The kidneys play a central role in the maintenance of homeostasis of the body. The kidneys stabilize extracellular fluid electrolyte composition, maintain acid–base balance, regulate volume status and blood pressure, secrete erythropoietin and renin, and excrete toxins and metabolic waste. These functions involve complex interactions within the kidneys and with other organ systems and are frequently altered during anesthesia. Hence, a thorough understanding of kidney function is important for the anesthesiologist before, during, and after patients receive care in the operating room.
Kidney Structure and Function
Basic Anatomy of the Kidney
The kidneys are paired organs located below the diaphragm in the retroperitoneal space, weighing between 115 and 160 g each. Each kidney has an outer portion (cortex) and inner portion (medulla). The renal arteries arise from the abdominal aorta, and the renal veins direct blood flow into the inferior vena cava. The kidneys are prominently innervated by the sympathetic nervous system from T4 through T12.
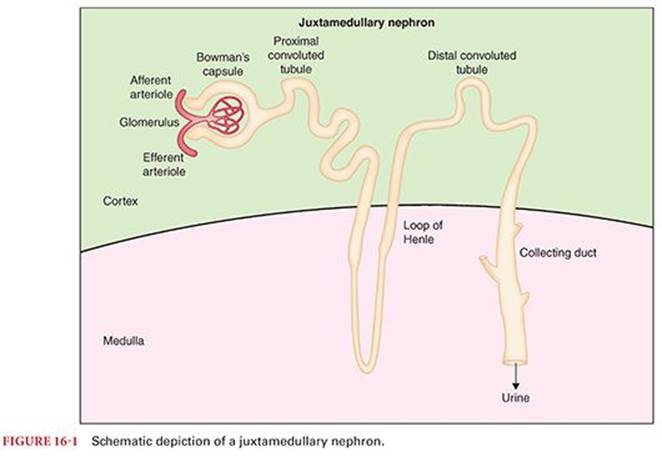
The nephron is the functional unit of the kidney. (Fig. 16-1) A nephron is composed of a capillary bed called the glomerulus surrounded by epithelial cells called Bowman’s capsule. Bowman’s capsule, in turn, is continuous with a long tubule that drains into the renal pelvis. Fluid is filtered through the glomerular capillaries and is converted, along the length of the renal tubule, into urine.1 There are two types of nephrons: Juxtamedullary nephrons have tubules that descend into the renal medulla, whereas cortical nephrons are closer to the surface of the kidney.
The Glomerulus
Structure and Function of the Glomerulus
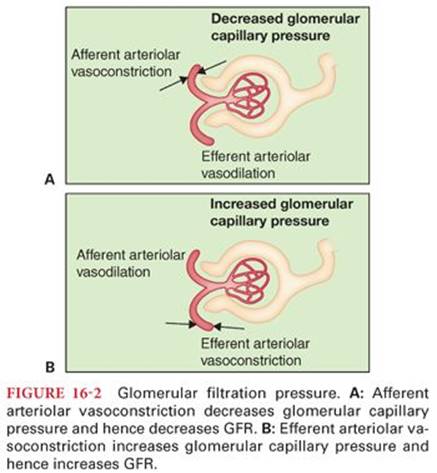
Glomeruli are found in the renal cortex and consist of a tuft of capillaries surrounded by Bowman’s capsule, the dilated blind end of the renal tubule. Glomerular capillaries are uniquely interposed between two sets of arterioles. Blood flows from the afferent arterioles through the glomerular capillaries and then on to the efferent arterioles. Hence, pressure in the glomerular capillaries is a function of the vascular activity of both the afferent and efferent arterioles (Fig. 16-2): Afferent vasoconstriction lowers glomerular capillary pressure, whereas efferent arteriolar vasoconstriction raises glomerular capillary pressure. Glomerular capillary pressure causes water and low-molecular-weight substances to be filtered into Bowman’s capsule and the renal tubule system.
Fluid that is filtered from the glomerular capillaries into the renal tubules is called glomerular filtrate. Because the glomerular capillary membrane contains pores, its permeability is much greater than that of the typical tissue capillary. Fluid, amino acids, and ions are rapidly filtered, whereas higher molecular weight proteins are retained within the capillary. The composition of glomerular filtrate, then, is similar to plasma but without protein.
Glomerular Filtration Rate
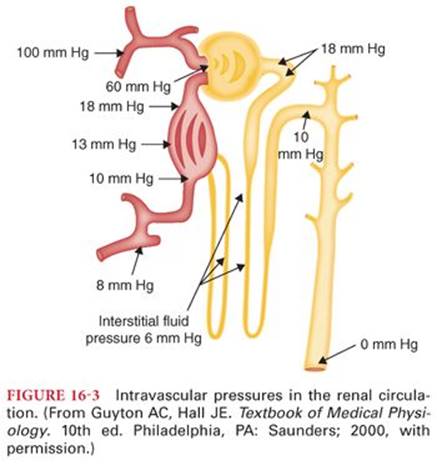
The volume of collective glomerular filtrate formed over time is called the glomerular filtration rate (GFR). In a normal person, the GFR is approximately 125 mL per minute or 180 L per day. Because 99% of this 180 L of glomerular filtrate is reabsorbed, daily urine output is 1 to 2 L. As described earlier, glomerular capillary pressure leads to filtration into the renal tubule. The normal filtration pressure of approximately 10 mmHg is calculated as glomerular capillary pressure (60 mm Hg) minus colloid osmotic pressure (32 mm Hg) and pressure in Bowman’s capsule (18 mm Hg) (Fig. 16-3). The filtration rate is influenced by several factors: Mean arterial pressure, cardiac output, and the sympathetic nervous system may each raise glomerular capillary pressure and increase the GFR.
The kidneys have an autoregulatory mechanism to modulate the effect of mean arterial pressure on the GFR. This process of autoregulation involves feedback from the distal renal tubules to the glomerulus. Specialized cells in the distal renal tubules, called the macula densa, signal the afferent or efferent renal arterioles to either vasoconstrict or vasodilate. Through the consequent adjustment of glomerular capillary pressure, a nearly constant filtration pressure leads to a consistent GFR across a range of mean arterial pressure, remaining relatively constant between mean arterial pressures of 60 and 160 mm Hg. Because even a small change in GFR can lead to wide variations in urine output, it is clear that tubule–glomerular feedback serves an important role in homeostasis.
The Renal Tubule
Structure of the Renal Tubule
The renal tubule is composed of the proximal convoluted tubule, the loop of Henle, and the distal convoluted tubule1 (see Fig. 16-1). As described earlier, the loops of Henle in juxtamedullary nephrons extend into the renal medulla before returning filtrate back to the renal cortex in the distal convoluted tubule. Glomerular filtrate, after passing along the length of the renal tubule, then enters the collecting duct, which is a confluence of flow from several nephrons into the renal pelvis.
Renal Tubular Function
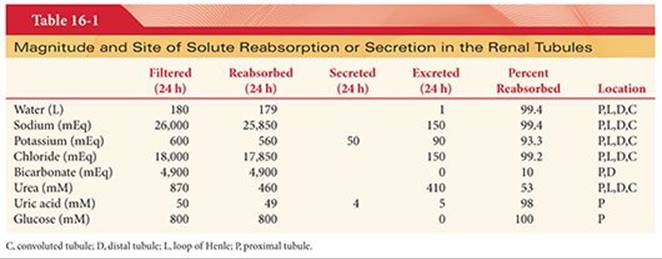
Glomerular filtrate is converted into urine along the course of the renal tubule2 (Table 16-1). The majority of water and various solutes that are filtered by glomerular capillaries are reabsorbed into peritubular capillaries. Metabolic waste products, also filtered by glomerular capillaries, are not reabsorbed. Other solutes are secreted by renal tubular epithelial cells into the lumen of the renal tubule. Thus, the urine found in the collecting duct is composed mainly of substances filtered through the glomerular capillaries and a small amount of secreted substances. The process of reabsorption determines the volume of urine formed, whereas secretion is particularly important in determining the nature of the urine, such as concentration of potassium and hydrogen ions. Various portions of the renal tubule have differing roles in reabsorption and secretion2 (see Table 16-1). Approximately two-thirds of all reabsorption and secretion in the renal tubules occurs in the proximal renal tubules. The most important factors influencing the reabsorption of sodium and water are aldosterone, arginine vasopressin (AVP), renal prostaglandins, and atrial natriuretic peptide.
Reabsorption of sodium involves moving this ion against a concentration gradient from the lumen of the proximal tubule into peritubular capillaries. This process requires energy, supplied by the sodium–potassium adenosine triphosphatase (ATPase) system and is appropriately called active transport. Other transport processes along the tubule, including glucose reabsorption, amino acid reabsorption, and organic acid secretion, share a common carrier with sodium. Hence, the great majority of transport across renal tubular cells is dependent on sodium–potassium ATPase activity. The proximal convoluted renal tubules, driving the ATPase enzyme for sodium reabsorption, consume approximately 80% of renal oxygen consumption.3
More than 99% of the water in the glomerular filtrate is reabsorbed into peritubular capillaries as it passes through renal tubules. The variation in permeability of epithelial cells lining the tubules is important in renal function. Rapid osmosis of water through proximal renal tubules means that the concentration of solutes on the capillary side of cell membranes is almost never more than a few milliosmoles greater than in the tubular lumen. However, the distal tubules are almost completely impermeable to water, allowing for control of the specific gravity of the urine. The permeability of the collecting ducts is variable and determined by the action of AVP. When AVP activates adenylate cyclase in the epithelial cells lining the collecting duct, the resulting cyclic adenosine monophosphate increases permeability of cell membranes to water. Hence, increased AVP leads to reabsorption of water from the collecting ducts, resulting in highly concentrated urine. Decreased AVP results in little water reabsorption and large amounts of dilute urine.
Countercurrent System
The ability of the kidneys to produce either dilute or concentrated urine depends on the gradient in osmolarity between the renal cortex and renal medulla that is created by the loop of Henle. Whereas the renal cortex has a relatively low osmolarity (300 mOsm/L), the renal medulla contains highly concentrated interstitial fluid (1,400 mOsm/L near the renal pelvis) due to active reabsorption of solutes in the loop of Henle3 (Fig. 16-4). The high medullary osmolarity is maintained in part by sluggish blood flow, preventing the removal of solutes.
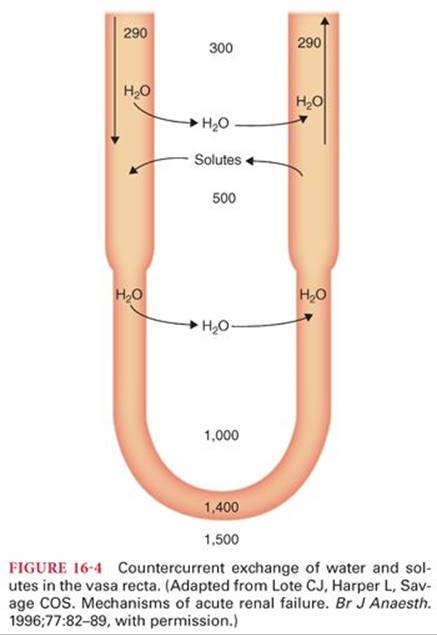
Just as the juxtamedullary loops of Henle carry glomerular filtrate from the cortex into the renal medulla and back to the cortex, the vascular supply has a similar structure. The U-shaped arrangement of peritubular capillaries, known as the vasa recta, parallels the loops of Henle. This forms a countercurrent system, in which capillary inflow runs parallel and in an opposite direction to capillary outflow.
Aquaporins
The high osmolarity in the renal medulla allows for the potential for quick reabsorption of water by osmosis as filtrate passes through the renal collecting ducts. This process is mediated by aquaporins, which are channels that facilitate rapid passage of water across lipid cell membranes at a velocity greater than possible by simple diffusion.4–6 They are tetramer protein structures and are found in the kidneys, brain, salivary and lacrimal glands, and respiratory tract. Five aquaporins in the kidney have a role in water balance.5 Aquaporin-1 is in the proximal renal tubules, while aquaporin-2 is found in the renal collecting ducts. In response to AVP, the channels in the tubular collecting duct edpithelium are opened, leading to reabsorption of water and formation of concentrated urine. In the absence of AVP, dilute urine traverses the collecting ducts without being affected by medullary osmolarity.
Tubular Transport Maximum
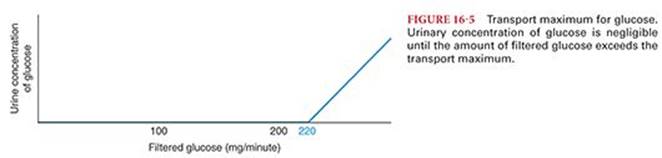
Tubular transport maximum (Tmax or Tmax) is the maximum amount of a substance that can be actively reabsorbed from the lumens of renal tubules each minute. The Tm depends on the amounts of carrier substance and enzyme available to the specific active transport system in the lining epithelial cells of renal tubules.
The Tmax for glucose is approximately 220 mg per minute. When the amount of glucose that filters through the glomerular capillary exceeds this amount, the excess glucose cannot be reabsorbed and passes into urine (Fig. 16-5). The usual amount of glucose in the glomerular filtrate entering proximal renal tubules is 125 mg per minute, and there is no detectable loss into urine. When the tubular load, however, exceeds approximately 220 mg per minute (threshold concentration), glucose begins to appear in urine. A blood glucose concentration of 180 mg/dL in the presence of a normal GFR results in delivery of 220 mg per minute of glucose into the renal tubular fluid. Loss of glucose in urine occurs at concentrations above the Tm for glucose. The presence of large amounts of unreabsorbed solutes in the urine such as glucose (or mannitol) produces osmotic diuresis by retaining water in the collecting system.
Transport of Urine to the Bladder
From the collecting ducts, urine travels into the renal pelvis. A ureter arises from the pelvis of each kidney. At its distal end, the ureter penetrates the bladder obliquely such that pressure in the bladder compresses the ureter, thereby preventing reflux of urine into the ureter when bladder pressure increases during micturition.
Each ureter is innervated by the sympathetic and parasympathetic nervous system. As urine collects in the renal pelvis, the pressure in the pelvis increases and initiates a peristaltic contraction that travels downward along the ureter to force urine toward the bladder. Parasympathetic nervous system stimulation increases the frequency of peristalsis, whereas sympathetic nervous system stimulation decreases peristalsis.
Obstruction of a ureter by a stone causes intense reflex constriction and pain. In addition, pain elicits a sympathetic nervous system reflex (ureterorenal reflex) that causes vasoconstriction of the renal arterioles and a concomitant decrease in urine formation in the kidney when there is obstruction of the ureter.
As the bladder fills with urine, stretch receptors in the bladder wall initiate micturition contractions. Sensory signals are conducted to the sacral segments of the spinal cord through the pelvic nerves and then back again to the bladder through parasympathetic nervous system fibers. The micturition reflex is a completely automatic spinal cord reflex that can be inhibited (tonic contraction of the external urinary sphincter) or facilitated by centers in the brain. Spinal cord damage above the sacral region leaves the micturition reflex intact but is no longer controlled by the brain.
Renal Blood Flow
Although the kidneys represent about 0.5% of total body weight, their blood flow is disproportionately large at 20% to 25% of the cardiac output.3 Renal blood flow is approximately 400 mL/100 g/minute, compared with 70 mL/100 g/minute for the heart and liver. The ability to autoregulate keeps renal blood flow relatively constant across a range of systemic mean arterial pressures. Because renal blood flow is large, the fraction of oxygen extraction is low despite high oxygen consumption, the PO2 decreasing from 95 mm Hg in the renal artery only to about 70 mm Hg in the renal vein.
Approximately 90% of the renal blood flow is distributed to the renal cortex, with less than 10% of renal blood flow going to the medulla. The generous delivery of blood to the cortex supports flow-dependent functions such as glomerular filtration and tubular reabsorption processes of the cortex. By contrast, low blood flow in the medulla maintains a high interstitial fluid osmolarity, which in turn permits concentration of the urine.7 Low blood flow also makes the medulla more susceptible to ischemia than the cortex.
Renal Cortex Blood Flow: Glomerular and Peritubular Capillaries
Blood enters the renal artery, whose branches ultimately supply the afferent arterioles of the glomeruli. As described earlier, afferent arterioles feed the glomerular capillary bed, out of which blood flows into the efferent arterioles1(see Fig. 16-1). The vascular resistance of the efferent arterioles raises glomerular capillary pressure, which promotes continuous filtration of fluid from glomerular capillaries into Bowman’s capsule. Blood flows from the arterioles into a second capillary network called peritubular capillaries. These capillaries have substantially lower pressure8 (see Fig. 16-3), promoting reabsorption of fluid from the tubules into the peritubular capillaries.
Renal Medulla Blood Flow: The Vasa Recta
Capillaries that descend with the loops of Henle are referred to as the vasa recta, which receive only 1% to 2% of renal blood flow. These capillaries, after descending into the renal medulla, return to the renal cortex and empty into veins. As described earlier, this countercurrent system minimizes the washout of solutes from the interstitial fluid of the medulla creating a high osmolarity that promotes the absorption of water from the collecting ducts and the formation of concentrated urine.
Autoregulation of Renal Blood Flow
Renal blood flow and GFR are kept relatively constant within a range of mean arterial pressure between approximately 60 and 160 mm Hg8 (Fig. 16-6). Because the GFR parallels renal blood flow, cardiac output has an important effect on the GFR. The mechanism for autoregulation is controversial.9 One theory is a myogenic response, whereby increased perfusion pressure leads to an increased wall tension in the afferent arterioles, resulting in automatic contraction of the smooth muscle fibers in the vessel wall. This increases resistance keeping flow constant despite the increase in perfusion pressure.3 An alternative hypothesis is that a tubuloglomerular feedback mechanism is responsible for autoregulation, whereby increased perfusion pressure will increase filtration, increasing the tubular fluid delivery to the macula densa, which then releases a factor or factors that cause vasoconstriction.
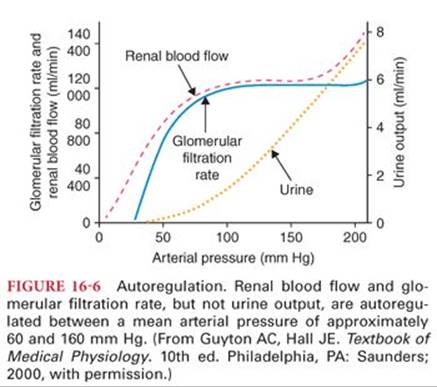
In the setting of decreased effective circulating volume, however, renal blood flow may be decreased despite adequate perfusion pressure. Activation of the sympathetic nervous system shunts cardiac output away from the kidneys. Hence, adequate systemic blood pressure does not necessarily indicate adequate renal perfusion in the presence of hypovolemia.
Juxtaglomerular Apparatus
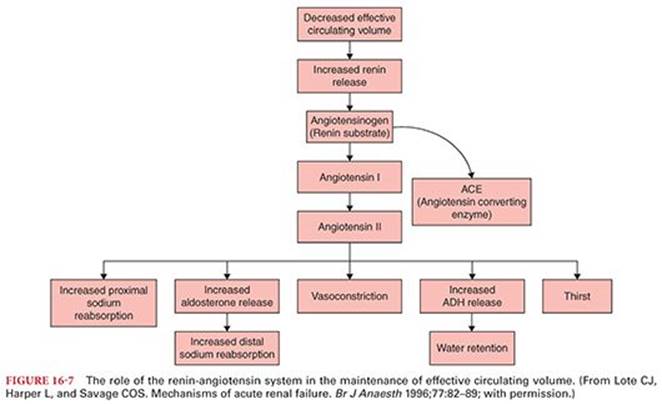
The juxtaglomerular apparatus is where the distal renal tubule passes between the afferent and efferent arterioles. Epithelial cells of the distal renal tubules that contact these arterioles are called the macula densa, whereas corresponding cells in the arterioles are known as juxtaglomerular cells. In response to decreased renal blood flow, juxtaglomerular cells release renin into the circulation3 (Fig. 16-7). Renin converts angiotensinogen to angiotensin I, which is then converted to angiotensin II by angiotensin-converting enzyme. Effects of angiotensin II include thirst, vasoconstriction, and salt and water reabsorption by the kidneys to maintain circulating volume and increase renal blood flow. Whether the initial cause of decreased renal blood flow is the result of hypovolemia, systemic hypotension, or sympathetic nervous system stimulation, the effect of renin is to maintain renal blood flow and GFR.
Regulation of Body Fluid
The kidneys have a primary role in the regulation of the amount and nature of body fluids. They control the following characteristics:
• Blood and extracellular fluid volume
• Osmolarity of body fluids
• Plasma concentration of ions and urea
Blood and Extracellular Fluid Volume
Blood volume is maintained over a narrow range despite large daily variations in fluid and solute intake or loss. The mechanism for control of blood volume also affects systemic blood pressure and cardiac output, as these are interrelated. An increase in blood volume increases the cardiac output, which usually increases the systemic blood pressure. Increased cardiac output and systemic arterial pressure will increase renal blood flow and GFR, resulting in an increase in urine output. The negative feedback loop is completed by a consequent decrease in circulating blood volume.
This basic regulatory mechanism is augmented by other factors. In the setting of decreased blood volume, an increased circulating concentration of AVP will increase water reabsorption, whereas an increase in aldosterone promotes sodium reabsorption and thus an osmotic reabsorption of water. This decreases urine volume and restores blood volume. Another factor is mediated through atrial stretch receptors and atrial natriuretic peptide to be described in the next section.
Regulation of normal circulating blood volume is impaired by factors directly affecting vascular capacitance. Persistent vasoconstriction associated with essential hypertension or sympathetic nervous system stimulation (from pheochromocytoma, for example) results in a decrease in blood volume. Conversely, blood volume may be increased by chronic drug-induced vasodilation or the effects of severe varicose veins.
The regulation of extracellular fluid volume is controlled indirectly through the maintenance of circulating blood volume. An increase in blood volume leads to an increase in extracellular fluid volume, whereas decreased extracellular fluid volume accompanies reduced blood volume. Although these volumes move in the same direction, their proportional change is affected by capillary permeability, which is commonly influenced by perioperative factors. The extracellular fluid space may be considered as a reservoir for excess intravenous fluid administered during the perioperative period.
Atrial and Renal Natriuretic Factors
Cardiac atrial muscle synthesizes and secretes a peptide hormone known as atrial natriuretic peptide (ANP), which is released in response to increased right and left atrial pressure and volume. ANP binds to receptors in the renal collecting ducts and, acting via transcellular second messenger systems, inhibits sodium reabsorption. Hence, atrial “stretch” promotes elimination of sodium and water, and a subsequent decrease in circulating volume. ANP additionally has vasodilatory properties, thereby lowering systemic blood pressure and eliciting renal artery vasodilation.
The renal analogue of ANP is renal natriuretic peptide (urodilatin), which is synthesized in renal cortical nephrons. It is likely that ANP is primarily a cardiovascular regulator and relatively unimportant for sodium excretion, whereas renal natriuretic peptide participates in the intrarenal regulation of sodium excretion.3,10 In mechanical ventilation, positive end-expiratory pressure reduces atrial distension and atrial transmural pressure. This reduces ANP release, which may contribute to sodium and water retention by the kidneys.
Osmolarity of Body Fluids
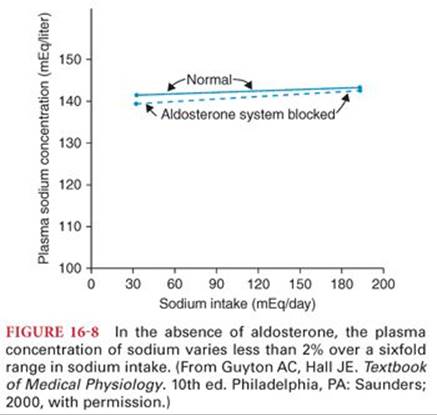
The primary determinant of body fluid osmolarity is the concentration of sodium in the extracellular fluid. Sodium ion concentration is largely controlled by two mechanisms: the osmoreceptor–AVP response and the thirst reflex. Aldosterone, by contrast, has a minimal role in the maintenance of sodium concentration and plasma osmolarity8 (Fig. 16-8). Aldosterone-induced reabsorption of sodium is accompanied by reabsorption of water. For this reason, patients with primary hyperaldosteronism typically have increased extracellular fluid volume but nearly normal serum sodium levels.
Osmoreceptor–Arginine Vasopressin Hormone
In response to increased extracellular fluid osmolarity, osmoreceptors in the hypothalamus signal the posterior pituitary to increase the release of AVP. AVP is also secreted in the setting of water deprivation and hemorrhage. Circulating AVP causes collecting ducts to retain water and thus decreases serum sodium levels. Because sodium is the primary determinant of serum osmolality, this results in a decrease in osmolality. The inverse is also true. Abnormally low extracellular fluid osmolality will lead to a decrease in the release of AVP, thereby increasing the production of dilute urine. This increases serum sodium and osmolality toward normal. A small change in osmolality (as little as 1%) can produce a large change in circulating AVP concentration, producing tight regulation of serum osmolality.
Thirst Reflex
The thirst reflex is primarily elicited by an increase in sodium concentration in the extracellular fluid. An increase in sodium as small as 2 mEq/L above normal (or an increase in osmolality of approximately 4 mOsm/L) will stimulate thirst, and water consumption decreases sodium concentration toward normal. In this way, extracellular fluid osmolality is maintained within a narrow range. In addition, angiotensin II promotes a thirst response, so circulatory changes that increase the production of angiotensin II, such as acute hemorrhage or congestive heart failure, will increase thirst.
Plasma Concentration of Ions and Urea
Sodium
The kidneys control the concentration of sodium through the process of reabsorption. As described earlier, active transport moves sodium ions from the tubular lumen into the peritubular capillaries. Two-thirds of the sodium in glomerular filtrate is reabsorbed in the proximal renal tubule, and less than 10% of sodium is expected to reach the distal renal tubule. The renin-angiotensin-aldosterone system modulates the sodium reabsorption by the renal tubules. Hypotension or decreased circulating blood volume leads to an increase in angiotensin, which is converted to angiotenin II in the lungs and ultimately results in increased sodium reabsorption. Angiotensin II leads to efferent arteriole vasoconstriction, increasing glomerular capillary pressure and GFR.
Lastly, angiotensin II increases secretion of aldosterone, which exerts its effect in the distal renal tubule. When aldosterone levels are increased, nearly all the remaining sodium is reabsorbed from the distal tubule, and urinary excretion of sodium is negligible. Typically, only 1% of the filtered sodium is excreted in the urine2 (see Table 16-1).
Potassium
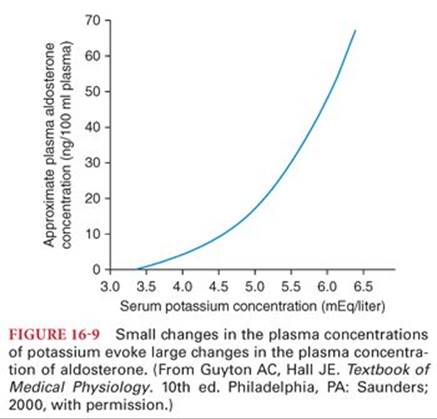
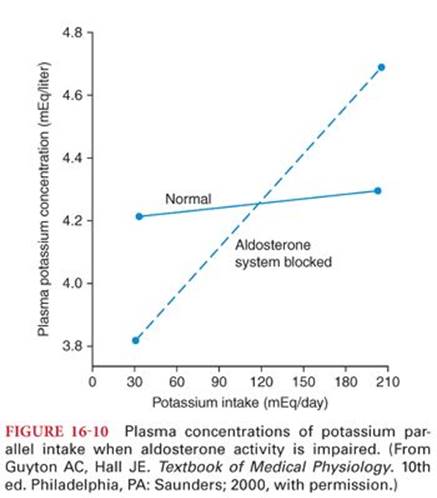
Potassium, after being filtered in the glomerulus, is then reabsorbed by the proximal tubule and loop of Henle. Potassium is either reabsorbed or secreted in the distal tubule and collecting duct, depending on the level of aldosterone. An increase in aldosterone increases potassium ion secretion into the renal tubules and consequently increases urinary potassium. The feedback mechanism allows for close regulation: Small changes in potassium concentration will lead to a substantial change in the concentration of aldosterone8 (Fig. 16-9). When aldosterone activity is blocked by certain diuretics, plasma potassium concentration depends more on dietary intake of potassium, making hypokalemia or hyperkalemia more likely8 (Fig. 16-10).
The regulation of sodium and hydrogen ion concentrations also has an effect on urinary excretion of potassium. Hydrogen ions compete with potassium for secretion into the renal tubules. In the presence of alkalosis (e.g., vomiting and loss of gastric acid), potassium is excreted in the urine in order to maintain acid–base balance. Conversely, a metabolic acidosis will lead to the secretion of hydrogen ions and retention of potassium, and plasma potassium concentration will increase. Sodium intake may influence plasma concentrations of potassium because sodium is transported through renal tubular epithelial cells in exchange for potassium.
Acid–Base Balance
The kidneys secrete excess hydrogen ions by exchanging a hydrogen ion for a sodium ion, thus acidifying the urine, and by the synthesis of ammonia, which combines with hydrogen to form ammonium. In the presence of hypovolemia, bicarbonate reabsorption by the kidneys will lead to acidification of the urine and a metabolic alkalosis.
Calcium and Magnesium
Calcium ion concentration is controlled principally by the effect of parathyroid hormone on bone reabsorption, which releases calcium. Additionally parathyroid hormone increases the reabsorption of calcium from the distal renal tubules and collecting ducts. Magnesium is reabsorbed by all portions of the renal tubules. Urinary excretion of magnesium parallels the plasma concentration of this ion.
Urea
Urea is the most abundant metabolic waste product. Without adequate clearance of urea, excessive accumulation in body fluids prevents normal function of multiple systems. Urea elimination depends on the plasma concentration of urea (blood urea nitrogen or BUN) and the GFR. Approximately 50% of the urea that is filtered into the renal tubules is eliminated in the urine; the remainder is reabsorbed. When the GFR is low, tubular filtrate flow is relatively slow, thus increasing the proportion of urea that is reabsorbed. This effectively increases the BUN by decreasing urinary elimination of urea. Conversely, when GFR increases, less urea is reabsorbed in the tubules, increasing its elimination in the urine, and BUN decreases.
Measuring Kidney Function
Formal measurement of kidney function requires labor-intensive studies such as collection of urine over time and measurement of blood and urine components. For clinical decision making, estimates of GFR are accessible and inexpensive, requiring only basic laboratory work. Serum creatinine (SCr), commonly used to measure changes in kidney function, is insensitive to small changes in GFR; GFR needs to decrease by about 50% before a rise in SCr is seen. For many years, the Cockcroft-Gault formula has been used to estimate GFR.11
GFRmen = (140 − age) × weight (kg) / serum creatinine (mg/dL) × 72
GFRwomen = (140 − age) × weight (kg) × 0.85 / serum creatinine (mg/dL) × 72
The difference in the formula between men and women accounts for the expected difference in muscle mass and creatinine production. The formula also takes into account the effect of age on GFR. Given an age-related decrease in muscle mass, GFR in an elderly person will typically be less than a younger person with the same weight and SCr.
The Modification of Diet in Renal Disease (MDRD) formula uses four variables to estimate GFR: age, SCr, ethnicity, and gender, and is independent of body weight.12 Developed in a population of patients with chronic kidney disease in the United States, it has been widely adopted as a useful estimate of GFR, although questions remain concerning its applicability to other populations. Both Cockcroft-Gault and MDRD assume steady state conditions: balanced production and clearance of SCr leading to a consistent concentration. Because an acute reduction in GFR will result in a rise in SCr over many hours or days, these formulas are a poor measure of GFR in acute kidney injury.
Acute Kidney Injury
Acute kidney injury results in an abrupt reduction in the kidney’s ability to eliminate nitrogenous waste products and maintain fluid and electrolyte homeostasis.3,13 Despite the kidney’s generous blood supply and large oxygen delivery, it remains at risk for ischemia in the perioperative period. Acute kidney injury may be classified by either the site of instigating pathology or the degree of injury sustained, as measured by changes in GFR and reduction in urine output. In light of the heterogeneous causes of acute kidney injury and the difficulty in comparing literature using disparate definitions,13 there has been a great deal of recent interest in standardizing and streamlining the classification of acute kidney injury.
Classification
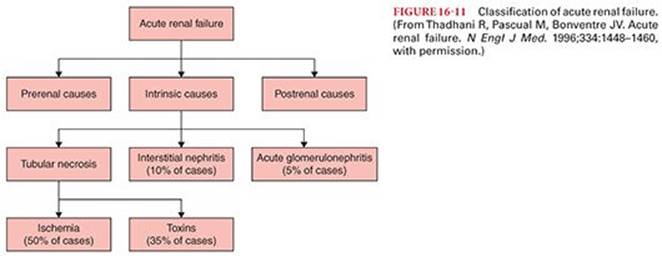
Acute kidney injury (AKI) has traditionally been classified by dividing the primary pathophysiology into prerenal, intrarenal, or postrenal causes13 (Fig. 16-11).
Prerenal Azotemia
Abnormalities of the systemic circulation that lead to a decrease of renal blood flow have the potential to impair renal function. The term azotemia refers to any condition characterized by abnormally high levels of nitrogen-containing compounds, such as urea, creatinine, and other nitrogen-rich compounds, in the blood. Prerenal azotemia refers to decreases in renal function due to hypoperfusion in the setting of intact glomeruli and tubules. Correcting the underlying problems in circulation will improve renal function. Common causes of prerenal azotemia in hospitalized patients include septic shock, heart failure, liver failure, and perioperative hemodynamic changes that lead to decreased renal perfusion.13 Medications may also be implicated in at-risk patients. Nonsteroidal antiinflammatory drugs (NSAIDs) are known to precipitate prerenal azotemia in hypovolemic patients or in patients with congestive heart failure.13 Calcineurin inhibitors, by causing vasoconstriction of afferent renal arteries, also reduce GFR and lead to prerenal azotemia.14,15
Intrinsic Causes of Acute Kidney Injury
The most common cause of intrinsic renal failure is acute tubular necrosis (ATN), caused by either ischemia or nephrotoxic agents. Less common causes seen perioperatively include acute glomerulonephritis and interstitial nephritis from such agents as β-lactam antibiotics and NSAIDs8 (see Fig. 16-11). In cases of ATN secondary to ischemia, prolonged decreases in renal blood flow stimulate epithelial cells to reabsorb sodium to restore renal blood flow. This increase in active transport in the renal medullary tubules exacerbates the mismatch of oxygen supply and demand, leading to injury and the expression of proteins that regulate the response to hypoxia.16,17 Thus, prerenal azotemia leads to ATN, and both entities may be considered to be on the continuum of ischemic renal disease.
Renal tubule cells are particularly susceptible to ischemia because of their transport-related oxygen requirements and the low baseline blood flow to the renal medulla. Injury is worsened both by hypoxemia and by endothelial cell swelling, which further decreases perfusion. Nephrotoxic agents such as aminoglycosides and iodinated radiocontrast media may also lead to ATN, either by direct injury to tubules or by processes mediated by free radicals.13,18
ATN secondary to renal medullary ischemia is the most common perioperative cause of AKI. In this setting, adequate urine output may be falsely reassuring. Ischemic ATN leads to failure of the sodium–potassium ATPase pump in the renal tubules; impairing their ability to concentrate urine. Thus, urine output does not correspond to the degree of cell damage or GFR in patients exposed to trauma, shock, or cardiovascular surgery.19
Postrenal Obstructive Nephropathy
Perioperative patients with AKI should also be evaluated for postrenal etiologies, particularly those with acute oliguria. Because the pathophysiology underlying this AKI involves hindrance to urine flow, this is also called obstructive nephropathy. Causes include renal stones, prostatic hypertrophy, and mechanical obstruction of urinary catheters.
Acute Kidney Injury Diagnosis
Diagnostic Criteria
As recently as 2004, there was no widespread consensus definition of acute renal failure (ARF). Comparison of the literature was challenging as definitions and categories varied between studies. In 2004, the Acute Dialysis Quality Initiative group conducted a systematic review and consensus conference, which produced a five-tiered classification scheme for ARF, called RIFLE (risk, injury, failure, loss of kidney function, and end-stage kidney disease)20 (Fig. 16-12). This scheme is based on easily measurable clinical variables and includes in its definition an accommodation for acute or chronic kidney disease.
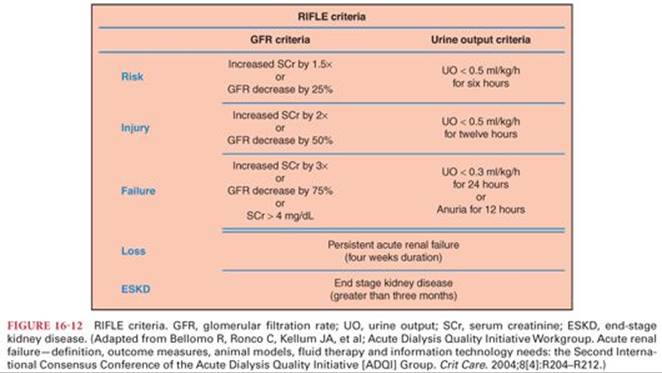
The RIFLE classification includes three levels of renal dysfunction and two clinical outcomes. The degrees of renal dysfunction are defined either by (a) changes in SCr or estimated GFR (eGFR) or (b) oliguria. A patient may be categorized by meeting one definition, the other, or both. The criterion that leads to the worst classification should be used.20
• R—risk of renal dysfunction: increased SCr by 1.5-fold (GFR decrease by 25%) or urine output (UOP) less than 0.5 mL/kg/hour for 6 hours
• I—injury to the kidney: increased SCr by two-fold (GFR decrease by 50%) or UOP less than 0.5 mL/kg/hour for 12 hours
• F—failure of kidney function: increased SCr by threefold (GFR decrease by 75%), SCr greater than 4 mg/dL, or UOP less than either 0.3 mL/kg/hour for 24 hours or anuria for 12 hours
Clinical outcomes include the following:
• L—Loss of kidney function is equivalent to persistent ARF, needing renal replacement therapy (RRT) for more than 4 weeks.
• E—end-stage kidney disease: need for dialysis for more than 3 months
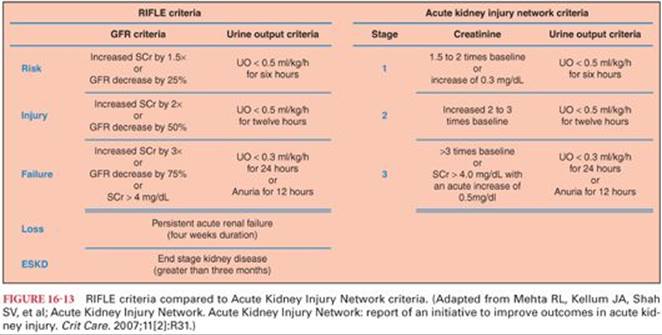
Proposed modifications to the RIFLE criteria were made by the Acute Kidney Injury Network (AKIN) in 200721 (Fig. 16-13). The first three stages of RIFLE (risk, injury, and failure) correspond to the first three stages of injury in AKIN definitions. In addition, stage 1 AKI includes patients with a rise in SCr as little as 0.3 mg/dL, as even small increases in SCr are associated with worse outcomes.21,22 AKIN further tightens the window to diagnosis of AKI by lab criteria or urine output to 48 hours as opposed to 7 days. Any patient with renal replacement therapy is included in stage 3, regardless of duration of therapy or concurrent urine output. Finally, “loss” and “end-stage kidney disease” have been removed, as they describe long-term outcomes rather than short-term categories. RIFLE and AKIN definitions have been compared and are in general concordance. It has been suggested that the AKIN definitions do not provide significant advantages, despite the increased sensitivity of stage 1 AKI.23,24
Biomarkers
As discussed earlier, the SCr and urine output are the most widely used diagnostic criteria to detect AKI. The use of SCr to estimate the GFR assumes steady-state conditions: Production of creatinine is equal to its clearance. It is therefore problematic to use SCr to estimate GFR in dynamic settings. Simply put, the rise in SCr lags an acute reduction in GFR. Furthermore, because even small changes in the GFR have significant impact on mortality and hospital length of stay,22 the need for early detection of AKI has led to the pursuit for biomarkers that may give real-time information. The hope is that earlier detection of AKI could lead to therapeutic interventions that improve outcomes, and research in this area has been very active for some time.
Neutrophil gelatinase–associated lipocalin (NGAL) has been identified as an early marker of AKI.25–27 Its expression is induced in renal tubular cells following ischemia-reperfusion injury, and it can be measured either in serum or urine soon after injury. Significant research has investigated this promising biomarker, although its clinical use has been slowed by a wide range of predictive value in various reports. An early meta-analysis suggested that NGAL is a useful predictor of early AKI across an array of clinical settings.24 When using standardized assays, a generally agreed upon cutoff upper value of 150 ng/mL may be suggestive of AKI. Despite questions regarding appropriate cutoff values and assay technique, more recent reviews have reaffirmed NGAL’s use in both the prediction of the presence and severity of AKI after cardiac surgery (AUCROC 0.82 to 0.83) and to predict delayed graft function after kidney transplantation (AUCROC 0.87).28
Other biomarkers of AKI are under ongoing investigation.29 Cystatin-C, a cysteine proteinase inhibitor produced by all nucleated cells, is small in size and easily filtered in the glomerulus. Because it is metabolized in the proximal renal tubules, urinary concentrations are insignificant. Furthermore, its short half-life means serum levels will reflect GFR. Interleukin-18 (IL-18) is synthesized both in the proximal tubular cells and in cells that mediate inflammatory response. Elevated levels are seen in patients with ATN, although its role as a biomarker of AKI is confounded by evidence indicating it may represent more closely the presence of general inflammatory processes. Lastly, kidney injury molecule (KIM)-1 is a membrane protein expressed in injured proximal tubular epithelial cells. Although associated with ATN, its predictive value is still being defined.29 Rather than relying on a single biomarker, a panel of biomarkers may be used in the future to predict AKI. Further research will also determine whether interventions in early AKI will affect clinical outcomes.
Anesthesia and the Kidneys
An understanding of kidney function is important for the anesthesiologist, as fundamental concepts of perioperative management include the maintenance of normal circulating volume, the regulation of electrolytes and acid–base status, and the clearance of metabolites and drugs. The perioperative time period is unique in that multiple potential insults, often concurrent or in rapid succession, challenge the kidney’s functional ability. Perioperative AKI has an estimated overall incidence of 1% and is associated with an eightfold increase in risk of mortality.30
Anesthesia and Renal Blood Flow
Several perioperative factors affect renal blood flow either directly by hemodynamic effects or indirectly through actions of the sympathetic nervous system or AVP. Regardless of the immediate cause, a fall in renal blood flow tends to decrease the GFR by diminishing blood flow to the renal cortex. Likewise, decreased renal blood flow puts the renal medulla at risk for ischemia because the blood supply to this region is already low at baseline. The sum effect of these changes is conservation of sodium and water and, consequently, a decrease in urine output.
Many perioperative factors influence renal blood flow through changes in cardiac output or systemic arterial pressure. Anesthetic drugs commonly have significant direct hemodynamic effects, either by reducing systemic vascular resistance, depressing myocardial function, or decreasing effective preload. Likewise, perioperative hypovolemia (from preoperative fasting, bowel preparation, fluid shifts, acute hemorrhage, or any combination of factors) will decrease cardiac output and systemic arterial pressure, ultimately leading to a similar direct effect on renal blood flow.
Because the kidney has rich autonomic innervation, renal blood flow is also highly sensitive to the action of the sympathetic nervous system. Sympathetic stimulation leads to increased renal vascular resistance, which has two significant effects. First, blood is shunted away from the kidneys to other organs, preserving perfusion of critical organs such as the brain and heart. Second, constriction of the afferent renal arterioles lowers glomerular capillary pressure and decreases the GFR. Whether the root cause is pain, surgical stimulation, or exogenous catecholamines, excessive sympathetic stimulation can decrease glomerular blood flow to the point that urine output drops to nearly zero. Furthermore, painful stimuli elicit the release of AVP, which increases water absorption from the collecting ducts, resulting in concentrated urine. Retention of sodium and water caused by positive end-expiratory pressure is not associated with changes in the circulating plasma concentrations of AVP.31
These direct and indirect mechanisms altering renal blood flow are not mutually exclusive. Any factor that decreases the cardiac output will also lead to a release of AVP and an increase in the activity of both the sympathetic nervous system and the renin-angiotensin-aldosterone system. AVP and aldosterone tend to restore both normal circulating volume and normal renal blood flow by retaining sodium and water. Hypovolemia from acute hemorrhage also increases sympathetic tone, again reducing renal blood flow.
The autoregulation of renal blood flow may also be affected by perioperative factors. Although most anesthetic drugs do not abolish autoregulation, it is impaired in the following circumstances: severe sepsis, acute kidney failure, and cardiopulmonary bypass. Sustained changes in mean arterial pressure (greater than 10 minutes) are associated with a decreased ability to autoregulate renal blood flow. Autoregulation of GFR, by contrast, is sustained over longer periods of time. Thus, the GFR may remain near normal despite a marked reduction in renal blood flow.
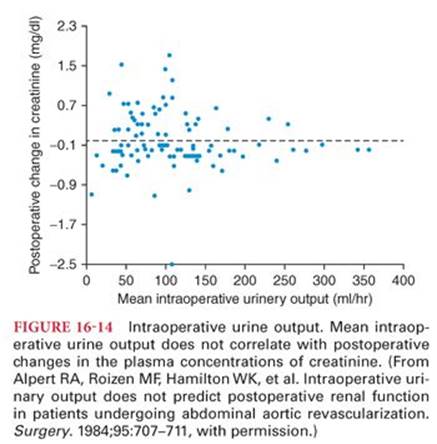
It is important to remember that normal systemic arterial pressure does not ensure adequate renal blood flow and that renal ischemia may occur even in the absence of hypotension. Also, intraoperative urine output is a poor predictor of postoperative changes in renal function.32 (Fig. 16-14). Hence, factors that commonly emerge in the perioperative setting including anesthetic agents, fluid shifts, stress response, changes in hemodynamics, and administration of catecholamines all have the potential to decrease renal blood flow and affect kidney function.
Perioperative Risk Assessment
An assessment of perioperative risk allows perioperative physicians to address this concern with patients and consultants, as well as to plan the anesthetic with the aim of avoiding AKI. In the patient population undergoing general surgery, the risk of AKI is believed to be about 1%. Patient risk factors include age older than 56 years, male gender, active congestive heart failure, ascites, diabetes, and hypertension (Table 16-2). Surgical factors increasing the likelihood of AKI are intraperitoneal surgery and emergent operations.30 The risk of perioperative AKI remains a significant concern in vascular and cardiac surgery.33–35 Sepsis and blood transfusions also increase the risk of AKI.

Patients undergoing cardiac surgery requiring cardiopulmonary bypass (CPB) are at particular risk. Alterations in renal blood flow, inflammatory response, microemboli, and direct toxicity are among the several mechanisms for kidney injury in these patients. Furthermore, clamping of the aorta in cardiac or major vascular surgery is associated with atheromatous emboli to the kidneys. Increased baseline SCr is the most significant risk factor for perioperative AKI after CPB. In patients with a baseline SCr between 2.0 and 4.0 mg/dL, the risk of AKI requiring dialysis is 10% to 20%. Other risk factors include ejection fraction less than 40% and preoperative use of an intraaortic balloon pump.36 Patients undergoing valve surgery are at higher risk than those undergoing coronary artery bypass grafting alone.37
Intraoperative Management
Although the ability of the anesthesiologist to prevent AKI remains limited, general management principles include identifying potential causes of kidney injury (and mitigating their impact), minimizing exposure to nephrotoxic agents (NSAIDs and radiographic contrast) and the maintenance of renal blood flow. This can be accomplished by prompt correction of intravascular volume depletion and the maintenance of adequate systemic arterial pressure. Preoperative optimization of congestive heart failure, as well as maintaining normal cardiac output perioperatively, will help maintain renal blood flow. The judicious use of positive end-expiratory pressure and the avoidance of unnecessary increases in mean airway pressure will help maintain cardiac output and adequate renal blood flow. In laparoscopic surgery, using the lowest possible abdominal insufflation pressure will promote renal blood flow, although this must be balanced with operative needs. Adequate analgesia will minimize sympathetic nervous system-mediated decreases in renal blood flow. This may be a potential benefit of regional anesthesia. Agents used to promote afferent arteriolar dilation have not been shown to improve perioperative renal outcomes. Low-dose dopamine does not prevent AKI or improve morality.38 Fenoldopam mesylate, a selective dopamine agonist, may reduce the risk of AKI in selected patient populations,39,40 but further studies are needed to confirm this potential benefit.
References
1. Pitts RF. Physiology of the Kidney and Body Fluids: An Introductory Text. 3rd ed. Chicago, IL: Year Book Medical Publishers; 1974.
2. Ganong WF. Review of Medical Physiology. 21st ed. New York, NY: McGraw-Hill; 2003.
3. Lote CJ, Harper L, Savage CO. Mechanisms of acute renal failure. Br J Anaesth. 1996;77(1):82–89.
4. Agre P, King LS, Yasui M, et al. Aquaporin water channels—from atomic structure to clinical medicine. J Physiol. 2002;542(pt 1):3–16.
5. Kortenoeven MLA, Fenton RA. Renal aquaporins and water balance disorders. Biochimica et Biophysica Acta. 2014;1840:1533–1549.
6. Kozono D, Yasui M, King LS, et al. Aquaporin water channels: atomic structure molecular dynamics meet clinical medicine. J Clin Invest. 2002;109(11):1395–1399.
7. Epstein FH, Brezis M, Silva P, et al. Physiological and clinical implications of medullary hypoxia. Artif Organs. 1987;11(6):463–467.
8. Guyton AC, Hall JE. Textbook of Medical Physiology. 10th ed. Philadelphia, PA: W.B. Saunders; 2000.
9. Steinhausen M, Endlich K, Wiegman DL. Glomerular blood flow. Kidney Int. 1990;38(5):769–784.
10. Shirakami G, Segawa H, Shingu K, et al. The effects of atrial natriuretic peptide infusion on hemodynamic, renal, and hormonal responses during gastrectomy. Anesth Analg. 1997;85(4):907–912.
11. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
12. Levey AS, Bosch JP, Lewis JB, et al; for Modification of Diet in Renal Disease Study Group. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Ann Intern Med. 1999;130(6):461–470.
13. Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334(22):1448–1460.
14. Finn WF. FK506 nephrotoxicity. Ren Fail. 1999;21(3–4):319–329.
15. Kahan BD. Cyclosporine. N Engl J Med. 1989;321(25):1725–1738.
16. Byric RJ, Rose DK. Pathophysiology and prevention of acute renal failure: the role of the anaesthetist. Can J Anaesth. 1990;37(4)(pt 1):457–467.
17. Conde E, Alegre L, Blanco-Sanchez I, et al. Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS One. 2012;7(3):e33258.
18. Barrett BJ, Parfrey PS. Clinical practice. Preventing nephropathy induced by contrast medium. N Engl J Med. 2006;354(4):379–386.
19. Kellen M, Aronson S, Roizen MF, et al. Predictive and diagnostic tests of renal failure: a review. Anesth Analg. 1994;78(1):134–142.
20. Bellomo R, Ronco C, Kellum JA, et al; Acute Dialysis Quality Initiative Workgroup. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204–R212.
21. Mehta RL, Kellum JA, Shah SV, et al; Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31.
22. Chertow GM, Burdick E, Honour M, et al. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16(11):3365–3370.
23. Bagshaw SM, George C, Bellomo R; ANZICS Database Management Committee. A comparison of the RIFLE and AKIN criteria for acute kidney injury in critically ill patients. Nephrol Dial Transplant. 2008;23(5):1569–1574.
24. Haase M, Bellomo R, Devarajan P, et al; NGAL Meta-analysis Investigator Group. Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;54(6):1012–1024.
25. Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14(10):2534–2543.
26. Wagener G, Jan M, Kim M, et al. Association between increases in urinary neutrophil gelatinase-associated lipocalin and acute renal dysfunction after adult cardiac surgery. Anesthesiology. 2006;105(3):485–491.
27. Wagener G, Minhaz M, Mattis FA, et al. Urinary neutrophil gelatinase-associated lipocalin as a marker of acute kidney injury after orthotopic liver transplantation. Nephrol Dial Transplant. 2011;26(5):1717–1723.
28. Haase-Fielitz A, Haase M, Devarajan P. Neutrophil gelatinase-associated lipocalin as a biomarker of acute kidney injury: a critical evaluation of current status. Ann Clin Biochem. 2014;51(pt 3):335–351.
29. McIlroy DR, Wagener G, Lee HT. Biomarkers of acute kidney injury: an evolving domain. Anesthesiology. 2010;112(4):998–1004.
30. Kheterpal S, Tremper KK, Heung M, et al. Development and validation of an acute kidney injury risk index for patients undergoing general surgery: results from a national data set. Anesthesiology. 2009;110(3):505–515.
31. Payen DM, Farge D, Beloucif S, et al. No involvement of antidiuretic hormone in acute antidiuresis during PEEP ventilation in humans. Anesthesiology. 1987;66(1):17–23.
32. Alpert RA, Roizen MF, Hamilton WK, et al. Intraoperative urinary output does not predict postoperative renal function in patients undergoing abdominal aortic revascularization. Surgery. 1984;95(6):707–711.
33. Loef BG, Epema AH, Smilde TD, et al. Immediate postoperative renal function deterioration in cardiac surgical patients predicts in-hospital mortality and long-term survival. J Am Soc Nephrol. 2005;16(1):195–200.
34. Thakar CV, Arrigain S, Worley S, et al. A clinical score to predict acute renal failure after cardiac surgery. J Am Soc Nephrol. 2005;16(1):162–168.
35. Wijeysundera DN, Karkouti K, Beattie WS, et al. Improving the identification of patients at risk of postoperative renal failure after cardiac surgery. Anesthesiology. 2006;104(1):65–72.
36. Kumar AB, Suneja M. Cardiopulmonary bypass-associated acute kidney injury. Anesthesiology. 2011;114(4):964–970.
37. Haase M, Bellomo R, Matalanis G, et al. A comparison of the RIFLE and Acute Kidney Injury Network classifications for cardiac surgery-associated acute kidney injury: a prospective cohort study. J Thorac Cardiovasc Surg. 2009;138(6):1370–1376.
38. Bellomo R, Chapman M, Finfer S, et al. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Lancet 2000;356(9248):2139–2143.
39. Landoni G, Biondi-Zoccai GG, Tumlin JA, et al. Beneficial impact of fenoldopam in critically ill patients with or at risk for acute renal failure: a meta-analysis of randomized clinical trials. Am J Kidney Dis. 2007;49(1):56–68.
40. Morelli A, Ricci Z, Bellomo R, et al. Prophylactic fenoldopam for renal protection in sepsis: a randomized, double-blind, placebo-controlled pilot trial. Crit Care Med. 2005;33(11):2451–2456.