Dianne Brundage
LEARNING OBJECTIVES
Upon completion of the chapter, the reader will be able to:
1. Describe the etiology of cancer.
2. Define the tumor, nodes, metastases (TNM) system of cancer staging.
3. Classify each drug used in the treatment of cancer, and compare and contrast the mechanisms of action, uses, and side effects.
4. Outline actions for all health care providers to prevent medication errors with cancer treatments.
5. Describe the role of the health care practitioner in the care of cancer patients.
KEY CONCEPTS
![]() The word cancer covers a diverse array of tumor types that affect a significant number of Americans and are a significant cause of mortality.
The word cancer covers a diverse array of tumor types that affect a significant number of Americans and are a significant cause of mortality.
![]() Numerous cellular changes occur in the genetic material of the cancer cell so that programmed cell death, or apoptosis, does not occur. Proliferation of cancer cells goes unregulated.
Numerous cellular changes occur in the genetic material of the cancer cell so that programmed cell death, or apoptosis, does not occur. Proliferation of cancer cells goes unregulated.
![]() Many tumors are staged according to the tumor, nodes, metastases (TNM) system. Metastases are cancer cells that have spread to sites distant from the primary tumor site and have started to grow. The most frequently-occurring sites of metastases of solid tumors are the brain, bone, liver, and lungs.
Many tumors are staged according to the tumor, nodes, metastases (TNM) system. Metastases are cancer cells that have spread to sites distant from the primary tumor site and have started to grow. The most frequently-occurring sites of metastases of solid tumors are the brain, bone, liver, and lungs.
![]() Each category of chemotherapy drugs has some similar side effects, usually on the most rapidly-growing cells of the body. However, there are unique toxicities of various pharmacologic categories of antineoplastic agents. Anthracyclines cause cardiac toxicity, which is related to the cumulative dose. Tubulin-interactive agents are associated with neuropathy and ileus. Alkylating agents are associated with secondary malignancies.
Each category of chemotherapy drugs has some similar side effects, usually on the most rapidly-growing cells of the body. However, there are unique toxicities of various pharmacologic categories of antineoplastic agents. Anthracyclines cause cardiac toxicity, which is related to the cumulative dose. Tubulin-interactive agents are associated with neuropathy and ileus. Alkylating agents are associated with secondary malignancies.
![]() Because of the severe toxicities associated with many of the chemotherapy agents, safety precautions must be in place to prevent chemotherapy errors, accidental chemotherapy exposures, and overdosages.
Because of the severe toxicities associated with many of the chemotherapy agents, safety precautions must be in place to prevent chemotherapy errors, accidental chemotherapy exposures, and overdosages.
![]() Clinicians should play a role in chemotherapy safety, patient education, and monitoring patient response to therapy. For example, cumulative doses of anthracyclines should be monitored along with signs and symptoms of heart failure. Clinicians also should monitor for drug interactions between other current medications and chemotherapy agents.
Clinicians should play a role in chemotherapy safety, patient education, and monitoring patient response to therapy. For example, cumulative doses of anthracyclines should be monitored along with signs and symptoms of heart failure. Clinicians also should monitor for drug interactions between other current medications and chemotherapy agents.
EPIDEMIOLOGY
![]() The word cancer covers a diverse array of tumors types that affect a significant number of Americans and are a significant cause of mortality. The term cancer actually refers to more than 100 diseases. What is common to all cancers is that the cancerous cell has uncontrolled growth so that it invades tissues and spreads to other parts of the body, called metastases. In 2008, it was projected that over 1.4 million Americans will be diagnosed with cancer, and more than 565,000 Americans will die from the cancer.1 Figure 88–1 describes cancers by gender, new cases, and deaths.
The word cancer covers a diverse array of tumors types that affect a significant number of Americans and are a significant cause of mortality. The term cancer actually refers to more than 100 diseases. What is common to all cancers is that the cancerous cell has uncontrolled growth so that it invades tissues and spreads to other parts of the body, called metastases. In 2008, it was projected that over 1.4 million Americans will be diagnosed with cancer, and more than 565,000 Americans will die from the cancer.1 Figure 88–1 describes cancers by gender, new cases, and deaths.
A cancer patient may encounter many different health care professionals: phlebotomists, pathologists, surgeons, medical and radiation oncologists, physician assistants, pharmacists, nurses, counselors, dieticians, social workers, and chaplains all may be involved with a single patient. The pharmacist’s role may include recommendations of various pharmacologic agents, education of patients and family members, education of staff about new agents and safety issues, preparation of therapies, resolution of reimbursement issues, development of order sets, and participation in clinical trials. Each patient should have access to an interdisciplinary team to assist him or her during treatment.
Cancer treatments have exploded due to advances in technology in the last couple of decades. The fields of radiat ion therapy, surgery, and pharmaceuticals have had numerous developments, so patients are receiving not only less toxic treatments but also treatments that have improved outcomes over those of 15 years ago. Supportive-care therapies have improved, so patients may be at less risk for toxicity and have a better quality of life than patients 10 to 15 years ago. In the early 1990s, most patients received chemotherapy in the hospital because of side effects. Today, most patients receive chemotherapy in the clinic and/or are taking oral agents at home.
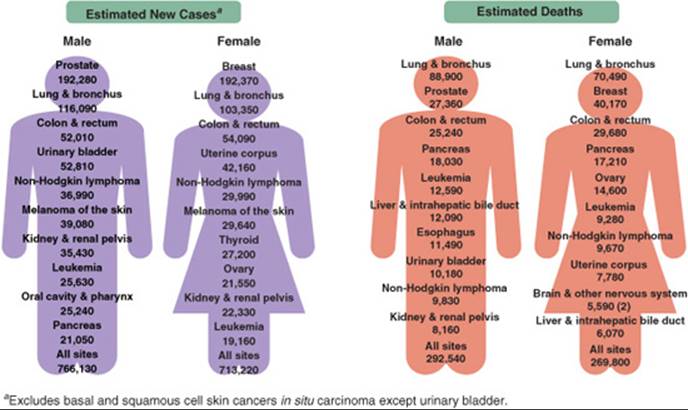
FIGURE 88–1. Cancer incidences (left) and deaths (right) in the United States for males and females estimated for 2009. (Reprinted with permission from American Cancer Society. Cancer facts and figures—2009. Atlanta: American Cancer Society; 2009.)
Cancer Prevention
Because most cancers are not curable in advanced stages, cancer prevention is an important avenue of exploration. Both lifestyle modifications and chemoprevention agents ultimately may reduce the risk of developing cancer.
Tobacco
Tobacco smoking increases the risk of developing not only lung cancer but also many other types of cancer, including cancer of the bladder, mouth, pharynx, larynx, and esophagus. While the immediate benefit of smoking cessation is minimal for lung cancer, documented benefit has been observed 6 or more years after stopping.
Sun Exposure
Ultraviolet light and increased skin exposure may increase the risk of skin malignancies, especially in individuals who are fair-skinned. Practitioners can counsel patients to minimize skin exposure to the sun and to use strong sunscreens on exposed areas.
CARCINOGENESIS
The exact cause of cancer remains unknown and probably is very diverse given the vast array of diseases called cancer. It is thought that cancer develops from a single cell in which the normal mechanisms for control of growth and proliferation are altered. Initiation occurs when a carcinogenic substance encounters a normal cell to produce genetic damage, or a mutated cell. Environmental or other factors that favor the growth of the mutated cell refer to promotion. Transformation occurs when the mutated cell becomes malignant, and progression occurs when cell proliferation takes over and the tumor spreads or develops metastases. Depending upon the type of cancer, many years may go by between the carcinogenic phases and the development of a clinically detectable tumor.
Carcinogenic agents include chemicals in the environment, such as aniline and benzene, which are associated with the development of bladder cancer and leukemia, respectively. Environmental factors, such as excessive sun exposure, also may result in cancer. Viruses, including the human papilloma virus and hepatitis B, may be associated with the development of cancer. Some of the chemotherapy agents cause secondary cancers after therapy has been completed. Numerous factors may contribute to the development of cancer. In addition to the carcinogenic agents mentioned, factors such as the patient’s age, gender, diet, and chronic irritation or inflammation may be considered to be promoters of carcinogenesis.
Cancer Genetics
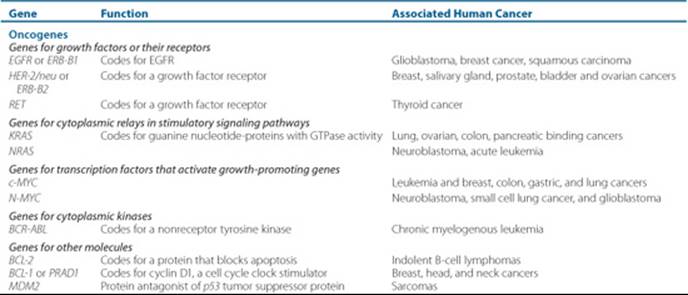
Because the human genome has been sequenced, and with the great improvements in genetic technology, there is an ever-increasing body of knowledge regarding the genetic changes of cancer. Currently, there are two major classes of genes involved in cancer: oncogenes and tumor-suppressor genes. Protooncogenes are normal genes that, through some genetic alteration caused by carcinogens, change into oncogenes. Protooncogenes are present in all normal cells and regulate cell function and replication. Genetic damage of the protooncogene may occur through point mutation, chromosomal rearrangement, or an increase in gene function, resulting in the oncogene. The oncogene produces abnormal or excessive gene product that disrupts normal cell growth and proliferation.2 This may cause the cell to have a distinct growth advantage, increasing its likelihood of becoming cancerous. Table 88–1 provides examples of oncogenes.
Tumor-suppressor genes inhibit inappropriate cellular growth and proliferation by gene loss or mutation. This results in loss of control over normal cell growth. The p53 gene is one of the most common tumor-suppressor genes, and mutations of p53 may occur in up to 50% of all malignancies. This gene stops the cell cycle to enable “repairs” of the cell. If p53 is inactivated, then the cell allows the mutations to occur. While mutations of the p53 gene are found in many tumors, such as breast, colon, and lung cancer, it is also associated with drug resistance of cancer cells. DNA-repair genes fix errors in DNA that occur because of environmental factors or errors in replication and sometimes are referred to as tumor-suppressor genes. Mutations in DNA-repair genes have been reported in hereditary nonpolyposis colon cancer and in some breast cancer syndromes.
![]() Numerous cellular changes occur in the genetic material of the cancer cell so that programmed cell death, or apoptosis, does not occur. Proliferation of cancer cells goes unregulated. If mutations persist and cells aren’t repaired or suppressed, cancer may develop. Apoptosis, or programmed cell death, may prevent the mutated cell from becoming cancerous. Loss of p53 and overexpression of bcl-2 are two examples of changes within the cell that occur to result in enhanced cell survival. Cellular senescence refers to cell death that occurs after a preset number of cell doublings. Telomeres are DNA segments at the ends of chromosomes that shorten with each replication to the point where senescence is triggered.
Numerous cellular changes occur in the genetic material of the cancer cell so that programmed cell death, or apoptosis, does not occur. Proliferation of cancer cells goes unregulated. If mutations persist and cells aren’t repaired or suppressed, cancer may develop. Apoptosis, or programmed cell death, may prevent the mutated cell from becoming cancerous. Loss of p53 and overexpression of bcl-2 are two examples of changes within the cell that occur to result in enhanced cell survival. Cellular senescence refers to cell death that occurs after a preset number of cell doublings. Telomeres are DNA segments at the ends of chromosomes that shorten with each replication to the point where senescence is triggered.
Table 88–1 Examples of Oncogenes and Tumor-Suppressor Genes

Cancer genetics may be done on the tumor itself to determine if a particular drug will be effective, or if the patient will suffer toxicity. Table 88–2 presents the genetic tests currently recommended for either tumor or patient.
Principles of Tumor Growth
It takes about 109 cancer cells to be clinically detectable by palpation. Figure 88–2 demonstrates the classic Gompertzian kinetics tumor-growth cycle. From the diagram, one can see that malignant cell growth occurs many times before a mass may be palpated. The number of malignant cells may plummet drastically because of surgery or in decreasing steps by each administration of chemotherapy. One dosing round, or cycle, of chemotherapy does not eliminate all malignant cells, and therefore, repeated cycles of chemotherapy are administered to eliminate tumor-cell burden. The cell kill hypothesis states that a fixed percentage of tumor cells will be killed with each cycle of chemotherapy. According to this hypothesis, the number of tumor cells will never reach zero. There are three assumptions to this theory: all cancers are equally responsive and drug resistance and metastases do not occur.
Table 88–2 RECIST Criteria
Metastases
A metastasis is a growth of the same cancer found at some distance from the primary tumor site.3 The metastasis may be large, or it may be just a few cells that may be detected through polymerase chain reaction (PCR); however, the presence of metastasis at staging usually is associated with a poorer prognosis than the patient with no known metastatic disease. As the technology to find malignant cells evolves, the dilemma exists on how to treat patients based on current guidelines that were not based on cellular detection technology.
Cancers spread usually by two pathways: hematogenous (through the bloodstream) or through the lymphatics (drainage through adjacent lymph nodes). The malignant cells that split from the primary tumor find a suitable environment for growth. It is believed that malignant cells secrete mediators that stimulate the formation of blood vessels for growth and oxygen, the process of angiogenesis.
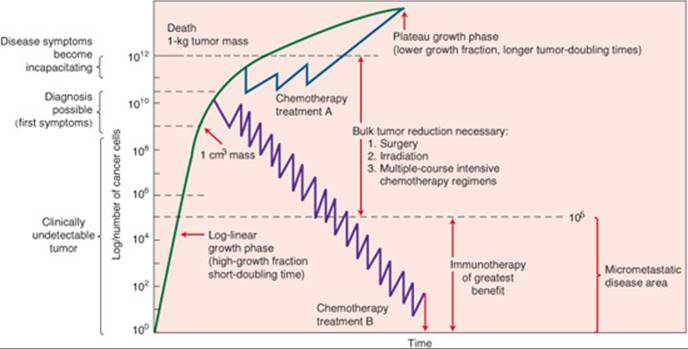
FIGURE 88–2. The Gompertzian growth curve demonstrating symptoms and treatments versus tumor volume. (From Buick RN. Cellular basis of chemotherapy. In: Dorr RT, Von Hoff DD, eds. Cancer Chemotherapy Handbook. 2nd ed. New York: Elsevier; 1994: 3–14.)
The usual metastatic sites for solid tumors are the brain, the bone, the lung, and the liver. It is important to realize and educate patients that breast cancer cells may metastasize to the brain, so the individual does not have brain and breast cancer but breast cancer with metastases to the brain.
PATHOPHYSIOLOGY
Tumor Characteristics
Tumors are either benign or malignant.4 Benign tumors often are encapsulated, localized, and indolent; they seldom metastasize; and they recur rarely once removed. Histologically, the cells resemble the cells from which they developed. Malignant tumors are invasive and spread to other locations, even if the primary tumor is removed. The cells no longer perform their usual functions, and their cellular architecture changes. This loss of structure and function is called anaplasia. Despite improvements in screening procedures, many patients have metastatic disease at the time of diagnosis. Usually, once distant metastases have occurred, the cancer is deemed to be incurable.
Tumor Origin
Tumors may arise from epithelial, connective (i.e., muscle, bone, and cartilage), lymphoid, or nerve tissue. The suffix -oma is added to the name of the cell type if the tumor cells are benign. A lipoma is a benign growth that resembles fat tissue.
Precancerous cells have cellular changes that are abnormal but not yet malignant and may be described as hyperplastic or dysplastic. Hyperplasia occurs when a stimulus is introduced and reverses when the stimulus is removed. Dysplasia is an abnormal change in the size, shape, or organization of cells or tissues.
Malignant cells are divided into categories based on the cells of origin. Carcinomas arise from epithelial cells, whereas sarcomas arise from muscle or connective tissue. Adenocarcinomas arise from glandular tissue. Carcinoma in situ refers to cells limited to epithelial origin that have not yet invaded the basement membrane. Malignancies of the bone marrow or lymphoid tissue, such as leukemias or lymphomas, are named differently.
DIAGNOSIS OF CANCER
Cancer can present as a number of different signs and symptoms as well as pain and loss of appetite. Unfortunately, many people fear a diagnosis of cancer, and may not seek medical attention at the first warning signs, when the disease is at its most treatable stage. After the initial visit with the physician, a variety of tests will be performed, which are somewhat dependent on the initial differential diagnosis. Appropriate blood work, radiologic scans, and tissue sample are necessary. The sample of tissue may be obtained by a biopsy, fine-needle aspiration, or exfoliative cytology. No treatment of cancer should be initiated without a pathologic diagnosis of cancer. During the pathologic workup, cytogenetics may be done. Depending on the type of cancer, the cytogenetics can provide the additional information on prognosis of the malignancy, and whether certain therapies may be appropriate.
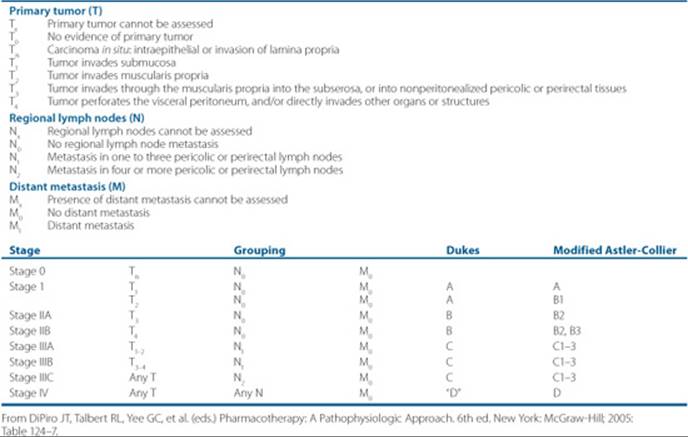
Once the pathology of cancer is established, then the staging of the disease is done before treatment is initiated. Each cancer disease chapter will discuss the specifics of staging of the disease. Cancer staging will be done according to the primary tumor size, extent of lymph node involvement, and the presence, or absence of metastases, or sometimes referred to the tumor, nodes, metastases (TNM) system (Table 88–3). The stage of the disease is a compilation of the primary tumor size, the nodal involvement, and metastases, and is usually referred to as stages I through IV. Not all cancers can be staged according to this system, but many of the solid tumors are classified this way.
Why are tumors staged? First, the stage of the disease is an important part of determining prognosis of the cancer. Second, staging of the cancers allows comparison of patient groups when examining data from clinical trials; staging reflects the extent of disease. Third, the clinician uses it as a guide to treatment, and may use restaging after treatment to guide further treatment.
Some cancers produce substances that are detected by a blood test, that may be useful in following response to therapy or detecting a recurrence; these are referred to as tumor markers. Unfortunately, some tumor markers are nonspecific and may be elevated from nonmalignant causes. Some tumors may express a marker in some patients, and not in others. The full role of tumor markers has not been fully elucidated.
TREATMENT
Desired Outcome
While at the time of surgery the surgeon may be able to remove all macroscopic disease, microscopic cells may be present near the surgical site or may have traveled to other parts of the body. When malignant cells have traveled to other parts of the body and become established there and are able to grow in this new environment, they are called metastatic cancer cells. Thus, for chemotherapy-sensitive diseases, systemic therapies may be administered after surgery to destroy these microscopic malignant cells; this is called adjuvant therapy. The goals of adjuvant therapy are to decrease recurrence of the cancer and to prolong survival. Chemotherapy may also be given prior to surgical resection of the tumor; this is referred to as neoadjuvant therapy. Chemotherapy given prior to surgery should decrease the tumor burden to be removed (which may result in a shorter surgical procedure) and make the surgery easier to perform because the tumor has shrunk away from vital organs or vessels. Neoadjuvant chemotherapy also gives the clinician an idea of the responsiveness of the tumor to that particular chemotherapy.
Chemotherapy may be given to cure cancers that are curable, or it may be given to help control the symptoms of an incurable cancer, which is referred to as palliative therapy.
Table 88–3 TNM Staging Classification System for Colorectal Cancer
Clinical Presentation and Diagnosis Cancer Chemotherapy and Treatment
Signs and Symptoms
The seven warning signs of cancer are:
• Change in bowel or bladder habits
• A sore that does not heal
• Unusual bleeding or discharge
• Thickening or lump in breast or elsewhere
• Indigestion or difficulty in swallowing
• Obvious change in wart or mole
• Nagging cough or hoarseness
The eight warning signs of cancer in children are:
• Continued, unexplained weight loss
• Headaches with vomiting in the morning
• Increased swelling or persistent pain in bones or joints
• Lump or mass in abdomen, neck, or elsewhere
• Development of a whitish appearance in the pupil of the eye
• Recurrent fevers not caused by infections
• Excessive bruising or bleeding
• Noticeable paleness or prolonged tiredness
Diagnostic Procedures
• Laboratory tests: CBC, lactate dehydrogenase (LDH), renal function, and liver function tests
• Radiologic scans: x-rays, CT scans, MRI, position-emission tomography (PET)
• Biopsy of tissue or bone marrow with pathologic evaluation
• Cytogenetics
• Tumor markers
• Staging determination of the primary tumor size, extent of lymph node involvement, and the presence or absence of metastases, or sometimes referred to the TNM system (Table 88–2). ![]() Many tumors are staged according to the TNM system. Metastases are cancer cells that have spread to sites distant from the primary tumor site and have started to grow. The most frequently-occurring sites of metastasis are the brain, bone, liver, and lungs.
Many tumors are staged according to the TNM system. Metastases are cancer cells that have spread to sites distant from the primary tumor site and have started to grow. The most frequently-occurring sites of metastasis are the brain, bone, liver, and lungs.
Response
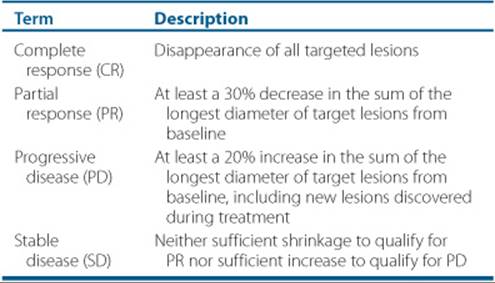
The responses to chemotherapy may be referred to as complete response (CR), partial response (PR), stable disease (SD), or disease progression. A cure in oncology implies that the cancer is completely gone, and the patient will have the same life expectancy as a patient without cancer. The World Health Organization response criteria were updated in 2000. The Response Evaluation Criteria in Solid Tumors (RECIST) is considered to be the standard criteria to evaluate a response to therapy (see Table 88–2). A CR refers to complete disappearance of all cancer for 1 month after treatment. A PR is defined as a 30% or greater decrease in tumor diameter along with no new disease for 1 month. The term overall objective response rate refers to the combination of PR and CR. SD occurs in a patient whose tumor size neither grows nor shrinks by the above criteria. Disease progression refers to tumor that has spread or the primary tumor that has increased in size by 20% while receiving treatment. Some cancers, such as leukemia, cannot be measured by size, so biopsy of the bone marrow provides a cellular indication of absence or presence of disease.
Cancer chemotherapy and the treatment of cancers are analogous to anti-infectives and the treatment of infections. Cancer cells may be sensitive to certain chemotherapy agents, but then with repeated exposure, the cells become resistant to treatment. The resistant cells then may grow and multiply. While tumors may be tested for chemotherapy sensitivity, this area is still developing. Today, tumor sensitivity can demonstrate tumor resistance so that needless exposure to an inadequate therapy and its toxicity can be avoided.
Tumor cells may become resistant when genetic changes occur during cell proliferation. Resistant cancer cells with the mdr-1 gene may possess a membrane-associated protein, p-glycoprotein, that facilitates efflux of chemotherapy agents out of the cells. Numerous attempts at blocking this efflux pump have been unsuccessful.
Nonpharmacologic Therapy
The three primary treatment modalities of cancer are surgery, radiation, and pharmacologic therapy. Surgery is useful to gain tissue for diagnosis of cancer and for treatment, especially those cancers with limited disease. Radiation plays a key role not only in the treatment and possible cure of cancer but also in palliative therapy. Together, surgery and radiation therapy may provide local control of symptoms of the disease. However, when cancer is widespread, surgery may play little or no role, whereas radiation therapy localized to specific areas may palliate symptoms.
Pharmacologic Therapy
Chemotherapy of cancer started in the early 1940s when nitrogen mustard was administered to patients with lymphoma. Since then, numerous agents have been developed for the treatment of different cancers.
Dosing of Chemotherapy
Chemotherapeutic agents typically have a narrow therapeutic index. Many chemotherapy agents have significant organ toxicities that preclude using larger and larger doses to treat the cancer. The doses of chemotherapy must be spaced out to allow the patient to recover from the toxicity of the chemotherapy; each period of chemotherapy dosing is referred to as a cycle. Each cycle of chemotherapy may have the same dosages, or the dosages may be modified based on toxicity, or a chemotherapy regimen may alternate from one set of drugs given during the first, third, and fifth cycles to another set of different drugs given during the second, fourth, and sixth cycles. Dose density of chemotherapy refers to shortening of the period between doses of chemotherapy. This can accomplish two things: First, the tumor has less time between doses of chemotherapy to grow, and second, patients receive chemotherapy over a shorter period of time and hopefully can get back to a normal life sooner. Usually dose-dense chemotherapy regimens require colony-stimulating factors to be administered to shorten the time of neutropenia. The chemotherapy regimens that are dose-dense tend to be adjuvant regimens, where the tumor burden is not measurable, and the cancer outcome is a cure. When a chemotherapy regimen is used as palliative therapy (to control symptoms), the dosages of chemotherapy should be decreased based on toxicity, or the interval between dosages should be lengthened to maintain quality of life.
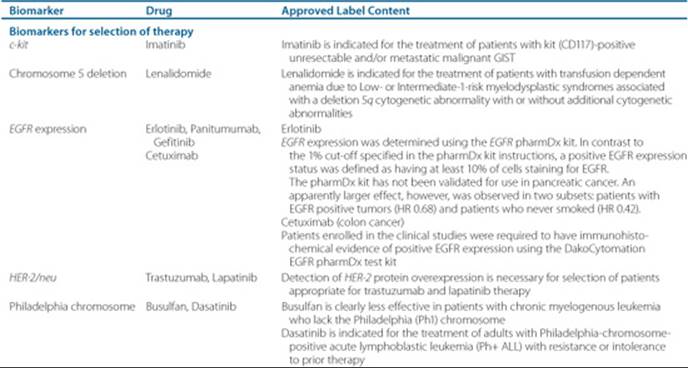
Patient and tumor biology also affect how cancer therapy is dosed. Patients with a uridine diphosphate–glucuronosyltransferease 1A1 enzyme deficiency can have life-threatening diarrhea and complications from irinotecan. The patient may have a blood test prior to therapy to determine if there is a genetic problem prior to receiving irinotecan (see Table 88–4). In the case of the some of the monoclonal antibodies, flow cytometry results will reveal whether the tumor has the receptor where the drug will bind and exert the pharmacologic effect.
Another consideration of chemotherapy administration is the patient. Factors that affect chemotherapy selection and dosing are age, concurrent disease states, and performance status. Performance status can be assessed through either the Eastern Cooperative Oncology Group Scale or the Karnofsky Scale (Table 88–5). The patient is evaluated on whether he or she is active to bedridden most of the day; performance status is a very important prognostic factor for many types of cancer. If a patient has kidney dysfunction, and the chemotherapy is eliminated primarily by the kidney, dosing adjustments will need to be made. If a patient has had a myocardial infarction recently, the clinician will weigh the risks of anthracycline therapy against the benefit of the treatment of the cancer.
Another important consideration for treatment of cancers is reimbursement by third-party payors for off-label use of chemotherapy agents because of the high expense. The American Association of Cancer Centers (www.accc-cancer.org) provides a drug compendium quarterly to which clinicians may refer to verify coverage by Medicare based on ICD-9 codes. The drugs used according to FDA-approved indications are almost always reimbursed. If sufficient literature exists, an insurer may pay for an off-label use.
Table 88–4 Oncology Drugs With Valid Genomic Biomarkers

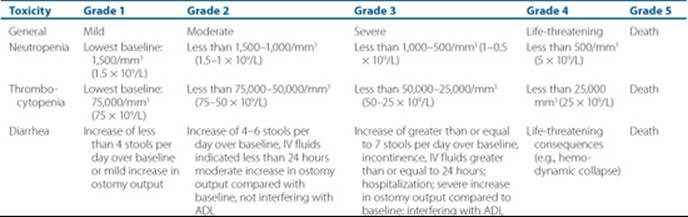
During the time of chemotherapy, patients will experience toxicity from it. The National Cancer Institute (NCI) has provided a standardized system for evaluating and grading the toxicity from chemotherapy to provide uniform grading of toxicity and evaluation of new agents and new regimens (see Table 88–6).
Combination Chemotherapy
Again, the analogy to antibiotic therapy can be made when deciding on monotherapy versus combination therapy for the treatment of cancer. The underlying principles of using combination therapy are to use (a) agents with different pharmacologic actions, (b) drugs with different organ toxicities, (c) agents that are active against the tumor and ideally synergistic when used together, and (d) agents that do not result in significant drug interactions (although these can be studied carefully and the interactions addressed). When two or more agents are used together, the development of resistance may be slowed, but increased toxicity may result. ![]() Each category of chemotherapy drugs has similar side effects. Anthracyclines cause cardiac toxicity, which is related to the cumulative dose. Tubulin-interactive agents are associated with neuropathy and ileus. Alkylating agents are associated with secondary malignancies.
Each category of chemotherapy drugs has similar side effects. Anthracyclines cause cardiac toxicity, which is related to the cumulative dose. Tubulin-interactive agents are associated with neuropathy and ileus. Alkylating agents are associated with secondary malignancies.
Table 88–5 Performance Status Scales
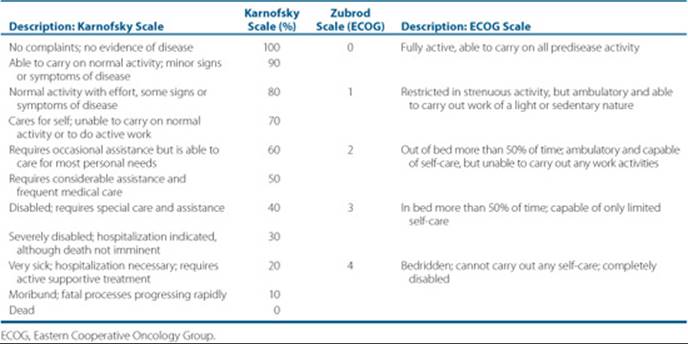
Table 88–6 Selected NCI Common Toxicity Criteria
Currently, anticancer agents are categorized by the mechanism of action. As depicted in Figure 88–3, different agents work in different parts of the cell.
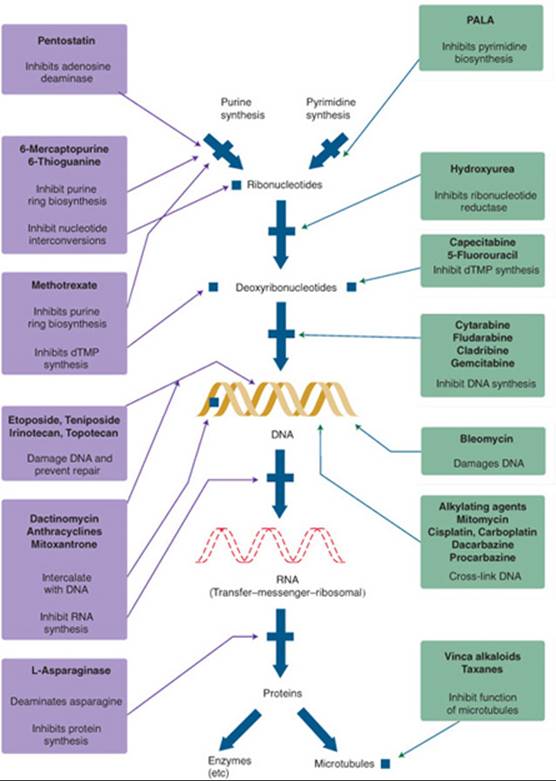
FIGURE 88–3. The mechanisms of action of antineoplastic agents. (From Chabner BA, Ryan DP, Paz-Ares L, et al. Antineoplastic agents. In: Hardman JG, Limbird LE, Gilman AG, eds. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 10th ed. New York: McGraw-Hill; 2001: 1381.)
Antimetabolites
Fluorouracil
5-Fluorouracil, commonly referred to as 5-FU, is an analog of the pyrimidine uracil. It is metabolized by dihydropyrimi-dine dehydrogenase. 5-FU ultimately is metabolized to fluorodeoxyuridine monophosphate (FdUMP), which interferes with the function of thymidylate synthase, which is required for synthesis of thymidine. The triphosphate metabolite of 5-FU is incorporated into RNA to produce the second cytotoxic effect of 5-FU. It appears that inhibition of thymidylate synthesis occurs with the continuous infusion regimens, whereas the triphosphate form is associated with bolus administration. Patients with low activity of dihydropyrimidine dehydrogenase appear to be at risk for life-threatening toxicities.5 Folates appear to increase the stability of the FdUMP-thymidylate synthase inhibition, which enhances the activity of the drug in certain cancers. 5-FU has shown to be useful in the treatment of cancers of the colon, rectum, gastric, head and neck, and breast. 5-FU is metabolized extensively by the liver, whereas up to 15% of a dose may be found unchanged in the urine. The clearance of 5-FU ranges from 155 L/m2/h (range 56–466 L/m2/h) in women to 179 L/m2/h (range 29–739 L/m2/hr) in men. Age does not appear to alter the pharmacokinetics of 5-FU. 5-FU has shown clinical activity in the treatment of colorectal, breast, esophageal, pancreas, stomach, anal, and head and neck cancers. Side effects of 5-FU include stomatitis, diarrhea, cardiac abnormalities, and rarely reported cerebellar toxicities. Esophagitis and gastric ulcerations also may occur. Some alopecia may occur, but hair regrowth may occur with subsequent doses. A recent study demonstrated that if the patient uses ice chips in the mouth for 30 minutes while receiving bolus 5-FU, mucositis may be decreased significantly. Neurotoxicity may consist of headaches, visual disturbances and cerebellar ataxia. Cardiac toxicity may consist of ST-segment elevation, which appears to be more common in patients with a prior history of coronary artery disease.
Capecitabine
Capecitabine is the prodrug of 5-FU and comes as oral tablets that are administered with food twice a day. Capecitabine has shown to be active in tumors of the colon, rectum, and breast. The toxicity profile of capecitabine is similar to that of 5-FU and includes diarrhea, mucositis, palmar-plantar erythrodyesthesia, nausea, and myelosuppression. Palmar-plantar erythrodyesthesia refers to redness, itching, and blistering of the palms of the hands and soles of the feet. Patients should be educated to notify the prescriber when palmar-plantar erythrodyesthesia occurs. Significant increases in International Normalization Ratio (INR) and prothrombin time may occur within several days when capecitabine is started in patients who are on warfarin, and the INR should be monitored closely, or the patient may be switched to a low–molecular weight heparin. Phenytoin levels may become elevated related to possible CYP2C9 inhibition by capecitabine. Patients should be instructed to take capecitabine within 30 minutes of a meal.
Cytarabine
Cytarabine, often referred to as Ara-C, is an analog of cytosine and is phosphorylated intracellularly to the active triphosphate form, which inhibits DNA polymerase. The triphosphate form also may be incorporated into DNA to result in chain termination to prevent DNA elongation. The drug may be administered as a low-dose continuous infusion, high-dose intermittent infusion, and into the subdural space via intrathecal or intraventricular administration. There is also a liposomal formulation available for less-frequent administration into the CNS. Cytarabine pharmacokinetics are best described by a two-compartment model, with an α-half-life of 15 minutes and a β-half-life of 2 hours. Cytarabine is eliminated by the kidney with a renal clearance of 90 mL/min. Cytarabine has shown efficacy in the treatment of acute leukemias and some lymphomas. The toxicities of cytarabine in high doses include myelosuppression, cerebellar syndrome (i.e., nystagmus, dysarthria, and ataxia), and eye irritation that requires round-the-clock steroid eye drop administration. The risk of CNS toxicity is increased with the high-dose cytarabine regimen with renal dysfunction; dosage modification is necessary with the high-dose regimen with renal dysfunction.
Gemcitabine
Gemcitabine is a deoxycytidine analog that is structurally related to cytarabine. Gemcitabine inhibits DNA polymerase activity and ribonucleotide reductase to result in DNA chain elongation. The pharmacokinetics of gemcitabine are best described by a two-compartment model, with a terminal half-life of 6 to 20 minutes. Approximately 5% of the dose is excreted unchanged by the kidney.6Gemcitabine has shown activity in cancers of the pancreas, breast, lung, ovary, and lung (nonsmall cell), along with some lymphomas. The toxicities include myelosuppression, flu-like syndrome with fevers during the first 24 hours after administration, rash that appears 48 to 72 hours after administration, and hemolytic uremic syndrome. While hemolytic uremic syndrome is uncommon, it is a life-threatening side effect. Patients should be counseled about using acetaminophen to treat the fevers during the first 24 hours; however, fevers occurring 7 to 10 days after gemcitabine are likely to be febrile neutropenias and need prompt treatment with broad-spectrum antibiotics.
Azacitidine
Azacitidine, a cytidine analog, causes hypomethylation of DNA, which normalizes the function of genes that control cell differentiation to promote normal-cell maturation. The suspension is administered as a subcutaneous injection daily for 7 days for the treatment of myelodysplastic syndrome, a preleukemia disease. The pharmacokinetics of azacitidine are best described by a two-compartment model, with a terminal half-life of 3.4 to 6.2 hours, whereas peak concentrations are achieved 30 minutes after a subcutaneous injection.7 Azacitidine has been shown to be clinically active in the treatment of myelodysplastic syndromes. The side effects include myelosuppression, renal tubular acidosis, renal dysfunction, and injection-site reactions.
Decitabine
Decitabine, approved by the FDA in 2006 for the treatment of myelodysplastic syndrome, is incorporated into DNA and directly inhibits DNA methyltransferase which causes hypomethylation of DNA. The pharmacokinetics of decitabine are best described by a two-compartment model, with a terminal half-life of 0.5 hours. Side effects include myelosuppression, constipation, edema, headache, and nausea.
Nelarabine
Nelarabine is indicated for the treatment of patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma, whose disease has been already treated with at least two other chemotherapy regimens. Nelarabine is a prodrug, which accumulates as the active 5’-triphosphate form in leukemic blasts to result in inhibition of DNA synthesis and cell death. The plasma half-life of nelarabine is approximately 30 minutes. Nelarabine is primarily metabolized by demethylation, with only 5% to 10% excreted unchanged by the kidney.
Purines and Purine Antimetabolites
6-Mercaptopurine
6-Mercaptopurine (6-MP) is an oral purine analog that is converted to a ribonucleotide to inhibit purine synthesis. Mercaptopurine is converted into thiopurine nucleotides, which are catabolized by thiopurine S-methyltransferase (TPMT), which is subject to genetic polymorphisms and may cause severe myelosuppression. TPMT status may be assessed prior to therapy to reduce drug-induced morbidity and the costs of hospitalizations for neutropenic events. Mercaptopurine is poorly absorbed, with a time to peak concentration of 1 to 2 hours after an oral dose. The half-life is 21 minutes in pediatric patients and 47 minutes in adults. Mercaptopurine is used in the treatment of acute lymphocytic leukemia and chronic myelogenous leukemia. Significant side effects include myelosuppression, mild nausea, skin rash, and cholestasis. When allopurinol is used in combination with 6-MP, the dose of 6-MP must be reduced by 66% to 75% of the usual dose because allopurinol blocks the metabolism of 6-MP.
6-Thiogaunine
6-Thioguanine (6-TG) is another oral purine analog that works similarly to 6-MP, and because of this, cross-resistance is observed. While little is known about the pharmacokinetics of thioguanine, it appears that absorption is incomplete and approximates 30% of the dose. Thioguanine may be used in the treatment of acute and chronic myelogenous leukemia. Side effects include myelosuppression, mild nausea, cholestasis, and rarely, veno-occlusive disease.
Fludarabine
Fludarabine is an analog of the purine adenine. It interferes with DNA polymerase to cause chain termination and inhibits transcription by its incorporation into RNA. Fludarabine is dephosphorylated rapidly and converted to 2-fluoro-Ara-AMP (2-FLAA), which enters the cells and is phosphorylated to 2-fluoro-Ara-ATP, which is cytotoxic. Fludarabine is converted rapidly to 2-FLAA. The pharmacokinetics of 2-FLAA are best described by a two-compartment model, with an α-half-life of 0.6 hours and a terminal half-life of 9.3 hours.8 Fludarabine is used in the treatment of chronic lymphocytic leukemia, some lymphomas, and refractory acute myelogenous leukemia. This drug is given IV usually daily for 5 days every 4 weeks. Significant myelosuppression may occur, along with immunosuppression, so patients are susceptible to opportunistic infections. Mild nausea and vomiting and diarrhea have been observed. Rarely, interstitial pneumonitis has occurred.
Cladribine
Cladribine (2-chlorodeoxyadenosine, or 2-CDA) is a purine nucleoside that once it is in the triphosphate form is incorporated into DNA, which results in inhibition of DNA synthesis and chain termination. The pharmacokinetics of cladribine are best described by a two-compartment model, with an α-half-life of 35 minutes and a terminal half-life of 6.7 hours.9 It may be administered as a continuous 7-day IV infusion or as a 2-hour infusion daily for 5 days; both regimens deliver the same total dose of drug. Cladribine is used to treat hairy cell leukemia and, therefore, is myelosuppressive. Unfortunately, one of the other side effects of the drug is fever, so the clinician struggles with the dilemma of whether the fever is due to the drug or an infection. Rash occurs in approximately 50% of patients with hairy cell leukemia. Cladribine also may be used to treat chronic lymphocytic leukemia, refractory low-grade non-Hodgkin’s lymphoma, and Waldenström’s macroglobulinemia.
Clofarabine
Clofarabine was developed based on the structures of fludarabine and cladribine, with the hope it would be resistant to deamination by adenosine deaminase. Clofarabine has shown activity in myeloid leukemia and myelodysplastic syndrome.10 The pharmacokinetics are best described by a two-compartment model with a terminal half-life of approximately 5.2 hours. Clofarabine is 47% bound to plasma proteins, primarily albumin. In children, 49% to 60% of the dose is excreted unchanged in the urine. No dosing adjustments are available for renal dysfunction. Side effects include bone marrow suppression, severe but transient liver dysfunction in 15% to 25% of patients, skin rashes, and hand-foot syndrome.
Pentostatin
Pentostatin is an inhibitor of adenosine deaminase, an enzyme important in purine base metabolism. Pentostatin irreversibly inhibits adenosine deaminase, which ultimately is believed to block DNA synthesis through inhibition of RNA ribonucleotide reductase. The pharmacokinetics of pentostatin are best described by a two-compartment model with a half-life of 2.6 to 6 hours. While the drug is primarily eliminated unchanged by the kidney, preliminary data suggest no dosage adjustments are necessary for renal dysfunction. Side effects include bone marrow suppression, myalgias, conjunctivitis, and rash.
Antifolates
Folates carry one-carbon groups in transfer reactions required for purine and thymidylic acid synthesis. Dihydrofolate reductase is the enzyme responsible for supplying reduced folates intracellularly for thymidylate and purine synthesis.
Methotrexate
Methotrexate inhibits dihydrofolate reductase of both malignant and nonmalignant cells. When high doses of methotrexate are given, leucovorin, a reduced folate, is administered to bypass the methotrexate inhibition of dihydrofolate reductase of normal cells and is usually initiated 24 hours after methotrexate administration. For safety purposes, the term folinic acid, another term used for leucovorin, should not be used because of medication errors where folic acid was given instead. Methotrexate concentrations should be monitored to determine when to stop leucovorin administration. Generally, leucovorin administration may be stopped when methotrexate concentrations decrease to 5 × 10–8 M, although this may vary by the chemotherapy regimen. High dosages of methotrexate may cause methotrexate to crystallize out in the kidney, which may result in renal dysfunction and decreased methotrexate clearance. IV hydration with sodium bicarbonate to maintain urinary pH greater than or equal to 7 helps to prevent methotrexate-induced renal dysfunction. The pharmacokinetics of methotrexate are best described by either a two- or three-compartment model. The α-half-life is less than 1 hour, whereas the β-half-life is 3 to 4 hours, and the γ-half-life is 8 to 10 hours or longer with impaired kidney function. Approximately 60% to 100% of methotrexate is eliminated primarily as unchanged drug by the kidney. Since methotrexate is eliminated by tubular secretion, concomitant drugs that may inhibit or compete for tubular secretion should be avoided. Methotrexate doses must be adjusted for renal dysfunction. A recommended dosing adjustment is to divide the creatinine clearance of the patient by 70 mL/minute, which is the average creatinine clearance of patients who received methotrexate during clinical trials, and then to multiple this fraction by the dosage recommended for that disease state. Again, close monitoring of methotrexate concentrations in patients with renal impairment is advised. Methotrexate has shown activity in lymphoma, gastric, esophageal, bladder, and breast cancer and acute lymphocytic leukemia. Side effects of methotrexate include myelosuppression, nausea and vomiting, and mucositis. Methotrexate also may be administered via the intrathecal route in very low doses as small as 12 mg to doses of 20 g IV, so it is crucial for the clinician to know the correct dose by the correct route in order to avoid substantial toxicity. Methotrexate is also administered as an intrathecal injection into the cerebrospinal fluid or directly into the ventricle via an Ommaya reservoir. The methotrexate used for intrathecal and intraventricular injection must be preservative free. Drugs that may block the tubular secretion of methotrexate include probenecid, salicylates, penicillin G, and ketoprofen.
Pemetrexed
Pemetrexed inhibits at least three pathways in thymidine and purine synthesis. Pemetrexed is excreted primarily as unchanged drug by the kidney, with 70% to 90% of a dose recovered in 24 hours as unchanged drug in the urine. Patients with normal kidney function have a half-life of 3.5 hours.11 Pemetrexed has shown activity in the treatment of mesothelioma and nonsmall cell lung cancer. Side effects include myelosuppression, rash, diarrhea, and nausea and vomiting. Patients should receive folic acid and cyanocobalamin to reduce bone marrow toxicity and diarrhea. Doses of folic acid of at least 400 mcg/day starting 5 days before treatment and continuing throughout therapy, as well as for 21 days after the last pemetrexed dose, have been used. Cyanocobalamin 1,000 mcg is given intramuscularly the week prior to pemetrexed and then every three cycles thereafter. Dexamethasone 4 mg twice daily the day before, the day of, and the day after pemetrexed administration helps to decrease the incidence and severity of rash.
Tubulin Active Agents
The periwinkle, or vinca plant, served as a source for the drugs vincristine and vinblastine, which are commonly referred to as the vinca alkaloids. The vinca alkaloids inhibit the assembly of microtubules, which interferes in the formation of the mitotic spindle. Care must be taken not to confuse the names and doses of vincristine and vinblastine.
Vincristine
Vincristine causes mitotic inhibition to arrest cells in metaphase. The pharmacokinetics of vincristine have been described by a three-compartment model, with an α-half-life of 0.8 minutes, a β-half-life of 7 minutes, and an α-half-life of 164 minutes.12 Biliary excretion accounts for a significant portion of elimination of vincristine and its metabolites, so doses need to be adjusted for obstructive liver disease. Vincristine has been useful in the treatment of sarcomas, Wilms’ tumor, many kinds of lymphoma, multiple myeloma, and acute lymphocytic leukemia. Vincristine is a vesicant that may cause significant neuropathy. Patients should be counseled regarding prevention of constipation and ileus caused by vincristine. Many clinicians cap IV vincristine doses at 2 mg to prevent severe neuropathic side effects, however, if the intent of chemotherapy is curative, the vincristine dose is often not capped at 2 mg. Several patients have died as a result of vincristine being administered intrathecally; it should only be administered IV and appropriate labeling should be placed on all doses. Itraconazole has been reported to cause severe neurotoxicity when administered to patients receiving vincristine. Patients have been reported to experience paralytic ileus, neurogenic bladder, absence of deep reflexes, and severe paralysis of the lower extremeties within 10 days of starting itraconazole. Clinicians need to be aware of the potential interactions of the newer azoles with vincristine and the agents should only be used in combination when the benefit clearly outweighs the risk In most cases, alternative antifungals can be administered.
Vinblastine
Vinblastine is another vesicant vinca alkaloid that causes myelosuppression and less neurotoxicity than vincristine. The pharmacokinetics of vinblastine are best described by a three-compartment model, with an α-half-life of 25 minutes, a β-half-life of 53 minutes, and a terminal half-life of 19 to 25 hours.13 Vinblastine has shown activity in the treatment of bladder, breast, and kidney cancer, as well as some lymphomas. The doses of vinblastine tend to be higher on a milligram per meter squared basis than vincristine. Nausea and vomiting are minimal with vinblastine. Other side effects include mild alopecia, rash, photosensitivity, and stomatitis.
Vinorelbine
The vesicant vinorelbine is structurally similar to vincristine and may cause many of the same side effects as vincristine. Vinorelbine is administered IV over 6 to 10 minutes, and patients should be counseled about neuropathy, ileus, and myelosuppression. The pharmacokinetics of vinorelbine are best described by a three-compartment model, with an α-half-life of 2 to 6 minutes, a β-half-life of 1.9 hours, and a γ-half-life of 40 hours. Vinorelbine has shown efficacy in the treatment of breast cancer and non-small cell lung cancer. Additional side effects include myelosuppression, paresthesias, and mild nausea and vomiting.
Paclitaxel
Paclitaxel, a taxane, binds to tubulin to promote microtubule assembly and to prevent microtubule disassembly. The pharmacokinetics of paclitaxel can be described by a two-compartment model, with an α-half-life of 30 to 45 minutes and a β-half-life of 4 to 8 hours. Hepatic metabolism and biliary excretion account for the majority of paclitaxel’s elimination. Paclitaxel has demonstrated activity in ovarian, breast, nonsmall cell lung, prostate, esophageal, gastric, and head and neck cancers. Considerable variability exists in paclitaxel dosing, from weekly 1-hour infusions to 24-hour infusions administered every 3 weeks. The diluent for paclitaxel, Cremophor EL, is composed of ethanol and castor oil. Infusions must be prepared and administered in non-PVC- containing bags and tubings, and solutions must be filtered. Patients receive dexamethasone, diphenhydramine, and an H2 blocker to prevent hypersensitivity reactions from paclitaxel/Cremophor EL. Patients also may have asymptomatic bradycardia (i.e., heart rates around 45 bpm) during the infusion. Approximately 3 to 5 days after administration, patients may complain of myalgias and arthralgias that may last several days. Myelosuppression, flushing, neuropathy, ileus, and total-body alopecia are other common side effects. Because paclitaxel is a substrate for CYP 3A4, steady-state concentrations of paclitaxel were 30% lower in patients receiving phenytoin than in patients not receiving phenytoin. Paclitaxel clearance was decreased by 33% when it was administered following cisplatin, so paclitaxel is administered before cisplatin.
Recently, a nanoparticle albumin-bound paclitaxel product became available commercially for the treatment of metastatic breast cancer. This product does not have the serious allergic reactions encountered with paclitaxel in Cremophor EL, so premedication with H1 and H2 blockers and steroids is not necessary. The dose is infused over 30 minutes and does not require a special IV bag, tubing, or filter. The dosing of this product is different from that of the original paclitaxel, so practitioners need to be aware of which product is being prescribed. The pharmacokinetics of the albumin-bound paclitaxel display a higher clearance and larger volume of distribution than paclitaxel. The drug is eliminated primarily via fecal excretion.14 The side effects of bone marrow suppression, neuropathy, ileus, arthralgias, and myalgias still occur.
Docetaxel
Docetaxel, a semi-synthetic taxane, binds to tubulin to promote microtubule assembly. The pharmacokinetics of docetaxel are best described by a three-compartment model, with an α-half-life of 0.08 hours, a β-half-life of 1.6 to 1.8 hours, and a terminal half-life of 65 to 73 hours.15 Docetaxel has activity in the treatment of breast, nonsmall cell lung, prostate, bladder, esophageal, stomach, ovarian, and head and neck cancers. Dexamethasone, 8 mg twice daily for 3 days starting the day before treatment, is used to prevent the fluid-retention syndrome associated with docetaxel and possible hypersensitivity reactions. The fluid-retention syndrome is characterized by edema and weight gain that is unresponsive to diuretic therapy and is associated with cumulative doses greater than 800 mg/m2. Myelosuppression, alopecia, and neuropathy are other side effects associated with docetaxel treatment.
Estramustine
Estramustine, an oral drug, also inhibits microtubule assembly and has weak estrogenic activity at the estradiol hormone receptors of the cell. Approximately 75% of a dose of estramustine is absorbed.16 The terminal half-life ranges between 20 and 24 hours, with nonrenal excretion as the major route of elimination. This drug is used primarily for the treatment of prostate cancer, but its use is limited by the side effects, which include nausea and vomiting, diarrhea, thromboembolic events, and gynecomastia.
Ixabepilone
Ixabepilone, an epothilone analog, bind to β-tubulin sub-units on microtubules which leads to suppression of microtubule dynamics. Ixabepilone is primarily eliminated by the liver by oxidation through the CYP3A4 system, with a terminal half-life of 52 hours. Approximately 5% of the drug is excreted unchanged by the kidney. Ixabepilone is indicated for the treatment of metastatic or locally advanced breast cancer after failures of anthracyclines and a taxane. Side effects include hypersensitivity reactions, myelosuppression, and peripheral neuropathy. To minimize the occurrence of hypersensitivity reactions, patients must receive both H1 and H2 antagonists prior to therapy. If a reaction still occurs, corticosteroids should be added to the premedications.
Topoisomerase Inhibitors
Topoisomerase is responsible for relieving the pressure on the DNA structure during unwinding by producing strand breaks. Topoisomerase I produces single-strand breaks, whereas topoisomerase II produces double-strand breaks.
Etoposide
Etoposide causes multiple DNA double-strand breaks by inhibiting topoisomerase II. The pharmacokinetics of etoposide are described by a two-compartment model, with an α-half-life of 0.5 to 1 hour and a β-half-life of 3.4 to 8.3 hours. Approximately 30% of the dose is excreted unchanged by the kidney.17 Etoposide has shown activity in the treatment of several types of lymphoma, testicular and lung cancer, retinoblastoma, and carcinoma of unknown primary. The IV preparation has limited stability, so final concentrations should be 0.4 mg/mL. IV administration needs to be slow to prevent hypotension. Oral bioavailability is approximately 50%, so oral dosages are approximate two times those of IV doses; however, relatively low oral daily dosages are used for 1 to 2 weeks. Side effects include mucositis, myelosuppression, alopecia, phlebitis, hypersensitivity reactions, and secondary leukemias.
Teniposide
Teniposide, a topoisomerase-II inhibitor, is administered as an infusion over 30 to 60 minutes to prevent hypotension. The pharmacokinetics are described by a three-compartment model, with an α-half-life of 0.75 hours, a β-half-life of 4 hours, and a terminal half-life of 20 hours. Considerable variability in clearance of teniposide in children has been reported.18 Teniposide has shown activity in the treatment of acute lymphocytic leukemia, neuroblastoma, and non-Hodgkin’s lymphoma. Side effects include myelosuppression, nausea, vomiting, mucositis, and venous irritation. Hypersensitivity reactions may be life-threatening.
Irinotecan
Irinotecan, a camptothecin analog, inhibits topoisomerase I to interfere with DNA synthesis through the active metabolite SN38, which is 100-fold more potent in vitro. The pharmacokinetics of irinotecan are best described by a three-compartment model, with an α-half-life of 0.07 hours, a β-half-life of 2.2 hours, and a terminal half-life of about 18 hours.19 Irinotecan has shown activity in the treatment of cancers of the colon, rectum, cervix, and lung. Irinotecan induced diarrhea may be life-threatening. IV atropine should be used to treat diarrhea that occurs during the first 24 hours of administration. Loperamide, 2 mg every 2 hours or 4 mg every 4 hours until diarrhea has stopped for 12 hours, should be used for diarrhea occurring for more than 24 hours after administration. Other side effects include myelosuppression, fatigue, and alopecia. Individuals homozygous for UGT1A1* 28 have an increased risk of febrile neutropenia and diarrhea and should be considered for an upfront dose reduction of one level; and heterozygotes should receive closer monitoring, including more frequent CBC.
Topotecan
Topotecan inhibits topoisomerase I to cause single-strand breaks in DNA. The pharmacokinetics of topotecan can be described by a two-compartment model, with a terminal half-life of 80 to 180 minutes, with renal clearance accounting for approximately 70% of the clearance.20 Topotecan has shown clinical activity in the treatment of ovarian and lung cancer, myelodysplastic syndromes, and acute myelogenous leukemia. The IV infusion may be daily for 5 days or once weekly. Side effects include myelosuppression, mucositis, and diarrhea.
Anthracyclines
All the anthracyclines contain a four-membered anthracene ring, a chromophore, with an attached sugar portion. Free radicals formed from the anthracyclines combine with oxygen to form superoxide, which can make hydrogen peroxide. Oxygen-free-radical formation is a cause of cardiac damage and extravasation injury, which is common to these drugs. Daunorubicin, doxorubicin, epirubicin, and idarubicin cause cardiac toxicity, as manifested by a congestive heart failure/cardiomyopathy symptomotology, mucositis, and myelosuppression. These drugs are vesicants; significant tissue damage may occur with extravasation.
Daunorubicin
Daunorubicin is an anthracycline that is sometimes referred to as an antitumor antibiotic. Daunorubicin inserts between base pairs of DNA to cause structural changes in DNA; however, the primary mechanism of cytotoxicity is the inhibition of topoisomerase II. The pharmacokinetics are best described by a two-compartment model, with a terminal half-life of about 20 hours. The predominant route of elimination of daunorubicin and hydroxylated metabolites is hepatobiliary secretion. Daunorubicin has shown clinical activity in the treatment of acute lymphocytic leukemia, non-Hodgkin’s lymphoma, neuroblastoma, and Ewing’s and Kaposi’s sarcomas. Myelosuppression is the major toxicity, along with alopecia, stomatitis, and mild to moderate nausea and vomiting, and it imparts a red to color to the urine so that patients need to be educated on side effects. Cardiac toxicity is dose related and manifested as congestive heart failure. To reduce the risk of cardiotoxicity, the maximum cumulative dose in children older than 2 years of age is 300 mg/m2, and the cumulative dose is 400 to 600 mg/m2 in adults. Ventricular ejection fractions should be measured before therapy, and periodically if therapy is continued. Therapy should be halted if there is a 10% to 20% decrease from baseline in ejection fraction. Daunorubicin is a vesicant also.
Doxorubicin
The addition of a hydroxyl group to daunorubicin, resulted in the drug hydroxydaunorubicin (the H in CHOP therapy), or doxorubicin, which inhibits topoisomerase II. The pharmacokinetics of doxorubicin may be described by either a two- or three-compartment model, with a terminal half-life of 30 to 40 hours. Doxorubicin is metabolized extensively, and doxorubicinol, a major metabolite, is also a cardiotoxin. Biliary excretion accounts for about 40% of a dose; patients with cholestasis experience greater toxicity from standard doses. Doxorubicin has shown clinical activity in breast, esophageal, bladder, lung, ovarian, and head and neck cancers, along with lymphomas and multiple myeloma. This red drug causes a red-orange discoloration of the urine. Cumulative doses greater than 550 mg/m2 are associated with cardiomyopathy. Doxorubicin is a vesicant and may cause significant pain when administered into the peritoneal cavity. Other side effects include myelosuppression, alopecia, mucositis, and nausea and vomiting.
Doxorubicin, Liposomal
Liposomal doxorubicin is an irritant, not a vesicant, and is dosed differently from doxorubicin, so clinicians need to be very careful when prescribing these two drugs. The pharmacokinetics of liposomal doxorubicin are best described by a two-compartment model, with a terminal half-life of 30 to 90 hours.21 Liposomal doxorubicin has shown significant activity in the treatment of breast and ovarian cancer, along with multiple myeloma and Kaposi’s sarcoma. Side effects include mucositis, myelosuppression, alopecia, and palmar-plantar erythrodysesthesia. The liposomal doxorubicin may be less cardiotoxic than doxorubicin.
Epirubicin
Epirubicin inhibits both DNA and RNA polymerases and thus inhibits nucleic-acid synthesis and topoisomerase-II enzymes. Epirubicin pharmacokinetics are best described by a three-compartment model, with an α-half-life of 4 to 5 minutes, a β-half-life of 2.4 hours, and a terminal half-life of 30 hours. Dosage alterations should be made in the presence of cholestasis because approximately 35% of a dose undergoes biliary excretion.22 Epirubicin has shown clinical activity in the treatment of breast, esophageal, lung, ovarian, and stomach cancers. Epirubicin also has been used in the treatment of lymphomas and soft-tissue sarcomas. Cumulative doses of epirubicin greater than 900 mg/m2 are associated with cardiomyopathy, so this drug may be less cardiotoxic on a milligram-per-milligram basis than doxorubicin. This topoisomerase-II inhibitor may cause red-orange urine, myelosuppression, alopecia, and significant nausea and vomiting.
Idarubicin
Idarubicin inhibits both DNA and RNA polymerase, as well as topoisomerase II. The pharmacokinetics of idarubicin can best be described by a three-compartment model, with an α-half-life of 13 minutes, a β-half-life of 2.4 hours, and a terminal half-life of 16 hours.23 Idarubicin is metabolized to an active metabolite, idarubicinol, which has a half-life of 41 to 69 hours. Idarubicin and idarubicinol are eliminated by the liver and through the bile. Idarubicin has shown clinical activity in the treatment of acute leukemias, chronic myelogenous leukemia, and myelodysplastic syndromes. Idarubicin causes cardiomyopathy at cumulative doses of greater than 150 mg/m2 and produces cumulative cardiotoxic effects with other anthracyclines. Idarubicin is a vesicant and causes red-orange urine, mucositis, mild to moderate nausea and vomiting, and bone marrow suppression.
Mitoxantrone
This royal-blue-colored drug is an anthracenedione that inhibits DNA topoisomerase II. The pharmacokinetics of mitoxantrone may best be described by a three-compartment model, with an α-half-life of 3 to 10 minutes, a β-half-life of 0.3 to 3 hours, and a median terminal half-life of 12 days. Biliary elimination appears to be the primary route of elimination, with less than 10% of the drug eliminated by the kidney.24 Mitoxantrone has shown clinical activity in the treatment of acute leukemias, breast and prostate cancer, and non-Hodgkin’s lymphomas. Myelosuppression, mucositis, nausea and vomiting, and cardiac toxicity are side effects of this drug. The total cumulative dose limit is 160 mg/m2 for patients who have not received prior anthracycline or mediastinal radiation. Patients who have received prior doxorubicin or daunorubicin therapy should not receive a cumulative dose greater than 120 mg/m2 of mitoxantrone. Patients should be counseled that their urine will turn a blue-green color.
Patient Encounter 1
AB is a 40-year-old white woman who went to urgent care because she has not been feeling well for a couple of days and has a fever. Her WBC results are greater than 100 × 109/L (100 × 103/μ L) with greater than 85% blast cells, which is indicative of acute leukemia. AB is admitted directly to the hospital to start chemotherapy.
The oncologist prescribes the normal doses of idarubicin 12 mg/m2 IV daily for 3 days and cytarabine 100 mg/m2/day by continuous infusion for 7 days to treat her acute myelogenous leukemia. Her baseline laboratory measurements are significant for an elevated WBC, a creatinine concentration of 2.5 mg/dL (221 μmol/L), and a bilirubin level of 1.6 mg/dL (27 μmol/L).
Should her chemotherapy dosages be adjusted? If so, how would you adjust the dosages?
What should be monitored while she is receiving her chemotherapy?
Alkylating Agents
The alkylating drugs are the oldest category of chemotherapy agents. Alkylating agents add a alkyl group to the DNA, which, inhibits DNA replication because interlinked strands do not separate.
Busulfan
Busulfan is an alkylating agent that forms DNA-DNA and DNA-protein cross-links to inhibit DNA replication. Oral busulfan is well absorbed, has a terminal half-life of 2 to 2.5 hours, and is eliminated primarily by metabolism. Busulfan has shown significant clinical activity in the treatment of acute myelogenous leukemia and chronic myelocytic leukemia. Side effects include bone marrow suppression, hyperpigmentation of skin creases, and rarely, pulmonary fibrosis. High doses used for bone marrow transplant preparatory regimens result in severe nausea and vomiting, tonic-clonic seizures, and sinusoidal obstruction syndrome (formerly known as veno-occlusive disease). Patients receiving high-dose busulfan should receive anticonvulsant prophylaxis.
Cyclophosphamide
Cyclophosphamide prevents cell division by cross-linking DNA strands. Cyclophosphamide is activated to phosphoramide mustard and acrolein. Acrolein, which has no tumor activity, causes hemorrhagic cystitis of the bladder. The pharmacokinetics of cyclophosphamide are best described by a two-compartment model, with a terminal half-life that ranges from 4 to 10 hours. Approximately 15% of the dose is excreted unchanged by the kidney. Cyclophosphamide has shown clinical activity in numerous types of cancer, ranging from leukemias to lymphomas to breast and ovarian cancer. Whether the drug is administered orally or IV, patients need to be counseled on the importance of good hydration and frequent voiding to prevent hemorrhagic cystitis. Nausea and vomiting may occur 12 hours after administration, so patients need to have antiemetics available after the acute treatment period. Other side effects include myelosuppression, alopecia, SIADH (usually with doses greater than 50 mg/kg), secondary malignancies (e.g., bladder cancers and acute leukemias), and infertility issues.
Ifosfamide
Hemorrhagic cystitis is such a predominant side effect of the alkylator ifosfamide that mesna always must be given with ifosfamide, along with hydration. Dosing regimens of mesna range from an equal milligram dose to the ifosfamide mixed in the same IV bag to 20% of the dose prior to ifosfamide and 20% of the dose repeated at 4 and 8 hours after the dose. The pharmacokinetics of ifosfamide are best described by a one-compartment model, with a terminal half-life of 7 to 15 hours. Approximately 50% of a dose is excreted unchanged by the kidney. Ifosfamide has shown clinical activity in the treatment of acute lymphocytic leukemia, lymphomas, and breast, ovarian, lung, and head and neck cancers. CNS side effects of confusion, delirium, and somnolence are associated with high doses infused quickly.
Patient Encounter 2
JP is receiving a highly myelosuppressive chemotherapy regimen for the next 3 days for his lymphoma. The chemotherapy orders specify ifosfamide, carboplatin, and etoposide. The goal of this cycle of chemotherapy is to put the cancer into remission so that his lymphoma can be cured with a bone marrow transplant.
What drug is missing from his ifosfamide orders?
What instructions should the patient receive about ifosfamide?
Carmustine
Carmustine, a nitrosurea, cross-links DNA strands to inhibit DNA replication. Carmustine, which is reconstituted with ethanol, crosses the blood-brain barrier when given IV. It also comes as a wafer formulation that may be implanted surgically for brain tumors. The pharmacokinetics are best described by a two-compartment model, with an α-half-life of 6 minutes and a terminal half-life of 21 minutes.25Carmustine has shown clinical activity in the treatment of lymphoma, melanoma, and brain tumors. Side effects include myelosuppression, severe nausea and vomiting, and pulmonary fibrosis with long-term therapy.
Lomustine
Lomustine is an orally available nitrosurea alkylating agent. Lomustine is converted rapidly to the cis- and trans-4-hydroxy metabolites; the range of half-lives of these two metabolites is 2 to 4 hours.26Lomustine has shown clinical activity in the treatment of non-Hodgkin’s lymphoma and melanoma. Side effects are similar to those of carmustine. Patients should receive only enough drug for one cycle at a time to prevent confusion and accidental overdose.
Dacarbazine
While the exact mechanism of action remains unclear, dacarbazine appears to inhibit DNA, RNA, and protein synthesis. Dacarbazine disappears rapidly from the plasma, with a terminal half-life of about 40 minutes. Dacarbazine has shown clinical benefit in the treatment of melanoma, Hodgkin’s lymphoma, and soft-tissue sarcomas. Side effects include myelosuppression, severe nausea and vomiting, and a flu-like syndrome that starts about 7 days after treatment and lasts 1 to 3 weeks.
Temozolomide
Temozolomide is an orally active agent with the same mechanism of action as dacarbazine. It is well absorbed and crosses the blood–brain barrier. Temozolomide is converted enzymatically to the active metabolite 5-(3-methyltriazeno)-imidazole-4-carboxamide. Temozolomide has a terminal half-life of 1.8 hours, with a mean time to peak concentrations of 1.4 hours, and a small amount of the drug is excreted unchanged by the kidney.27 The active metabolite has a terminal half-life of 1.5 hours and is metabolized primarily. Temozolomide may be used in the treatment of melanoma, refractory anaplastic astrocytoma, and glioblastoma multiforme. Nausea may be minimized by administering the drug at bedtime. Because patients receiving temozolomide may have confusion secondary to their brain tumor, and because dosing can consist of multiple capsule sizes, care must be taken by all providers to simplify regimens to prevent chemotherapy overdose.
Procarbazine
While the exact mechanism of action of procarbazine is unknown, it does inhibit DNA, RNA, and protein synthesis. The pharmacokinetics have never been fully characterized, but it is known that the drug is metabolized extensively. Procarbazine is used most often in the treatment of lymphoma. Myelosuppression is the major side effect. Nausea, vomiting, and a flu-like syndrome occur initially with therapy. Patients must be counseled to avoid tyramine-rich foods because procarbazine is a monoamine oxidase inhibitor. Patients should be provided a list of foods and beverages to avoid to prevent a hypertensive crisis. A disulfiram-like reaction can occur with the ingestion of alcohol.
Bendamustine
Bendamustine has three chemically active groups: a 2-chlorethyl group, a butyric acid side chain, and a benzi-midazole ring. The 2-chloroethyl group, which confers alkylating properties, is shared with chlorambucil and other agents from the nitrogen mustard class. The butyric acid side chain, also common to chlorambucil, confers water solubility. The benzimidazole ring, structurally similar to a purine ring, occupies the position of the benzene ring in chlorambucil. Bendamustine has shown activity in chronic lymphocytic leukemia and non-Hodgkin’s lymphoma. The recommended dose of bendamustine is 100 mg/m2 given IV over 30 minutes on days 1 and 2 of a 28-day cycle. Bendamustine is oxidized by the liver to two weakly active metabolites with 45% of the dose excreted unchanged by the kidney. The terminal half-life of bendamustine is about 40 minutes. Side effects include nausea, vomiting, bone marrow suppression, headache and dyspnea.
Thiotepa
Thiotepa is an alkylating agent that reacts with DNA phosphate groups to produce chromosomal cross-linkage. The pharmacokinetics of thiotepa are best described by a two-compartment model, with an α-half-life of 6 to 24 minutes and a terminal half-life of 78 to 160 minutes. Thiotepa has shown clinical activity in the treatment of breast, bladder, and ovarian cancer, along with carcinomatous meningitis and malignant effusions, and usually is administered IV or as an intravesicular infusion. It also may be used intrathecally. Side effects include myelosuppression, nausea and vomiting, and venous irritation.
Heavy-Metal Compounds
Platinum drugs form reactive platinum complexes that bind to cells, so the pharmacokinetics of the individual drug may be of the platinum, both free and bound, rather than of the parent drug.
Cisplatin
Cisplatin forms inter- and intrastrand DNA cross-links to inhibit DNA synthesis. The pharmacokinetics are best described by a three-compartment model, with an α-half-life of 20 minutes, a β-half-life of 48 to 70 minutes, and a terminal half-life of 24 hours. Ninety percent of the drug is removed by the kidney by glomerular filtration and tubular secretion. Cisplatin has shown clinical activity in the treatment of numerous tumor types, from head and neck cancers to anal cancer, including many types of lymphoma and carcinoma of unknown primary. Cisplatin is highly emetogenic, even when low doses are given daily for 5 days, and causes delayed nausea and vomiting as well; patients require aggressive antiemetic regimens for both delayed and acute emesis. Significant nephrotoxicity and electrolyte abnormalities can occur if inadequate hydration occurs. Ototoxicity, which manifests as a high-frequency hearing loss, and a glove-and-stocking neuropathy may limit therapy.
Carboplatin
While carboplatin has the same mechanism of action as cisplatin, it has a much less toxic side-effect profile than cisplatin. The pharmacokinetics of carboplatin are best described by a two-compartment model, with an α-half-life of 90 minutes and a terminal half-life of 180 minutes. Carboplatin is eliminated almost entirely by the kidney by glomerular filtration and tubular secretion. Many chemotherapy regimens dose carboplatin based on an area under the curve (AUC), which is referred to as the Calvert equation. According to the Calvert equation, the dose in milligrams = (CrCl + 25) × AUC desired.28Carboplatin has shown clinical activity in the treatment of ovarian, lung, breast, testicular, esophageal, and head and neck cancers, as well as lymphomas. Thrombocytopenia, nausea and vomiting, and hypersensitivity reactions are side effects.
Oxaliplatin
The pharmacokinetics of oxaliplatin are best described by a three-compartment model, with an α-half-life of 0.28 hours, a β-half-life of 16.3 hours, and a terminal half-life of 273 hours.29 Oxaliplatin has shown clinical activity in the treatment of colorectal cancer. Oxaliplatin, while similar in action to cisplatin and carboplatin, causes a cold-induced neuropathy. Patients should be counseled to avoid cold beverages, to use gloves to remove items from the freezer, and to wear protective clothing in cold climates for the first week after treatment. A glove-and-stocking neuropathy also occurs with long-term dosing. Hypersensitivity reactions and moderate nausea and vomiting are also side effects.
mTOR Inhibitors
The mammalian target of rapamycin (mTOR) is a downstream mediator in the phosphatidylinositol 3-kinase/Akt signaling pathway which controls translation of proteins that regulate cell growth and proliferation, but also angiogenesis and cell survival.
Temsirolimus
The mTOR is an intracellular component which stimulates protein synthesis by phosphorylating translation regulators, and contributes to protein degradation and angiogenesis. Temsirolimus is approved for the treatment advanced renal cell carcinoma. The pharmacokinetics of temsirolimus are best described by a two-compartment model, with a terminal half-life of 13 to 25 hours. Elimination is primarily via the feces. Temsirolimus, and its metabolite sirolimus, are substrates of the cytochrome P4503A4/5 isoenzyme system. The primary side effects of temsirolimus include mucositis, diarrhea, maculopapular rash, nausea, leucopenia, thrombocytopenia, and hyperglycemia.
Miscellaneous Agents
Altretamine
Altretamine, formerly known as hexamethylmelamine, is similar in structure to alkylating agents but is known to have anticancer activity in cancer cells resistant to alkylating agents. Altretamine is well absorbed after oral administration and undergoes rapid and extensive demethylation in the liver. Peak plasma concentrations were observed 0.5 to 3 hours after administration. The terminal half-life is 4.7 to 10.2 hours. Altretamine has shown activity in the treatment of ovarian and lung cancer. This orally administered drug has the dose-limiting side effects of anorexia, nausea, vomiting, diarrhea, and abdominal cramping. Other side effects include neuropathy, agitation, confusion, and depression.
Bleomycin
Bleomycin is a mixture of peptides with drug activity expressed in units, where 1 unit equals 1 mg. Bleomcyin causes DNA strand breakage. The pharmacokinetics of bleomycin are best described by a two-compartment model, where the α-half-life is 10 to 20 minutes and a terminal elimination half-life of 2 to 3 hours, which can be prolonged to 21 hours in patients with renal impairment.30 Bleomycin has shown clinical activity in the treatment of testicular cancer and malignant effusions, squamous cell carcinomas of the skin, and Kaposi’s sarcoma. Hypersensitivity reactions and fever may occur, so premedication with acetaminophen may be required. The most serious side effect is the pulmonary toxicity that presents as a pneumonitis with a dry cough, dyspnea, rales, and infiltrates. Pulmonary function studies will show decreased carbon monoxide diffusing capacity and restrictive ventilatory changes. “Bleomycin lung” is associated with cumulative dosing greater than 400 units and occurs rarely with a total dose of 150 units. The pulmonary toxicity is potentiated by thoracic radiation and by hyperoxia. Additional side effects include fever with or without chills, mild to moderate alopecia, and nausea and vomiting. Bleomycin has been used to manage malignant effusions at doses of 15 to 60 units through installation into the affected area. The drainage tube of the effusion is clamped off for some period of time after administration of the bleomycin (time varies based on the location of the effusion), and then the amount of drainage is monitored to determine efficacy of the bleomycin treatment.
Hydroxyurea
Hydroxyurea is an oral drug that inhibits ribonucleotide reductase, which converts ribonucleotides into the deoxyribuoncleotides used in DNA synthesis and repair. The time to peak concentrations of hydroxyurea is 1 to 2 hours after oral administration. Approximately 50% is degraded by the liver to form urea and respiratory carbon dioxide. The remainder is excreted by the kidney. The half-life ranges from 3.5 to 4.5 hours. Hydroxyurea has shown clinical activity in the treatment of chronic myelocytic leukemia, polycythemia vera, and thrombocytosis. The major side effects are myelosuppression, nausea and vomiting, diarrhea, and constipation. Rash, mucositis, and renal tubular dysfunction occur rarely.
L-Asparaginase
L-Asparaginase is an enzyme that may be produced by Escherichia coli. Asparaginase hydrolyzes the reaction of asparagines to aspartic acid and ammonia to deplete lymphoid cells of asparagine, which inhibits protein synthesis. The pharmacokinetics of L-asparaginase are best described by a two-compartment model, with an initial half-life of 4 to 9 hours and a terminal half-life of 1.4 to 1.8 days. L-Asparaginase has shown clinical activity in the treatment of acute lymphocytic leukemia and childhood acute myeloid leukemia. Severe allergic reactions may occur when the interval between doses is 7 days or greater, so while a skin test may be negative, patients should be observed closely after asparaginase administration. Pancreatitis and fibrinogen depletion also may occur during therapy. Repletion of fibrinogen should be done to prevent disseminated intravascular coagulation and fatal bleeding. If the patient suffers an allergic reaction to L-asparaginase, pegaspargase, which is L-asparaginase modified through a linkage with polyethylene glycol, which extends the half-life and allows for lower doses and less frequent administration, may be given. Cost and limited availability are the reasons why pegaspargase is not used first.
Arsenic Trioxide
Arsenic trioxide, which was approved recently for the treatment of acute promyelocytic leukemia, induces the growth of cancer cells into mature, more normal cells, as well as induces programmed cell death, or apoptosis. The pharmacokinetics of arsenic trioxide are best described by a two-compartment model, with an α-half-life of 0.89 hours and a β-half-life of 12.1 hours. Less than 10% of a dose is excreted by the kidney.31 Arsenic trioxide causes QT-interval prolongation, so frequent ECGs need to be done prior to each dose, and other drugs that may prolong the QT interval need to be avoided during therapy. Monitoring of potassium and magnesium should be done, and active replacement undertaken to prevent QT prolongation. Other side effects include dry skin with itching, nausea and vomiting, loss of appetite, and elevations of serum hepatic enzymes. An uncommon but serious side effect is a syndrome similar to the retinoic acid syndrome, which appears similar to pneumonia but is related to the arsenic therapy.
Mitomycin C
Mitomycin C is an alkylating agent that forms cross-links with DNA to inhibit DNA and RNA synthesis. The pharmacokinetics of mitomycin C are best described by a two-compartment model, with an α-half-life of 8 minutes and a terminal half-life of 48 minutes.32 Liver metabolism is the primary route of elimination. Mitomycin C has shown clinical activity in the treatment of anal, bladder, cervix, gallbladder, esophageal, and stomach cancer. Side effects consist of myelosuppression and mucositis, and it is a vesicant.
Tretinoin
Tretinoin, also referred to ATRA, which stands for all transretinoic acid, is a retinoic acid that is not cytotoxic but promotes the maturation of early promyelocytic cells and is specific to the t(15;17) cytogenetic marker. The time to peak concentrations is 1 to 2 hours after an oral dose. The elimination half-life is 21 to 51 minutes.33 These maroon-and-gold capsules are dosed at 45 mg/m2/day divided into two doses. The most significant side effect is the retinoic acid syndrome, which may occur anywhere from the first couple of days of therapy until the end of therapy and consists of symptoms of fever, respiratory distress, and hypotension. Chest radiographs are consistent with a pneumonia-like process. The syndrome can be confused easily with pneumonia in a patient with possible neutropenia. The treatment for retinoic acid syndrome is dexamethasone 10 mg IV every 12 hours; the syndrome may resolve within 24 hours of the start of dexamethasone therapy. However, the use of steroids in a febrile neutropenic patient may further compromise the treatment of infection.
Thalidomide
Thalidomide was introduced into the market on October 1, 1957, as a sedative-hypnotic, and when it was taken by pregnant women, it resulted in severe limb deformities (phocomelia). Thalidomide is used for the treatment of leprosy but now is part of the treatment of multiple myeloma. While thalidomide is believed to be an angiogenesis inhibitor, the mechanism of action is still unknown. Possible mechanisms of action include free-radical oxidative damage to DNA, inhibiting tumor necrosis factor α production, altering the adhesion of cancer cells, and altering cytokines that affect the growth of cancer cells. The pharmacokinetics of thalidomide demonstrate a time to peak concentration of 2 to 3 hours and a terminal half-life of 4 to 7 hours.34 Thalidomide has shown clinical activity in the treatment of multiple myeloma and is still being studied for the treatment of several other cancers. Because of thalidomide’s potential to cause phocomelia, each patient must be counseled on the risks of thalidomide not only for the patient but also the patient’s reproductive partner. Physicians must be registered to prescribe thalidomide. Pharmacists can fill prescriptions only where both the patient and the physician have completed surveys on a monthly basis.
Lenalidomide
Lenalidomide is approved for the treatment of myelodysplastic syndrome where the 5q deletion is present and multiple myeloma. Since lenalidomide is an analog of thalidomide, all the same precautions must be taken to prevent phocomelia. The time to maximum lenalidomide concentrations occurs 0.5 to 4 hours after the dose. The terminal half-life ranges from 3 to 9 hours. Approximately 65% of lenalidomide is eliminated unchanged in the urine, with clearance exceeding the glomerular filtration rate. Dosing adjustments are necessary for renal dysfunction.35 Lenalidomide is used in the treatment of myelodysplastic syndrome and multiple myeloma. Other side effects are neutropenia, thrombocytopenia, deep vein thrombosis, and pulmonary embolus.
Bexarotene
Bexarotene is a retinoid that selectively activates retinoid X receptors which affects cellular differentiation and proliferation. The time of maximum concentration after an oral dose of bexarotene is about 2 hours, while the terminal half-life is about 7 hours. Bexarotene is eliminated primarily by the hepatobilliary system. Bexarotene is indicated for the treatment of cutaneous manifestations of cutaneous T-cell lymphoma in patients who are refractory to other therapy. Side effects include hypercholesterolemia, elevations in triglycerides, pancreatitis, hypothyroidism, and leukopenia, headache, and dry skin.
Vorinostat
Vorinostat is indicated for the treatment for cutaneous T-cell lymphoma in patients with progressive, persistent, or recurrent disease after treatment with other drugs. Vorinostat inhibits the activity of histone deacetylases which results in repression of gene transcription. Vorinostat is eliminated primarily by glucuronidation and hydrolysis to pharmacologically inactive metabolites, with a terminal half-life of 2 hours. Side effects include diarrhea, fatigue, nausea and anorexia, hypercholesterolemia, hypertriglyceridemia, and hyperglycemia. Despite anemia, thrombocytopenia and neutropenia, patients have developed pulmonary embolism and deep vein thromboses while on therapy.
Immune Therapies
Interferons
The categories of α, β and γ interferons exist; the α interferons are used in the treatment of cancer. Interferon enhances the immune system’s attack on cancer cells, can decrease new blood vessel formation, and can augment expression of antigen on tumor cell surfaces. Interferon has an elimination half-life of 3.7 to 8.5 hours. Interferon is filtered through the glomeruli and then degraded during tubular reabsorption. Interferon has shown clinical activity in the treatment of melanoma, kidney cancer, Kaposi’s sarcoma, and chronic myelocytic and lymphocytic leukemia. Unfortunately, interferon is not well tolerated by patients because it causes a flu-like syndrome that consists of fevers and chills; depression, malaise, and fatigue are other side effects. Premedication with acetaminophen will help alleviate the flu-like symptoms, which will decrease with chronic administration.
Aldesleukin
Aldesleukin, commonly referred to as interleukin 2, is a lymphokine that promotes B- and T-cell proliferation and triggers a cytokine cascade to attack the tumor. The pharmacokinetics are best described by a two-compartment model, with an α-half-life of 13 minutes and a terminal half-life of 85 minutes. Aldesleukin is eliminated by both glomerular filatration and peritubular extraction in the kidney. Aldesleukin has shown clinical activity in the treatment of kidney cancer and melanoma. Side effects of interleukin 2 vary by dose and route. IV high-dose interleukin 2 causes a drug-induced shock-like picture. Patients may develop hypotension despite aggressive IV hydration. Patients develop a red, itching skin; liver and kidney function tests change; fluid and electrolyte imbalances occur; and high fevers occur while receiving scheduled acetaminophen and nonsteroidal anti-inflammatory agents. Severe rigors and chills may require IV meperidine for symptom control. All the side effects reverse within 24 hours of stopping the drug. The toxicity profile is much less with subcutaneous administration. However, with subcutaneous administration, little nodules form at the injection site and may take months to resolve. Corticosteroids should not be administered to patients while receiving aldesleukin unless a life-threatening emergency should occur. Steroids will reverse all the symptoms and the antitumor effect, even with topical administration. The itching, red skin may be treated with topical creams and antihistamines.
Denileukin Diftitox
Denileukin diftitox is a combination of the active sections of interleukin 2 and diphtheria toxin. It binds to high-affinity interleukin 2 receptors on the cancer cell (and other cells), and the toxin portion of the molecule inhibits protein synthesis to result in cell death. The pharmacokinetics of denileukin diftitox are best described by a two-compartment model, with an α-half-life of 2 to 5 minutes and a terminal half-life of 70 to 80 minutes. Denileukin diftitox is used for the treatment of persistent or recurrent cutaneous T-cell lymphoma whose cells express the CD25 receptor. Side effects include vascular leak syndrome, fevers/chills, hypersensitivity reactions, hypotension, anorexia, diarrhea, and nausea and vomiting.
Monoclonal Antibodies
The cell surface contains antigens, which are referred to as CD, which stands for “cluster of differentiation.” The antibodies are produced against a specific antigen. When administered, usually by an IV injection, the antibody binds to the antigen, which may trigger the immune system to result in cell death through complement-mediated cellular toxicity, or the antigen-antibody cell complex may be internalized to the cancer cell, which results in cell death. Monoclonal antibodies also may carry radioactivity, sometimes referred to as hot antibodies, and may be referred to as radioimmunotherapy, so the radioactivity is delivered to the cancer cell. Antibodies that contain no radioactivity are referred to as cold antibodies.
All monoclonal antibodies end in the suffix -mab. The syllable before -mab indicates the source of the monoclonal antibody (see Table 88–7). When administering an antibody for the first time, one should consider the source. The less humanized an antibody, the greater is the chance for the patient to have an allergic-type reaction to the antibody. The more humanized the antibody, the lower is the risk of a reaction. The severity of the reactions may range from fever and chills to life-threatening allergic reactions (which have resulted in death). Premedication with acetaminophen and diphenhydramine is common before the first dose of any antibody. If a severe reaction occurs, the infusion should be stopped and the patient treated with antihistamines, corticosteroids, or other supportive measures.
Alemtuzumab
Alemtuzumab is the antibody to the CD52 receptor present on B and T lymphocytes. The pharmacokinetics of alemtuzumab demonstrate a terminal half-life of 7 days. Alemtuzumab has shown clinical activity in the treatment of chronic lymphocytic leukemia. Severe and prolonged (6 months) immunosuppression may result, which necessitates prophylaxis with cotrimoxazole and antivirals to prevent opportunistic infections.
Table 88–7 Syllable Source Indicators for Monoclonal Antibodies

Bevacizumab
Bevacizumab is a humanized monoclonal antibody that binds to vascular endothelial growth factor, which prevents it from binding to its receptors, ultimately resulting in inhibition of angiogenesis. The pharmacokinetics of bevacizumab demonstrate a terminal half-life of 21 days, with a volume of distribution consistent with limited extravascular distribution.36 Bevacizumab has shown clinical activity in the treatment of colorectal, kidney, lung, breast, and head and neck cancer. Patients may develop hypertension requiring chronic medication during therapy. Impaired wound healing, thromboembolic events, proteinuria, bleeding, and perforation are serious side effects.
Cetuximab
Cetuximab is a chimeric antibody that binds to the epidermal growth factor receptor (EGFR) to block its stimulation. Recently investigators found that colorectal tumors that have KRAS mutations do not respond to treatment with cetuximab; therefore tumors should be tested for KRAS mutations prior to initiating therapy. The pharmacokinetics of cetuximab demonstrate a volume of distribution that approximates the vascular space and a terminal half-life of 70 to 100 hours. Cetuximab has shown clinical activity in the treatment of colorectal and head and neck cancers. An acne-like rash may appear on the face and upper torso 1 to 3 weeks after the start of therapy. Other side effects include hypersensitivity reactions, interstitial lung disease, fever, malaise, diarrhea, abdominal pain, and nausea and vomiting.
Gemtuzumab Ozogamicin
Gemtuzumab ozogamicin is a humanized antibody to the CD33 receptor present on about 80% of acute myelogenous leukemia cells. The antibody is linked to calicheamicin, a cellular toxin that is released intracellularly after the antigen-antibody complex is internalized. The pharmacokinetics of gemtuzumab ozogamicin show a terminal half-life of 67 to 78 hours of the antibody portion of the drug and a terminal half-life of about 45 hours of the calicheamicin.37 Gemtuzumab ozogamicin has shown clinical activity in the treatment of acute myeloid leukemia. Severe rigors and chills, which may occur after the infusion is completed, respond to meperidine IV. Premedications should include acetaminophen, diphenhydramine, and methylprednisolone to prevent rigors and chills.
Ibrotumomab Tiuxetan
This “hot antibody” is linked to yttrium and binds to the CD20 receptor of B lymphocytes (see Rituximab below). Hematologic toxicity may occur several weeks after administration and may take weeks to resolve.
Panitumumab
Panitumumab binds to the EGFR to prevent receptor auto-phosphorylation and activation of receptor-associated kina-ses, which results in inhibition of inhibition of cell growth and induction of apoptosis. Recently data were presented that demonstrated that colorectal tumors without KRAS mutations responded to panitumumab therapy, and had a longer median time to progression. Panitumumab demonstrates nonlinear pharmacokinetics with a terminal half-life of 7.5 days. Panitumumab has demonstrated activity against tumors of the colon and rectum. Side effects include dermatitis, pruritus, exfoliative rash, infusion reactions, pulmonary fibrosis, diarrhea, hypomagnesemia, hypocalcemia, and photosensitivity.
Rituximab
Rituximab is a monoclonal antibody to the CD20 receptor expressed on the surface of B lymphocytes; the presence of the antibody is determined during flow cytometry of the tumor cells. Cell death results from antibody-dependent cellular cytotoxicity. The pharmacokinetics of rituximab are best described by a two-compartment model, with a terminal half-life of 76 hours after the first infusion and a terminal half-life of 205 hours after the fourth dose.38Rituximab has shown clinical activity in the treatment of B-cell lymphomas that are CD20 positive. Side effects include hypersensitivity reactions, hypotension, fevers, chills, rash, headache, and mild nausea and vomiting.
Tositumomab
This “hot antibody” is linked to radioactive iodine and binds to the CD20 receptor present on B lymphocytes (see Rituximab above). Tositumomab has shown activity in non-Hodgkin’s lymphoma. Hematologic toxicity occurs several weeks after administration and may persist for months. Because radioactive iodine may have adverse effects on the thyroid, all patients must receive thyroid-blocking agents.
Trastuzumab
Trastuzumab is the antibody directed against human epidermal receptor 2 (HER-2), which is overexpressed by 25% to 30% of breast cancers and is associated with aggressive disease and decreased survival. Breast cancer tissue must be tested for the presence of HER-2, as patients that do not expresses HER-2 do not respond to trastuzumab. The pharmacokinetics of trastuzumab are best described by a two-compartment model, with a terminal half-life of 19 to 28 days.39 Severe congestive heart failure may occur with concurrent anthracycline administration. Cardiac toxicity may be seen when the drug is administered months after anthracycline administration, so patients must be counseled on the signs and symptoms of heart failure. Other side effects include hypersensitivity reactions, fever, diarrhea, infections, chills, cough, headache, rash, and insomnia.
Tyrosine-Kinase Inhibitors
There are more than 100 different types of tyrosine-kinases present in the body. Sometimes tyrosine-kinase inhibitors are referred to as small-molecule inhibitors. Each of the following drugs was developed to block either several or a specific tyrosine kinase.
Imatinib
Imatinib was the first FDA-approved tyrosine-kinase inhibitor. The drug was designed to block the breakpoint cluster region tyrosine kinase (BCR:ABL) produced by the Philadelphia chromosome associated with chronic myelogenous leukemia and acute lymphocytic leukemia. The pharmacokinetics of imatinib demonstrate a mean time to maximum concentration of 2 to 4 hours, with a terminal half-life of 15 hours.40 Imatinib also has shown activity against GI stroma tumors (GIST) that are positive for c-kit (CD117). Numerous drug interactions have been reported for imatinib. CYP450 3A4 inducers, such as rifampicin and St. John’s wort, increase the clearance of imatinib.41,42Ketoconazole, a CYP450 3A4 inhibitor, has been shown to decrease imatinib clearance by almost 30%.43 Imatinib also may increase the exposure of simvastatin, a CYP450 3A4 substrate.44
Dasatinib
Dasatinib is a second generation tyrosine kinase inhibitor that shares the same binding site on the BCR:ABL cluster region as imatinib, but maintains activity despite imatinib resistance, with more higher potency than imatinib. Dasatinib also inhibits SRC kinases, which are tyrosine kinases that mediate cellular differentiation, proliferation and survival. The terminal half-life of dasatinib is 3 to 5 hours, with elimination primarily via the liver and feces. Dasatinib is used in the treatment of chronic myeloid leukemia with resistance or intolerance to imatinib, and for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Side effects of dasatinib include myelosuppression, nausea and vomiting, headache, fluid retention, hypocalcemia and pleural effusions.
Nilotinib
Nilotinib also blocks the breakpoint cluster region tyrosine kinase (BCR:ABL) produced by the Philadelphia chromosome associated with chronic myelogenous leukemia and acute lymphocytic leukemia. Nilotinib is primarily eliminated by oxidation and hydroxylation, with a terminal half-life of 17 hours. Nilotinib should be administered on an empty stomach, or 2 hours after a meal. Nilotinib is indicated for the treatment of Philadelphia-chromosome-positive chronic myelogenous leukemia also. Side effects include QT prolongation, bone marrow suppression, elevations in lipase, and hepatotoxicity.
Erlotinib
Erlotinib, whose pharmacology is not entirely understood, is believed to inhibit the intracellular phosphorylation of the EGFR. Erlotinib is about 60% absorbed after oral administration; food increases bioavailability to almost 100%, however this is variable and experts recommend administering erlotinib on an empty stomach. The time to peak concentrations is approximately 4 hours after a dose. The half-life of erlotinib is about 36 hours, and it is eliminated predominately by CYP450 3A4. Smoking increases the clearance of erlotinib by 24%, which may result in treatment failure.45 Erlotinib is used in the treatment of nonsmall cell lung cancer and cancer of the pancreas. Side effects include interstitial lung disease, rash, diarrhea, anorexia, pruritus, conjunctivitis, and drug skin. Again, significant drug interactions have been documented with CYP450 3A4 inducers and inhibitors.
Lapatinib
Lapatinib inhibits the intracellular kinase domains of both EGFR and HER-2, and has been shown to retain activity against breast cancer cells that have become resistant to trastuzumab. The pharmacokinetics of lapatinib with repeated dosing display a time-dependent increase in systemic exposure. Absorption is enhanced when administered with food. A single-dose study demonstrated a terminal half-life of 14 hours, while repeated dosing studies indicated an effective half of 24 hours. Patients with Child-Pugh class C liver disease should have a dosage reduction to 750 mg daily to adjust the AUC to the normal range. Lapatinib is indicated for the treatment of patients with breast cancer whose tumors overexpress HER-2. Side effects of lapatinib include decreased left-ventricular ejection fraction, diarrhea, hepatotoxicity, rash, and QT prolongation.
Sorafenib
Sorafenib is a multikinase inhibitor that inhibits both intracellular and extracellular kinases to decrease renal cell cancer proliferation. The half-life of sorafenib is 25 to 48 hours, with a bioavailability of 38% to 49% and a time to peak concentration of 3 hours. Sorafenib is metabolized primarily by the liver by CYP450 3A4. Sorafenib is used for the treatment of renal cell cancer. The primary side effects of sorafenib include rash, hand-foot skin reaction, diarrhea, pruritus, and elevations in serum lipase.
Sunitinib
Sunitinib blocks several tyrosine kinases, so it inhibits platelet-derived growth factor, vascular endothelial growth factor receptor, stem cell factor receptor, fms-like receptor growth factor, colony-stimulating growth factor receptor type 1, and glial-cell-line-derived neurotrophic factor receptor. The active metabolite of sunitinib blocks these same enzymes with similar potency. The pharmacokinetics of sunitinib demonstrate a time of peak concentration of about 5 hours, with a half-life of 41 to 86 hours.46 It is indicated for the treatment of GISTs after disease progression or intolerance to imatinib. It is also indicated for the treatment of advanced renal cell cancer. Significant side effects include left ventricular dysfunction, hemorrhage, asthenia, hypertension, nausea and vomiting, and diarrhea. Approximately one-third of patients may develop a yellow color of the skin, along with dryness and cracking of the skin. Also, hair may become depigmented with doses of 50 mg/day or more, and the depigmentation is reversible when therapy is stopped. Clinically significant drug interactions exist with drugs metabolized via the CYP450 3A4 system; ketoconazole has been shown to increase concentrations of sunitinib, whereas rifampin has been shown to decrease concentrations of sunitinib.
Hormonal Therapies
Hormonal therapies have shown activity in the treatment of cancers whose growth is affected by gonadal hormonal control. Hormonal treatments either block or decrease the production of endogenous hormones.
Antiandrogens: Bicalutamide, Flutamide, and Nilutamide
The antiandrogens block androgen receptors to inhibit the action of testosterone and dihydrotestosterone in prostate cancer cells. Unfortunately, prostate cancer cells may become hormone refractory.
Flutamide is an androgen receptor antagonist that achieves peak concentrations approximatley 2 to 4 hours after an oral dose. Flutamide is metabolized extensively, with a terminal half-life of about 8 hours. Bicalutamide achieves peak concentrations approximately 6 hours after the dose, with a terminal half-life of 6 to 10 days. Bicalutamide undergoes stereospecific metabolism, where the S-enantiomer is cleared more rapidly by the liver than the R-enantiomer. Nilutamide achieves peak serum concentrations between 1 and 4 hours after an oral dose and has a terminal half-life of 38 to 60 hours. Nilutamide is metabolized extensively, with less than 2% excreted as unchanged drug by the kidney. Side effects common to these agents are hot flashes, gynecomastia, and decreased libido. Flutamide tends to be associated with more diarrhea and requires three-times-daily administration, whereas bicalutamide is dosed once daily. Nilutamide may cause interstitial pneumonia and is associated with the visual disturbance of delayed adaptation to darkness.
Luteinizing Hormone–Releasing Hormone Agonists: Goserelin and Leuprolide
Initially, luteinizing hormone–releasing hormone (LHRH) agonists increase levels of leutinizing hormone and follicle-stimulating hormone, but testosterone and estrogen levels are decreased because of continuous negative-feedback inhibition. Major side effects are testicular atrophy, decreased libido, gynecomastia, and hot flashes. Leuprolide is well absorbed, with a terminal half-life of 2.9 hours, whereas goserelin has a terminal half-life of 4.9 hours. Goserelin is injected as a pellet under the skin, so subcutaneous injection of lidocaine prior to administration helps to decrease the pain associated with goserelin administration. Numerous dosage forms are available for leuprolide with varying strengths and dosing intervals. Antiandrogens may be administered during initial therapy to decrease symptoms of tumor flare (e.g., bone pain and urinary tract obstruction).
Ketoconazole
While ketoconazole is an antifungal agent, it has been used for treatment of prostate cancer. In high doses of 400 mg three times daily, ketoconazole blocks the production of testosterone.
LHRH Antagonist: Abarelix
Abarelix is a gonadotropin-releasing hormone antagonist that is associated with life-threatening hypersensitivity reactions. Patients must be observed for 30 minutes after administration. The drug is limited to men who cannot risk tumor flare and refuse orchiectomy.
Aminoglutethimide
Aminoglutethimide blocks the conversion of androgens to estrogens and decreases the synthesis of glucocorticoids. Adrenocorticoid suppression may occur, so replacement therapy with hydrocortisone may be required. Other side effects include rash (which usually resolves in 5–8 days), lethargy, and anorexia.
Anastrozole
Anastrozole is a selective nonsteroidal aromatase inhibitor that lowers estrogen levels. The pharmacokinetics of anastrozole demonstrate good absorption, with hepatic metabolism the primary route of elimination and only 10% excreted unchanged by the kidney. The elimination half-life is approximately 50 hours. Anastrozole is used for the adjuvant treatment of postmenopausal women with hormone-positive breast cancer and in breast cancer patients who have had disease progression following tamoxifen. Side effects include hot flashes, arthralgias, osteoporosis/bone fractures, and thrombophlebitis.
Exemestane
Exemestane is an irreversible aromatase inactivator that binds to the aromatase enzyme to block the production of estrogen from androgens. Exemestane is absorbed rapidly after oral administration, with a terminal half-life of 24 hours. The drug is eliminated primarily by the liver and feces, with less than 1% of the dose excreted unchanged in the urine. Exemestane is indicated for the treatment of advanced breast cancer in postmenopausal women who have had disease progression following tamoxifen therapy. Side effects include hot flashes, fatigue, osteoporosis/bone fractures, and flu-like symptoms.
Fulvestrant
Fulvestrant is an estrogen receptor antagonist that binds to the estrogen receptor and is given as a monthly intramuscular injection. Fulvestrant has a terminal half-life of 40 days and a large volume of distribution of 3 to 5 L/kg. Fulvestrant is metabolized primarily, with excretion primarily via feces. Fulvestrant is used for the treatment of hormone-receptor-positive metastatic breast cancer in postmenopausal women with disease progression following antiestrogen therapy. Side effects are hot flashes, abdominal pain, depression, and myalgias.
Letrozole
Letrozole is another selective aromatase that inhibits the conversion of androgens to estrogen. Maximum plasma concentrations occur 1 hour after oral dosing; concomitant food has not been shown to have an effect on the extent of absorption of letrozole. The terminal half-life is approximately 2 days. Letrozole is used in the treatment of postmenopausal women with hormone-receptor-positive or unknown advanced breast cancer. Side effects include bone pain, hot flushes, back pain, nausea, arthralgia, osteoporosis/bone fractures, and dyspnea.
Megestrol Acetate
Megestrol is a synthetic progestin with antiestrogen properties that is used for breast cancer and in higher doses for weight gain. Side effects include fluid retention, hot flashes, vaginal bleeding and spotting, breast tenderness, and thrombosis.
Tamoxifen
Tamoxifen is an estrogen receptor antagonist. Most of a dose of tamoxifen is eliminated primarily by metabolism, with significant enterohepatic recirculation. The time to a peak concentration is 6 hours after an oral dose, and the terminal half-life is 7 days. Tamoxifen is used primarily for the treatment of postmenopausal hormone-receptor-positive breast cancer patients and the prevention of breast cancer in postmenopausal women. Side effects include hot flashes, fluid retention, mood swings, thrombosis, endometrial and uterine cancer, and corneal changes and cataracts. Since tamoxifen is a substrate of CYP450 3A4, decreased tamoxifen levels have occurred with use of St. John’s wort, and decreased tamoxifen levels have been observed with use of rifampin. Tamoxifen is also a substrate for CYP450 2D6, and recent evidence suggests that those who are CYP2D6*4/*4 may have a poorer response and more toxicity with tamoxifen.47 Significant drug interactions exist with antidepressants which may be used to treat depression or help relieve hot flashes.
Toremifene
Toremifene is an estrogen receptor antagonist. The pharmacokinetics of toremifene are best described by a two-compartment model, with an α-half-life of 4 hours and an elimination half-life of 5 days. Peak plasma concentrations are achieved approximately 3 hours after an oral dose. Toremifene is metabolized extensively, with metabolites found primarily in the feces. Toremifene is used for the treatment of metastatic breast cancer in postmenopausal women with estrogen-receptor-positive or unknown tumors. Toremifene causes hot flashes, vaginal bleeding, thromboembolism, and visual acuity changes.
ADMINISTRATION ISSUES
Extravasation
Another issue of chemotherapy safety is extravasation. Antineoplastic agents that cause severe tissue damage when they escape from the vasculature are called vesicants. Some examples of vesicants are the anthracyclines and the vinca alkaloids. The tissue damage may be severe, with tissue sloughing and loss of mobility, depending on the area of extravasation. Patients need to be educated to notify the nurse immediately if there is any pain on administration. If extravasation of a vesicant occurs, the injection should be stopped, and any fluid aspirated out of the injection site. If an antidote should be administered, such as with nitrogen mustard extravasation, the pharmacy should be notified immediately so that the sodium thiosulfate can be prepared and delivered quickly. Cold compresses should be applied (heat should be used for vincas) to the affected area. The area of extravasation should be recorded and inked and followed closely to detect early signs of infection and to treat pain. Obviously, prevention of extravasation is very important. Good IV access is key, which may include the placement of a central venous catheter, along with free-flowing IV fluids. During administration of a vesicant, blood return should be checked to be sure that the drug is going into the vein. Central venous catheters can be placed for patients needing infusions of vesicants or for patients with small, friable veins to prevent extravasation.
Hypersensitivity Reactions
Recently more attention has been focused on hypersensitivity reactions of cancer treatments because of cross-reactivity between agents, and the desire to continue active therapies against the cancer.48Bleomycin and asparaginase have skin tests suggested to be administered prior to administration. However, a negative skin test does not preclude an allergic reaction. For documented immediate hypersensitivity reactions to a particular agent, further administration of the agent may be achieved through extensive premedication with H1 and H2 antihistamines and corticosteroids, and through use of escalating doses of the offending agent given at doses of one-hundredth, one-tenth, and the balance of the dose (so the total dose administered is equivalent to the normally prescribed dose) administered over a much longer period of time. These treatments must be given in an environment where resuscitation is readily available in case of medical emergency.
Secondary Malignancies
Chemotherapy and radiation therapy treatments may cause cancers later in life; these are referred to as secondary cancers. The most common type of secondary cancer is myelodysplastic syndrome, or acute myeloid leukemia. The antineoplastic agents most commonly associated with secondary malignancies are alkylating agents, etoposide, teniposide, and anthracyclines. While the risk for secondary cancers is extremely low, it must outweigh the risk of survival produced by treatment of the primary malignancy. Because secondary malignancies may not occur for several years after treatment, patients with relatively short-term survival owing to the primary malignancy should consider the more immediate benefits of chemotherapy. Radiation therapy rarely may cause solid tumors as secondary cancers decades after treatment. The most common example of radiation therapy–induced secondary malignancy is breast cancer, which rarely occurs after mantle field radiation therapy for Hodgkin’s disease.
CHEMOTHERAPY SAFETY
One of the first Institute of Medicine reports starts out with a patient who died from an overdose of chemotherapy; the patient did not have an immediately life-threatening cancer, so her death was hastened by a medication error. Chemotherapy agents may cause harm to patients, health care workers, and the environment if not handled correctly. ![]() Because of the severe toxicities associated with many of the chemotherapy agents, safety precautions must be in place to prevent chemotherapy errors, accidental chemotherapy exposures, and overdosages. The Oncology Nursing Society and the American Society of Health-System Pharmacists have information to assist in the safe handling of chemotherapy agents.49 National, state, and local regulations regarding the safe disposal of chemotherapy agents and the equipment used to administer them need to be followed to protect the environment.
Because of the severe toxicities associated with many of the chemotherapy agents, safety precautions must be in place to prevent chemotherapy errors, accidental chemotherapy exposures, and overdosages. The Oncology Nursing Society and the American Society of Health-System Pharmacists have information to assist in the safe handling of chemotherapy agents.49 National, state, and local regulations regarding the safe disposal of chemotherapy agents and the equipment used to administer them need to be followed to protect the environment.
Table 88–8 Dosing Adjustment Guidelines for Chemotherapy for Renal Dysfunction


Each organization should have chemotherapy safety checks built into the prescribing, preparation, and administration of chemotherapy.50 Dosing based on patient-specific body information should be included on every order for chemotherapy, whether it is oral or parenteral. Many chemotherapy regimens are acronyms; these should not be allowed in the prescribing of chemotherapy. Also, abbreviations for the names of chemotherapy agents should be avoided because one abbreviation may stand for two different drug entities. For drugs such as doxorubicin and liposomal doxorubicin, the names should be written out fully, and in this case, the addition of the brand name may help to prevent a mistake.
The measured height and weight, along with the body surface area, if applicable, should be readily available, along with the dosage in milligrams per meter squared or kilogram, so that the dosage may be checked. If a chemotherapy regimen is a continuous infusion of 800 mg/m2/day for 4 days, an added safety feature would be to include the total dosage of 3,200 mg in order to prevent any ambiguity. In cases where the clinician wants to decrease the dosage based on a laboratory value or side effect, it is recommended that the clinician include that information with the order so that everyone understands what the correct dosage is for that patient. Chemotherapy dosages should be checked for route and dose to determine that the dosages prescribed are correct according to the regimen and do not exceed dosing guidelines. Appropriate laboratory values should be checked to verify that dosages are correct for any organ dysfunction present, and drug interactions should be scrutinized closely (Tables 88–8 and 88–9). Health care practitioners administering chemotherapy should check the dosage calculation for the body size, along with the five Rs of administering medication (i.e., right patient, right medication, right dose, right route, at the right time). If there is any question about the safe dosage or safe administration of a chemotherapy agent, the chemotherapy should not be administered until the question is resolved.
An area of controversy with chemotherapy dosing: What weight should be used for patients who are morbidly obese? Currently, there is no consensus. Some clinicians will cap BSA at some value; some clinicians will use an adjusted weight for the BSA calculation even though there is no recommendation on what adjust weight to use. This is an area for future research.
Many of the newer agents used to treat cancer are orally administered agents. The clinician needs to be aware of drug-herbal interactions, timing of agents with respect to meals, patient education on correct dosing when multiple dosage forms are used, and reimbursement issues. Table 88–10 presents a summary of timing of oral chemotherapy with respect to food. Information should be provided to the patient both in writing and verbally, and to family caregivers if brain metastases are present in order to avoid mistakes. Particular attention should be paid to educating patients on regimens given Monday through Friday only, daily times 4 weeks and then off 2 weeks. Patients should be encouraged to bring in oral medications with visits so adherence can be checked.
Table 88–9 Dosing Adjustment Guidelines for Chemotherapy for Hepatic Impairment
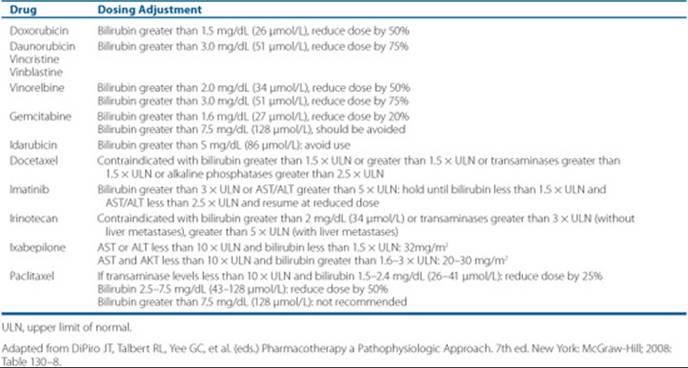
Table 88–10 Oral Chemotherapy Administration with Respect to Food
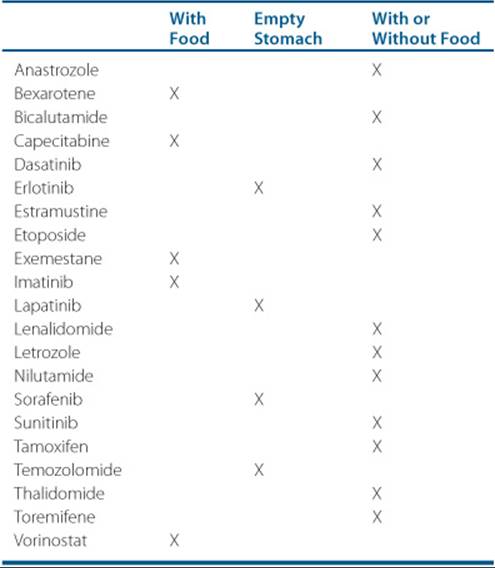
OUTCOME EVALUATION
Once a pathologic diagnosis of cancer is made, the patient may be evaluated by a radiation oncologist, a surgical oncologist, and a medical oncologist. Options for treatment are presented that may include surgery, radiation, chemotherapy, or some combination of these modalities. The goals of treatment will vary by the cancer and the stage of disease. For example, the patient who has metastatic kidney cancer could be cured by high-dose aldesleukin or receive palliative therapy with sorafenib or sunitinib or may decline any therapy because of fears of significant toxicity that would decrease quality of life. In this case, if the patient’s performance status is poor, such as an Eastern Cooperative Oncology Group (ECOG) performance status 3, then the patient would not be a candidate for aldesleukin therapy because of significant toxicity or even death from treatment for a patient who has a performance status of 3. The patient with the poor performance status will receive palliative therapy to control symptoms of the disease to improve the quality of life at the end of life. For the patient with a poor performance status and extensive metastatic disease, no treatment of the cancer may be appropriate, and the patient may be enrolled in a hospice program or provided comfort care.
Patient Encounter 3
DQ is a 57-year-old man who came to the emergency department because of shortness of breath. A radiographic examination demonstrates a large mass pressing on the bifurcation of the main stem bronchus. A biopsy demonstrates an aggressive lymphoma; a five-drug chemotherapy regimen with both IV and intrathecal chemotherapy will start today.
What steps should be taken to make sure that DQ gets the correct dosages of chemotherapy?
Patient Care and Monitoring
![]() Clinicians should play a role in chemotherapy safety, patient education, and monitoring patient response to therapy.
Clinicians should play a role in chemotherapy safety, patient education, and monitoring patient response to therapy.
Chemotherapy Administration
1. Evaluate the chemotherapy regimen to ensure the correct dose and route of administration.
2. Check patient laboratory values to ensure that the CBC and organ function studies are normal prior to administering chemotherapy.
3. If a patient has renal or hepatic impairment or a poor metabolizer genotype, adjust chemotherapy doses if necessary.
4. Ensure appropriate antiemetic therapy for the degree of emetogenicity.
5. Assess the regimen to determine if primary prophylaxis with colony-stimulating factors is needed.
6. Assess the patient’s concurrent medications for the possibility of drug interactions, and discontinue potentially interacting medications, if possible.
7. Employ safe handling and disposal methods.
Monitoring
1. Efficacy: Monitor CT scans or other imaging studies, and categorize patient with a complete response (CR), partial response (PR), stable disease (SD), or progressive disease.
2. Toxicity:
• Monitor CBC and other lab tests
• Monitor patient for other known toxicities of the chemotherapy regimen, and decrease the dose or discontinue the regimen if necessary
Abbreviations Introduced in This Chapter
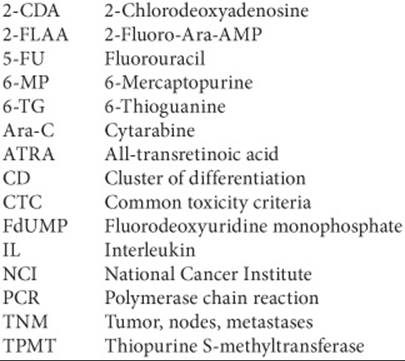
 Self-assessment questions and answers are available at http://www.mhpharmacotherapy.com/pp.html.
Self-assessment questions and answers are available at http://www.mhpharmacotherapy.com/pp.html.
REFERENCES
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin 2008;58:71–96.
2. Blagosklonny MV. Molecular theory of cancer. Cancer Biol Ther 2005 Jun;4(6):621-627.
3. Folkman J. Angiogenesis. Annu Rev Med 2006;57:1-18.
4. Mountford CE, Doran S, Lean CL, Russell P. Cancer pathology in the year 2000. Biophys Chem 1997 Oct;68(1-3):127.
5. Paolo A, Danesi R, Vannozzi F, et al. Limited sampling model for the analysis of 5-fluorouracil pharmacokinetics in adjuvant chemotherapy for colorectal cancer. Clin Pharmacol Ther 2002;72: 627-637.
6. Abbruzzese JL, Grunewald R, Weeks, EA, et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 1991;9(3):491-498.
7. Israili ZH, Vogler WR, Mingioli ES, et al. The disposition and pharmacokinetics in humans of 5-azacytidine administered intravenously as a bolus or by continuous infusion. Cancer Res 1976;36:1453-1461.
8. Hersh MR, Kuhn JG, Phillips JL, et al. Pharmacokinetic study of fludarabine phosphate. Cancer Chemother Pharmacol 1986; 17: 277-280.
9. Lilliemark J, Juliusson G. On the pharmacokinetics of 2’-chloro-2-deoxyadenosine in humans. Cancer Res 1991;51:5570-5572.
10. Kantarjian H, Gandhi V, Cortes J, et al. Phase 2 clinical and pharmacologic study of clofarabine in patients with refractory or relapsed acute leukemia. Blood 2003;1202:2379-2386.
11. Villela LR, Stanford BL, Shah SR. Pemetrexed, a novel antifolate therapeutic alternative for cancer chemotherapy. Pharmacotherapy 2006;26(5):641-654.
12. Bender R, Castle M, Margileth D, et al. The pharmacokinetics of [3H] vincristine in man. Clin Pharmacol Ther 1977;22(4):430-438.
13. Owellen RJ, Hartke CA, Hains FO. Pharmacokinetics and metabolism of vinblastine in humans. Cancer Res 1977;37:2597-2602.
14. Sparreboom A, Scripture CD, Trieu V, et al. Comparative preclinical and clinical pharmacokinetics of a Cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res 2005;11(11):4136–4143.
15. Tije AJ, Verweij J, Carducci MA, et al. Prospective evaluation of the pharmacokinetics and toxicity profile of docetaxel in the elderly. J Clin Oncol 2005;23:1070–1077.
16. Nilsson T, Jonsson G. Clinical results with estramustine phosphate: A comparison of the intravenous and oral preparations. Cancer Chemother Rep 1975;59:229–232.
17. Dorr RT, Von Hoff DD, eds. Cancer Chemotherapy Handbook. 2nd ed. New York: Elsevier; 1994:464.
18. Rodman JH, Abromowitch M, Sinkule JA, et al. Clinical pharmacodynamics of continuous infusion teniposide: Systemic exposure as a determinant of response in a phase I trial. J Clin Oncol 1987;5(7):1007–1014.
19. Chabot GG, Bariler I, Armand JP, et al. Pharmacokinetics of the camptothecin analog CPT-11 and its active metabolite SN-38 in cancer patients. Proc Am Assoc Cancer Res 1991;32:175.
20. Rowinsky E, Grochow L, Hendricks C, et al. Phase I and pharmacologic study of topotecan: A novel topoisomerase I inhibitor. Proc Am Soc Clin Oncol 1991;10:93.
21. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin: Review of animal and human studies. Clin Pharmacokinet 2003;42(5):419–436.
22. Ormrod D, Holm K, Goa K, Spencer C. Epirubicin: A review of its efficacy as adjuvant therapy and in the treatment of metastatic disease in breast cancer. Drugs Aging 1999;5:389–416.
23. Robert J. Clinical pharmacokinetics of idarubicin. Clin Pharmacokinet 1993;24(4):275–288.
24. Faulds D, Balfour JA, Chrisp P, Langtry HD. Mitoxantrone: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the chemotherapy of cancer. Drugs 1991; 41(3):400–449.
25. Levin VA, Hoffman W, Weinkam RJ. Pharmacokinetics of BCNU in man: A preliminary study of 20 patients. Cancer Treat Rep 1978;62:1305–1312.
26. Lee FYF, Workman P, Roberts JT, Bleechen NM. Clinical pharmaco-kinetics of oral CCNU. Cancer Chemother Pharmacol 1985; 14:125–131.
27. Rudek MA, Donehower RC, Statkevich P, et al. Temozolomide in patients with advanced cancer: Phase I and pharmacokinetic study. Pharmacotherapy 2004;24(1):16–25.
28. Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: Prospective evaluation of a simple formula based on renal function. J Clin Oncol 1989;7(11):1748–1756.
29. Graham MA, Lockwood GF, Greenshade D, et al. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin Cancer Res 2000;6:1205–1218.
30. Dorr RT. Bleomycin pharmacology: Mechanism of action and resistance, and clinical pharmacokinetics. Semin Oncol 1992;19(2 suppl 5):3–8.
31. Zhi-Xiang S, Chen, G, Ni J, et al. Use of arsenic trioxide in the treatment of acute promyelocytic leukemia: II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997;89(9): 3354–3360.
32. Van Hazel GA, Scott M, Rubin J, et al. Pharmacokinetics of mitomycin C in patients receiving the drug alone or in combination. Cancer Treat Rep 1983;67(9):805–810.
33. Avvisati G, Tallman MS. All-trans retinoic acid in acute promyelocytic leukaemia. Best Pract Res Clin Haematol 2003;16(3):419–432.
34. Wohl DA, Aweeka FT, Schmitz J, et al. Safety, tolerability, and pharmacokinetic effects of thalidomide in patients infected with human immunodeficiency virus: AIDS clinical trials group 267. J Infect Dis 2002;185:1359–1363.
35. Chen N, Lau H, Kong L, et al. Pharmacokinetics of lenalidomide in subjects with various degrees of renal impairment and in subjects on hemodialysis. J Clin Pharmacol 2007;47:1466–1475.
36. Gordon MS, Margolin K, Talpaz M, et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol 2001;19(3):843–850.
37. Korth-Bradley JM, Dowell JA, King SP, et al. Impact of age and gender on the pharmacokinetics of gemtuzumab ozogamicin. Pharmacotherapy 2001;21(10):1175–1180.
38. Wood AM. Rituximab: An innovative therapy for non-Hodgkin lymphoma. Am J Health Syst Pharm 2001;58(3):215–232.
39. Leyland-Jones B, Gelman K, Ayoub J, et al. Pharmacokinetics, safety, and efficacy of trastuzumab administered every three weeks in combination with paclitaxel. J Clin Oncol 2003;21:3965–3971.
40. Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol 2004;22:935–942.
41. Bolton AE, Peng B, Hubert M, et al. Effect of rifampicin on the pharmacokinetics of imatinib mesylate in healthy subjects. Cancer Chemother Pharmacol 2004;53(2):102–106.
42. Frye RF, Fitzgerald SM, Lagattuta TF, et al. Effect of St. John’s Wort on imatinib mesylate pharmacokinetics. Clin Pharmacol Ther 2004;76(4):323–329.
43. Dutreix C, Peng B, Mehring G, et al. Pharmacokinetic interaction between ketoconazole and imatinib mesylate in healthy subjects. Cancer Chemother Pharmacol 2004;54(4):290–294.
44. O’Brien SG, Meinhardt P, Bond E, et al. Effects of imatinib mesylate on the pharmacokinetics of simvastatin, a cytochrome p450 3A4 substrate, in patients with chronic myeloid leukaemia. Br J Cancer 2003;89(3):1855–1859.
45. Hamilton M, Wolf JL, Rusk J, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res 2006;12:2166–2171.
46. Faivre S, Delbaldo C, Vera K, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 2006;24:25–35.
47. Goetz MP, Knox SK, Suman VJ, et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res Treat 2007;101:113–121.
48. Gonzalez ID, Saez RS, Rodilla EM, Yges EL, Toledano FL. Hypersensitivity reactions to chemotherapy drugs. Alergo Immunol Clin 2000;15:151–181.
49. Cohen MR, Anderson RW, Attilio RM, et al. Preventing medication errors in cancer chemotherapy. Am J Health Syst Pharm 1996;53: 737–746.
50. ASHP technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm 1990;47:1033–1049.