, Levine MN; American College of Chest Physicians. Hemorrhagic complications of anticoagulant and thrombolytic treatment: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:257S-298S.
4. Hirsh J, Bauer KA, Donati MB, et al. Parenteral anticoagulants: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:141S-159S.
5. Ansell J, Hirsh J, Hylek E, et al. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:160S-198S.
6. Haines ST, Nutescu EA. Current and emerging treatment options for venous thrombosis: A case discussion. Am J Health Syst Pharm 2005; 62:593-605.
7. Spencer FA, Emery C, Lessard D, et al. The Worcester venous thromboembolism study: A population-based study of the clinical epidemiology of venous thromboembolism. J General Intern Med 2006;21:722-727.
8. Johnson CM, Mureebe L, Silver D. Hypercoagulable states: A review. Vasc Endovascular Surg 2005;39:123-133.
9. Khorana AA. The NCCN Clinical Practice Guidelines on venous thromboembolic disease: Strategies for improving VTE prophylaxis in hospitalized cancer patients. Oncologist 2007;12:1361-1370.
10. Canonico M, Plu-Bureau G, Lowe GDO, Scarabin P-Y. Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: Systematic review and meta-analysis. BMJ 2008;336:1227-1231.
11. Bates SM, Greer IA, Pabinger I, Sofaer S, Hirsh J. American College of Chest Physicians. Venous thromboembolism, thrombophilia, antithrombotic therapy, and pregnancy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:844S-886S.
12. Aird WC. Coagulation. Crit Care Med 2005;33:S485-S487.
13. Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol 2005;129:307-321.
14. Kearon C. Natural history of venous thromboembolism. Circulation 2003;107:122-130.
15. Wells PS, Owen C, Doucette S, Fergusson D, Tran H. Does this patient have deep vein thrombosis? JAMA 2006;295:199-207.
16. Sinert R, Foley M. Clinical assessment of the patient with a suspected pulmonary embolism. Ann Emerg Med 2008;52:76-79.
17. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:454S-545S.
18. Kanne JP, Lalani TA. Role of computed tomography and magnetic resonance imaging for deep venous thrombosis and pulmonary embolism. Circulation 2004;109:115-121.
19. Zierler BK. Ultrasonography and diagnosis ofvenous thromboembolism. Circulation 2004;109:19-114.
20. Bates SM, Ginsberg JS. Clinical practice. Treatment of deep-vein thrombosis. N Engl J Med 2004;351:268-277.
21. Decousus H, Leizorovicz A, Parent F, et al. A clinical trial of vena caval filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med 1998;338: 409-415.
22. Turpie AG, Bauer KA, Eriksson BI, Lassen MR. Fondaparinux vs enoxaparin for the prevention of venous thromboembolism in major orthopedic surgery: A meta-analysis of 4 randomized double-blind studies. Arch Intern Med 2002;162:1833-1840.
23. Buller HR, Sohne M, Middeldorp S. Treatment of venous thromboembolism. J Thromb Haemost 2005;3:1554-1560.
24. Nutescu EA. Emerging options in the treatment of venous thromboembolism. Am J Health Syst Pharm 2004;61:S12-S17.
25. Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004:CD002783.
26. Tapson VF. Acute pulmonary embolism. N Engl J Med 2008;358: 1037-1052.
27. Agnelli G, Becattini C, Kirschstein T. Thrombolysis vs heparin in the treatment of pulmonary embolism: A clinical outcome-based meta-analysis. Arch Intern Med 2002;162:2537-2541.
28. Wood KE. Major pulmonary embolism: Review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest 2002;121:877-905.
29. Nutescu EA, Shapiro NL, Chevalier A, Amin AN. A pharmacologic overview of current and emerging anticoagulants. Cleve Clin J Med 2005;72(Suppl 1):S2-S6.
30. Raschke RA, Reilly BM, Guidry JR, et al. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. Ann Intern Med 1993;119:874-881.
31. Segal JB, Streiff MB, Hofmann LV, et al. Management of venous thromboembolism: A systematic review for a practice guideline. Ann Intern Med 2007;146(3):211-222.
32. Kearon C, Ginsberg JS, Julian JA, et al. Comparison of fixed-dose weight-adjusted unfractionated heparin and low-molecular-weight heparin for acute treatment of venous thromboembolism. JAMA 2006;296(8):935-948.
33. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulation therapy. Laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782-788.
34. Monagle P, Chalmers E, Chan A, et al. Antithrombotic therapy in neonates and children: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:887S-968S.
35. Nutescu EA, Wittkowsky AK, Dobesh PP, Hawkins DW, Dager WE. Choosing the appropriate antithrombotic agent for the prevention and treatment of VTE: A case-based approach. Ann Pharmacother 2006;40(9):1558-1571.
36. Akl EA, Rohilla S, Barba M, et al. Anticoagulation for the initial treatment of venous thromboembolism in patients with cancer. Cochrane Database Syst Rev 2008(1):CD006649.
37. Nutescu EA. Assessing, preventing, and treating venous thromboembolism: Evidence-based approaches. Am J Health Syst Pharm 2007;64(11 Suppl 7):S5-S13.
38. ScarvelisD, Wells PS. Diagnosis and treatment of deep-vein thrombosis. CMAJ 2006;175(9):1087-1092.
39. Laposata M, Green D, Van Cott EM, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: The clinical use and laboratory monitoring of low-molecular-weight heparin, danaparoid, hirudin and related compounds, and argatroban. Arch Pathol Lab Med 1998;122:799-807.
40. Duhl AJ, Paidas MJ, Ural SH, et al., Pregnancy and Thrombosis Working Group. Antithrombotic therapy and pregnancy: Consensus report and recommendations for prevention and treatment of venous thromboembolism and adverse pregnancy outcomes. Am J Obstet Gynecol 2007;197(5):457.el-e21.
41. Weitz J, Hirsh J, Samama MM. New antithrombotic drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines, 8th ed. Chest 2008;133:234S-256S.
42. Gulseth MP, Michaud J, Nutescu EA. Rivaroxaban: An oral direct inhibitor of factor Xa. Am J Health Syst Pharm 2008;65(16)1520-1529.
43. Buller HR, Davidson BL, Decousus H, et al. Subcutaneous fondaparinux versus intravenous unfractionated heparin in the initial treatment of pulmonary embolism. N Engl J Med 2003;349:1695-1702.
44. Buller HR, Davidson BL, Decousus H, etal. Fondaparinux or enoxaparin for the initial treatment of symptomatic deep venous thrombosis: A randomized trial. Ann Intern Med 2004;140:867-873.
45. Nutescu EA, Shapiro NL, Chevalier A. New anticoagulant agents: Direct thrombin inhibitors. Cardiol Clin 2008;26(2):169-187.
46. Lenzini PA, Grice GR, Milligan PE, et al. Laboratory and clinical outcomes of pharmacogenetic vs. clinical protocols for warfarin initiation in orthopedic patients. J Thromb Haemost 2008;6(10):1655-1662.
47. Garcia DA, Witt DM, Hylek E, et al. Delivery of optimized anticoagulant therapy: Consensus statement from the Anticoagulation Forum. Ann Pharmacother 2008;42(7):979-988.
48. Holbrook AMMDPMF, Pereira JAM, Labiris RP, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005;165:1095-1106.
49. Nutescu EA, Shapiro NL, Ibrahim S, West P. Warfarin and its interactions with foods, herbs and other dietary supplements. Expert Opin Drug Saf 2006;5(3):433-451.