This chapter will review the pharmacology of agents commonly encountered in anesthetic practice that are either administered to treat pulmonary diseases or administered into the airway or systemically for action at end organs other than the lung but have effects on the airway and the pulmonary circulation. The pharmacology of the airways will be considered first, then the pharmacology of the pulmonary circulation, and finally, the intrinsic action of the lungs on a variety of exogenous and endogenous substances.
Pharmacology of the Airways
Pharmacologic agents administered via the lungs take advantage of the interface between air and blood allowing for rapid uptake of drugs into the bloodstream or immediate use by cells that populate the airway.1 The delivery of medications to the lungs can have systemic effects, direct effects on the airway, or both. For example, inhaled anesthetics are delivered via the lungs to act in the brain and have bronchodilatory effects. Conversely, β-adrenergic agonists delivered via aerosol exert direct effects on bronchial smooth muscle with few systemic effects. Drugs administered to the airway take advantage of the rapid exposure to blood and pulmonary parenchymal cells, making them advantageous for treating pulmonary parenchymal diseases such as asthma and chronic obstructive pulmonary disease (COPD).
Influence of the Autonomic Nervous System on the Airways
Traditionally, the autonomic nervous system (ANS) has been divided into two major parts, the parasympathetic and sympathetic nervous systems. The parasympathetic nervous system regulates airway caliber, airway glandular activity, and airway microvasculature.2–4 The vagus nerve provides the preganglionic fibers, which synapse with postganglionic fibers in airway parasympathetic ganglia. Acetylcholine activates the muscarinic (M3) receptor of postganglionic fibers of the parasympathetic nervous system to produce bronchoconstriction.5 Anticholinergics can provide bronchodilation even in the resting state because the parasympathetic nervous system produces a basal level of resting bronchomotor tone.6
Although the sympathetic nervous system plays no direct role in control of airway muscle tone, β2-adrenergic receptors are present on airway smooth muscle cells and cause bronchodilation via stimulatory G mechanisms. The abundance of these receptors in the airway allows for pharmacologic manipulation of airway tone.7
The ANS also influences bronchomotor tone through the nonadrenergic noncholinergic (NANC) system.8,9 The exact role of NANC in humans is not well defined; it has excitatory and inhibitory neuropeptides that influence inflammation and smooth muscle tone, respectively. Vasoactive intestinal peptide (VIP) and nitric oxide (NO) are the main inhibitory transmitters thought to be responsible for airway smooth muscle relaxation. Substance P (SP) and neurokinin A (NKA) are the main excitatory transmitters and have been shown to cause neurogenic inflammation, including bronchoconstriction. The precise role of NANC in healthy and diseased human lung is unclear.
Inhaled Adrenergic Agonists
The mainstay of therapy for bronchospasm, wheezing, and airflow obstruction is β-adrenergic agonists. β-adrenergic agonists used in clinical practice are typically delivered via inhalers or nebulizers, are β2selective, and are divided into short- and long-acting therapies.10 Short-acting β2 agonist therapy is effective for the rapid relief of wheezing, bronchospasm, and airflow obstruction. Longer acting β2 agonists are used as maintenance therapy providing improvement in lung function and reduction in symptoms and exacerbations (Table 25-1).
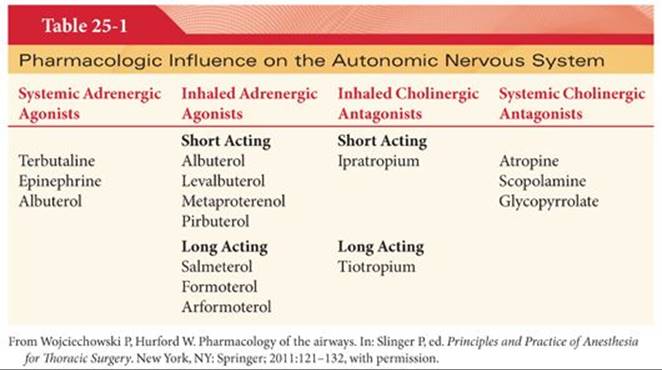
Short-acting β2 agonists bind to the β2-adrenergic receptor located on the plasma membrane of smooth muscle cells, epithelial, endothelial, and many other types of airway cells.11 This causes a stimulatory G protein to activate adenylate cyclase converting adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP). It is unknown precisely how cAMP causes smooth muscle relaxation; however, decreases in calcium release and alterations in membrane potential are the most likely mechanisms. Longer acting β2 agonists have the same mechanism of action as short-acting β2 agonists; however, they have unique properties that allow for a longer duration of action. For example, salmeterol has a longer duration of action because a side chain binds to the β2-receptor and prolongs the activation of the receptor.12 The lipophilic side chain of formoterol allows for interaction with the lipid bilayer of the plasma membrane and a slow, steady release prolonging its duration of action.
β2 agonists have a central role in the management of obstructive airway diseases allowing for control of symptoms and improvement in lung function. Short-acting β2 agonists such as albuterol, levalbuterol, metaproterenol, and pirbuterol are prescribed for the rapid relief of wheezing, bronchospasm, and airflow obstruction. Clinical effect is seen in a matter of minutes and lasts up to 4 to 6 hours. Scheduled, daily use of short-acting β2 agonists has largely fallen out of favor and they are now used primarily as rescue therapy.13–15 Long-acting β2 agonists are prescribed for control of symptoms when rescue therapies (i.e., short-acting β2 agonists) are used greater than two times per week.16 Combination therapy including a long-acting β2 agonist and an inhaled corticosteroid are effective in reducing symptoms, reducing the risk of exacerbation, and improving lung function while minimizing the dose of inhaled corticosteroid.17
Systemic absorption of inhaled β2 agonists is responsible for a myriad of side effects, most of which are not serious. Most commonly, β2 agonist therapy leads to tremors and tachycardia secondary to direct stimulation of the β2-adrenergic receptor in skeletal muscle or vasculature, respectively.18,19 In severe asthma, β2 agonists may cause a temporary reduction in arterial oxygen tension of 5 mm Hg or more, secondary to β2-mediated vasodilation in poorly ventilated lung regions.20 Hyperglycemia, hypokalemia, and hypomagnesemia also can occur with β2 agonist therapy but the severity of these side effects tends to diminish with regular use. Tolerance to β2 agonists can occur with regular use over a period of weeks and, while not affecting peak bronchodilation, can be evidenced by a decrease in the duration of bronchodilation and the magnitude of side effects (tremor, tachycardia, etc.).21,22 Tolerance likely reflects β2-adrenergic receptor downregulation. β2 agonist therapy withdrawal after regular use can produce transient bronchial hyperresponsiveness.
Evidence has associated the use of long-acting β2 agonist therapy without concomitant use of a steroid inhaler with fatal and near-fatal asthma attacks.23 In light of this evidence, it seems prudent to reserve long-acting β2 agonists for patients that are poorly controlled on inhaled steroids alone or for those patients with symptoms sufficiently challenging to warrant the potential extra risk associated with use of the agents.
Systemic Adrenergic Agonists
Systemic administration of adrenergic agonists for asthma was used more frequently in the past. Oral, intravenous (IV), or subcutaneous administration of β-specific or nonspecific adrenergic agonists is now reserved for rescue therapy. The mechanism of action of systemically administered adrenergic agonists is the same as it is for inhaled agents. Binding of the drug to the β2-adrenergic receptor on smooth muscle cells in the airway is responsible for the bronchodilatory effects. Specifically, β2-receptor stimulation induces a stimulatory G protein to convert ATP to cAMP and in turn reduces intracellular calcium release and alters membrane potential.
Terbutaline can be given orally, subcutaneously, or intravenously, albuterol (salbutamol) can be given intravenously, and epinephrine is usually given subcutaneously or intravenously. Regardless of the route of administration, all three will produce bronchodilation. Comparison of IV and inhaled formulations of terbutaline failed to demonstrate any difference in bronchodilation and, with the propensity for IV formulations to cause side effects, inhaled therapy should be considered the first-line treatment.24,25 This principle not only applies to terbutaline but all β-adrenergic agonists that are available in IV and inhaled forms. If inhaled therapy is not readily available or if inhaled therapy is maximized and symptoms persist, then subcutaneous epinephrine or terbutaline can be administered with improvement in symptoms and spirometry values.26 In summary, subcutaneous or IV β agonists should be reserved only for rescue therapy.
The side effect profile of systemic adrenergic agonists is similar to the side effect profile for inhalational adrenergic agonists. The most common side effects are tremor and tachycardia. Arterial oxygen tension can be transiently decreased and hyperglycemia, hypokalemia, and hypomagnesemia can also be present. Escalating oral, subcutaneous, or IV doses can be associated with a greater incidence of side effects for the same degree of bronchodilation compared to inhaled β-adrenergic agonists.
Inhaled Cholinergic Antagonists
The use of anticholinergics for maintenance therapy and treatment of acute exacerbations in obstructive airway diseases is common. The parasympathetic nervous system is primarily responsible for bronchomotor tone and inhaled anticholinergics act on muscarinic receptors in the airway to reduce tone. The use of inhaled anticholinergics (see Table 25-1) in COPD as maintenance and rescue therapy is considered standard treatment.27 Anticholinergics are not used for maintenance therapy in asthma and are only recommended for use in acute exacerbations.28 The targets of therapy for anticholinergics are the muscarinic receptors located in the airway. There are three subtypes of muscarinic receptors found in the human airway.29 Muscarinic 2 (M2) receptors are present on postganglionic cells and are responsible for limiting production of acetylcholine and protect against bronchoconstriction. M2 is not the target of inhaled anticholinergics but is antagonized by them. Muscarinic 1 (M1) and muscarinic 3 (M3) receptors are responsible for bronchoconstriction and mucus production and are the targets of inhaled anticholinergic therapy. Acetylcholine binds to the M3 and M1 receptors and causes smooth muscle contraction via increases in cyclic guanosine monophosphate (cGMP) or by activation of a G protein (Gq).29 Gq activates phospholipase C to produce inositol triphosphate (IP3), which causes release of calcium from intracellular stores and activation of myosin light chain kinase causing smooth muscle contraction. Anticholinergics inhibit this cascade and reduce smooth muscle tone by decreasing release of calcium from intracellular stores.
There are two inhaled anticholinergics specifically approved for the treatment of obstructive airway diseases. Ipratropium is classified as a short-acting anticholinergic and is commonly used as maintenance therapy for COPD and as rescue therapy for both COPD and asthmatic exacerbations. It is not indicated for the routine management of asthma. Patients treated with ipratropium experience an increase in exercise tolerance, decrease in dyspnea, and improved gas exchange. Tiotropium is the only long-acting anticholinergic available for COPD maintenance therapy. Tiotropium has been shown to reduce COPD exacerbations, respiratory failure, and all-cause mortality.30
Inhaled anticholinergics are poorly absorbed and therefore serious side effects are uncommon. Most commonly, patients experience dry mouth, urinary retention, and can experience pupillary dilation and blurred vision if the eyes are inadvertently exposed to the drug. Some initial data suggested an increase in cardiovascular and stroke complications with tiotropium; however, additional studies did not consistently demonstrate these complications. In general, anticholinergics are safe and effective treatment for patients with obstructive airway diseases.
Systemic Cholinergic Antagonists
The systemically administered anticholinergics atropine and glycopyrrolate act via the same mechanisms as inhaled anticholinergics. While these anticholinergics can be administered by IV or inhalation, significant systemic absorption occurs and their use is generally limited by side effects. Atropine, in particular, is limited in use because of its tertiary ammonium structure. It has a tendency to cause tachycardia, gastrointestinal upset, blurred vision, dry mouth, and central nervous system effects secondary to its ability to cross the blood–brain barrier. Glycopyrrolate has a quaternary ammonium structure and is insoluble in lipids, similar to ipratropium and tiotropium, and has fewer systemic side effects than atropine. IV glycopyrrolate is also clinically limited in use secondary to side effects.31 Glycopyrrolate has been studied as inhaled therapy, however, and is an effective bronchodilator with an intermediate duration of action.32–35 Clinically, it has never been popular as a mainstay of therapy for obstructive airway diseases.
Influence of Inflammation on the Airway
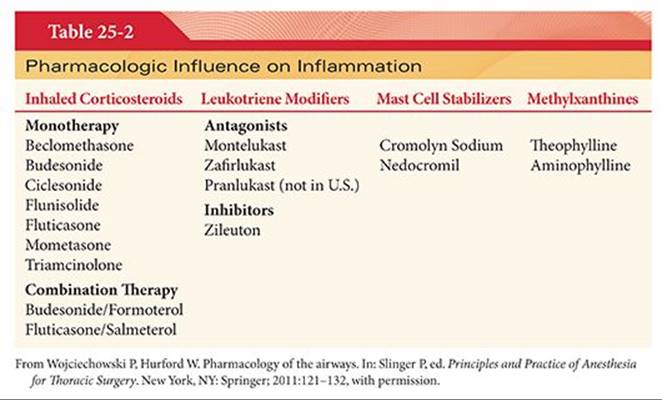
Asthma and COPD, the most common obstructive airway diseases, have a component of inflammation as part of their pathogenesis. Although inflammation is a common pathogenesis, the characteristics and prominent cellular elements involved in the inflammatory process for each disease are distinct.36 In COPD, neutrophils, macrophages, CD8+ T lymphocytes, and eosinophils are more prominent in the inflammatory composition. In asthma, eosinophils play a more prominent role followed by mast cells, CD 4+ T lymphocytes, and macrophages in the inflammatory composition. Inflammatory cell types present in sputum, biopsy specimens, and bronchoalveolar lavage fluid can help predict the response to antiinflammatory therapy. For example, eosinophilia in induced sputum of a patient presenting with a COPD exacerbation predicts an increase in steroid responsiveness.37,38 Patients presenting to the operating room with obstructive airway diseases have a high likelihood of taking one of the antiinflammatory therapies in Table 25-2 for control of their disease.
Inhaled Corticosteroids
In the treatment of asthma, the use of inhaled corticosteroids (ICS) reduces the inflammatory changes associated with the disease, thereby improving lung function and reducing exacerbations that result in hospitalization and death.39–41 On the contrary, the use of ICS as monotherapy in COPD is discouraged. In COPD, ICS are used as a part of combination therapy along with long-acting β-adrenergic agonists (LABA). The combination of drugs acts synergistically and is useful for reducing inflammation. Currently, combination therapy of ICS and LABA is recommended for use in severe to very severe COPD.42The glucocorticoid receptor alpha (GRα) located in the cytoplasm of airway epithelial cells is the primary target of ICS.43,44 Passive diffusion of steroids into the cell allows for binding of the steroid ligand to GRα, dissociation of heat shock proteins, and subsequent translocation to the nucleus. The complex can bind to promoter regions of DNA sequences and either induce or suppress gene expression. Additionally, the steroid-receptor complex can interact with transcription factors already in place, such as the ones responsible for proinflammatory mediators, without binding to DNA and repress expression of those genes. The steroid-receptor complex also can affect chromatin structure by association with transcription factors that influence the winding of DNA around histones, reducing access of RNA polymerase and other transcription factors, and thus reducing expression of inflammatory gene products.
ICS are used in asthma as part of a multimodal treatment regimen and are added to a therapeutic regimen when there is an increase in severity or frequency of asthma exacerbations. There is good evidence to show that ICS can reduce both hospitalizations and death in asthma. The use of ICS in COPD is limited to use in severe to very severe COPD and in combination with LABA. Although no improvement in mortality has been consistently demonstrated with combination therapy (ICS/LABA), there are reported improvements in health status and lung function along with a reduction in exacerbations.
Side effects have been reported with the use of ICS in asthma and COPD. A meta-analysis reported an increase in pneumonia and serious pneumonia but not deaths when ICS was used in the treatment of COPD.45 Other reported side effects in COPD and asthma include oropharyngeal candidiasis, pharyngitis, easy bruising, osteoporosis, cataracts, elevated intraocular pressure, dysphonia, cough, and growth retardation in children. As with any pharmacotherapy, the risks and benefits of therapy must be weighed, and the patient must be carefully monitored for adverse effects. This is especially true with the use of ICS in obstructive lung diseases.
Systemic Corticosteroids
Systemic corticosteroids given in IV or oral form are used for treatment of asthma and COPD exacerbations. The mechanism of action is the same as it is for ICS, activation or suppression of gene products at a transcriptional level and alteration of chromatin structure. Patients that are hospitalized with a COPD exacerbation will typically receive IV corticosteroids to suppress any inflammatory component that may be contributing to the flare up. A study done at the Veterans Affairs medical centers in the United States published in 1999 reported that corticosteroid therapy shortened hospital length of stay and improved forced expiratory volume in 1 second versus placebo.46 The study also compared a 2-week regimen versus an 8-week regimen of corticosteroids and found no difference, concluding that the duration of therapy should last only 2 weeks. In asthma, corticosteroids are recommended for exacerbations that are either severe, with a peak expiratory flow of less than 40% of baseline, or a mild to moderate exacerbation with no immediate response to short-acting β-adrenergic agonists. The recommended duration of therapy is 3 to 10 days without tapering. Alternatively, some patients with asthma and COPD will be receiving long-term oral corticosteroid therapy because their disease is difficult to manage. Side effects of systemic corticosteroids are well described and numerous. Hypertension, hyperglycemia, adrenal suppression, increased infections, cataracts, dermal thinning, psychosis, and peptic ulcers are reported complications of corticosteroid therapy.47
Leukotriene Modifiers
Leukotriene modifiers are used for the treatment of asthma. They are prescribed primarily for long-term control in addition to short-acting β-adrenergic agonists or in conjunction with ICS and short-acting β agonists. Leukotriene modifiers are taken by mouth, produce bronchodilatation in hours, and have maximal effect within days of administration. Their role in the management of COPD is not defined.48Arachidonic acid is converted to leukotrienes via the 5-lipoxygenase pathway.49 Leukotrienes C4, D4, and E4 are the end products of the pathway and cause bronchoconstriction, tissue edema, migration of eosinophils, and increased airway secretions. Leukotriene modifiers come in two different varieties, leukotriene receptor antagonists and leukotriene inhibitors.49 The leukotriene inhibitor zileuton antagonizes 5-lipoxygenase inhibiting the production of leukotrienes.
Leukotriene modifiers improve lung function, reduce exacerbations, and are used as long-term asthma therapy.50,51 Clinical trials have reported that ICS are superior to leukotriene modifiers for long-term control and should be the first-line choice.52,53 Leukotriene modifiers provide an additional pharmacologic option for the control of asthma. Addition of leukotriene modifiers to ICS will improve control of symptoms of asthma as opposed to ICS alone.54
Leukotriene antagonists are usually well tolerated without significant side effects. Links between Churg-Strauss syndrome and the use of leukotriene antagonists have been reported, but it is not clear whether these reports reflect unmasking of a preexisting condition or whether there is a direct link between the two. Zileuton is known to cause a reversible hepatitis in 2% to 4% of patients.
Mast Cell Stabilizers
Cromolyn sodium and nedocromil are the two agents in this category that are used in the treatment of asthma. These agents are delivered by powder inhaler and are not first-line therapy for asthma. They do provide an alternative treatment when the control of asthma is not optimal on other conventional therapies. Cromolyn sodium and nedocromil stabilize submucosal and intraluminal mast cells.55 These drugs interfere with the antigen-dependent release of mediators, such as histamine and slow-reacting substance of anaphylaxis, that cause bronchoconstriction, mucosal edema, and increased mucus secretion.
Systematic reviews of the available literature and consensus statements favor the use of ICS over cromolyn sodium or nedocromil as first-line agents to control symptoms of asthma.56 Alternatively, cromolyn sodium and nedocromil may be used as preventative treatment before exercise or known allergen exposure causing symptoms of asthma. There are no major side effects reported with the use of cromolyn sodium and nedocromil. The most commonly reported side effects are gastrointestinal upset and coughing or irritation of the throat.
Methylxanthines
The role of theophylline, a methylxanthine, has changed since the introduction of ICS and LABA. Theophylline was a common choice for the control of asthma and COPD because of its bronchodilatory and antiinflammatory effects.57 Currently, theophylline is recommended only as an alternative therapy and is not a first-line choice for asthma or COPD.58,59 Theophylline acts via multiple pathways causing improvement in symptoms in obstructive lung diseases. Theophylline is a nonselective inhibitor of phosphodiesterase and increases levels of cAMP and cGMP causing smooth muscle relaxation. Antagonism of the A1 and A2 adenosine receptors also causes smooth muscle relaxation via inhibition of the release of histamine and leukotrienes from mast cells, another reported action of theophylline. In asthma, theophylline reduces the number of eosinophils in bronchial specimens and, in COPD, reduces the number neutrophils in sputum, having an antiinflammatory effect in both conditions. In addition, theophylline activates histone deacetylase and reduces the expression of inflammatory genes. Theophylline and aminophylline are reported to improve diaphragmatic function; however, data have not demonstrated this effect consistently.60
Theophylline has been relegated to an alternative therapy in both asthma and COPD. This has occurred largely because of its significant side effect profile and the subsequent need for monitoring of blood level. Patients that are already on an ICS and a LABA and still have symptoms may benefit from the addition of theophylline, especially if leukotriene modifiers and other alternatives are not tolerated. Theophylline can cause significant and life-threatening side effects if not dosed carefully and monitored appropriately. Side effects tend to be more prominent when blood levels exceed 20 mg/L. The most common side effects include headache, nausea, vomiting, restlessness, abdominal discomfort, gastroesophageal reflux, and diuresis. The most significant side effects include seizures, cardiac arrhythmias, and death. Adverse effects from theophylline may be avoided if the clinician follows the patient carefully, monitors blood levels regularly, and educates the patient on the signs and symptoms of overdose.
Influence of Anesthetics on the Airways
Volatile Anesthetics
Volatile anesthetics have a host of effects on the respiratory system. Volatile anesthetics reduce bronchomotor tone and all commonly used volatile anesthetics (Table 25-3), except desflurane, produce a degree of bronchodilatation that may be helpful in patients with obstructive lung disease or in patients that experience any degree of bronchoconstriction.61 Rooke and colleagues62 in 1997 reported that sevoflurane produced a greater reduction in respiratory system resistance than isoflurane or halothane. Volatile anesthetics likely induce bronchodilation by decreasing intracellular calcium, partly mediated by an increase in intracellular cAMP and by decreasing the sensitivity of calcium mediated by protein kinase C.63 The effect is seen to a greater degree in distal airway smooth muscle secondary to the T-type voltage-dependent calcium channel, which is sensitive to volatile anesthetics.64

Volatile anesthetics are administered to provide amnesia and blunt the response to surgical stimulation but can be of use in patients that have obstructive airway diseases or experience bronchoconstriction in the operating room. Multiple case reports provide examples of how volatile agents were used solely for the treatment of status asthmaticus.65–68 The main concern with the use of volatile anesthetics is the rare occurrence of malignant hyperthermia. Hypotension can also be a concern with volatile anesthetics; however, the blood pressure is usually easily restored with small amounts of vasopressors. Deep levels of anesthesia associated with high concentrations of volatile anesthetics may be undesirable, and prolonged administration outside the operating room is problematic.
Intravenous Anesthetics
IV anesthetics can decrease bronchomotor tone when used for induction or IV anesthesia in the operating room. Ketamine, propofol, and midazolam (see Table 25-3) have relaxant effects on airway smooth muscle.69 Etomidate and thiobarbiturates do not affect bronchomotor tone to the same extent.70 The choice of IV anesthetics for induction and maintenance of anesthesia may be important for a patient with reactive airway disease. The mechanism of reduction of bronchomotor tone for the IV anesthetics is largely unknown. Ketamine is thought to have a direct relaxant effect on smooth muscle.71 Propofol is thought to reduce vagal tone and have a direct effect on muscarinic receptors by interfering with cellular signaling and inhibiting calcium mobilization.72,73 The preservative metabisulfite in propofol prevents the inhibition of vagal-mediated bronchoconstriction.74
Choosing an agent such as propofol or ketamine can be beneficial in patients with bronchospasm or obstructive airway disease. The use of these IV agents for induction or maintenance of anesthesia over other agents can be useful in minimizing the intraoperative effects of bronchospasm. Although each of the IV anesthetics carries a unique side effect profile, the major effects are not related to the airway. The use of ketamine is associated with increased salivation and coadministration of a small dose of anticholinergic can attenuate secretion production. Propofol is associated with hypotension that usually is easily corrected with vasopressors.
Local Anesthetics
Local anesthetics are primarily used to suppress coughing and blunt the hemodynamic response to tracheal intubation.75,76 Although animal models have demonstrated some ability of local anesthetics to relax bronchial smooth muscle, in clinical practice the use of local anesthetics as pure bronchodilators is limited by toxicity and the ready availability of more potent bronchodilators such as short-acting β-adrenergic agonists.
Influence of Adjunctive Agents on the Airway
Helium (administered as a mixture of helium and oxygen [heliox]) has the advantage of having a low Reynolds’ number and less resistance during turbulent airflow especially in large airways (see Chapter 24). A trial in patients with COPD exacerbations failed to demonstrate a statistically significant reduction in the necessity for endotracheal intubation in patients treated with noninvasive ventilation and helium-oxygen mixtures.77 Helium-oxygen mixtures may be useful as short-term temporizing therapy to decrease the work of breathing in patients with upper airway obstruction. The use of helium-oxygen mixtures is limited by a progressive reduction in efficacy at higher inspired oxygen concentrations.
Antihistamines: Histamine release from mast cells and basophils is responsible for airway inflammation and bronchoconstriction in asthma.78 Antihistamines are not standard therapy for asthma, but the use of antihistamines and leukotriene modifiers for allergen-induced bronchoconstriction has shown promise for diminishing the early and late responses to allergens.78,79 Patients that have allergen-induced asthma or patients that experience an allergic reaction in the operating room may benefit from antihistamines to attenuate the role that histamine plays in bronchoconstriction.
Magnesium sulfate is not standard therapy for asthma exacerbations. Magnesium sulfate is thought to produce additional bronchodilation when given in conjunction with standard therapy for asthma exacerbations. Currently, IV magnesium therapy is reserved as an alternative therapy when the patient has not responded to standard therapy.80 The combination of nebulized magnesium sulfate and β-adrenergic agonists have also been studied and show potential benefit in asthma exacerbations.81 Overall, magnesium sulfate, IV or nebulized, is not a first-line therapy for asthma exacerbations and should be reserved for situations when the patient is not responding to conventional therapy.
Pharmacology of the Pulmonary Circulation
Patients with pulmonary hypertension (PHTN) are high-risk candidates for both cardiac and noncardiac surgery. They have poor cardiorespiratory reserve and are at risk of having perioperative complications including pulmonary hypertensive crises with resultant heart failure, respiratory failure, and dysrhythmias.82,83 Anesthetic management of these patients can be complex and challenging. Drugs affecting the pulmonary vascular bed are routinely administered during anesthesia, and their effects are of particular interest in patients with PHTN. Reducing the consequences of an elevated pulmonary vascular resistance and the resulting right ventricular dysfunction should be considered as the primary goal of therapy with pulmonary vasodilators. Owing to the contractile properties of the naive right ventricle, attempts at improving its contractility are generally not effective. Therefore, principles of management of PHTN center on reducing right ventricular afterload while preserving coronary perfusion by avoiding reductions in systemic blood pressure.84
Anesthetic Drugs
Evaluating the effects of anesthetic drugs on the pulmonary vasculature is challenging. In clinical practice and research, these drugs are rarely administered in isolation. Their administration can lead to concurrent changes in nonpulmonary hemodynamic parameters such as cardiac output (CO) that ultimately affect pulmonary artery pressure (PAP). An increase in PAP may be the result of increased pulmonary vascular resistance (PVR), increased CO, or an increase in left atrial pressure (LAP) (PAP = [PVR × CO] + LAP). In addition, general anesthesia involves manipulation of variables that affect PVR, including fraction of inspired oxygen (FIO2), carbon dioxide (CO2), and positive pressure ventilation (PPV).
Ketamine
Historically, ketamine has occupied a controversial position in anesthesia for patients with PHTN. Despite its current widespread use in these challenging patients, it has been classically taught that ketamine causes pulmonary vasoconstriction and should be used with extreme caution in this group. The mechanism of action of ketamine is not fully elucidated. It is an N-methyl-D-aspartic acid (NMDA) receptor antagonist and also binds to opioid receptors and muscarinic receptors.85 It appears to stimulate release as well as inhibit neuronal uptake of catecholamines which may account for its cardiostimulatory and bronchodilatory effects. Some animal studies have shown an endothelium-independent vasodilatory response to ketamine in the pulmonary bed.
The effects of ketamine on the human pulmonary vasculature appear to be complex and the clinical literature reveals a vast heterogeneity in regard to results. Factors known to affect pulmonary vasoreactivity such as FIO2, CO2, presence of PHTN, and presence of premedicants are not reported or acknowledged in many studies. The hemodynamic effects of a bolus of ketamine can be attenuated or abolished with premedicants such as droperidol, dexmedetomidine, or benzodiazepines.86 Early study of the drug’s hemodynamic profile in adult patients showed increases of PAP and PVR in the range of 40% to 50%. This, combined with increases in variables contributing to myocardial oxygen consumption, raised concern about the use of ketamine in patients with coronary artery disease (CAD) and PHTN. More recently in the pediatric literature, Williams et al.87 showed no change in PVR or mean pulmonary artery pressure (mPAP) after ketamine administration in spontaneously breathing children with severe PHTN undergoing cardiac catheterization. In another pediatric study, ketamine maintained pulmonary to systemic blood flow and did not affect pulmonary pressure or resistance in children with intracardiac shunt undergoing cardiac catheterization. Propofol, on the other hand, decreased systemic vascular resistance (SVR) leading to increased right to left shunting.88 In adult patients undergoing one-lung ventilation (OLV) for lung resection, ketamine did not significantly increase PAP or PVR compared to enflurane. Other case reports highlight the value of the relative cardiostability of the drug in patients with minimal cardiorespiratory reserve.89,90 Many clinicians incorporate this drug into their routine inductions for patients with severe PHTN (e.g., pulmonary endarterectomy or lung transplantation). The advantages, in particular maintenance of stable hemodynamics and coronary perfusion pressure, seem to outweigh the potential disadvantages.
Propofol
Propofol is commonly used in anesthesia, including for patients with PHTN. It is frequently used to maintain anesthesia during and after lung transplantation. The effects of propofol are thought to be primarily mediated by γ-aminobutyric acid (GABA) receptors. The concerning hemodynamic effect of propofol in the context of PHTN is a decrease in SVR, which can not only have effects on intracardiac shunting, if present, but can lead to decreased coronary artery perfusion of the right ventricle and resultant right ventricular dysfunction. In regard to direct effects on the pulmonary vasculature, animal studies have shown that during increased tone conditions in the pulmonary vasculature, propofol may act as a pulmonary vasoconstrictor.91 Propofol has also been shown to interfere with acetylcholine-induced pulmonary vasodilation in dogs.92 On the other hand, in isolated pulmonary arteries from human and chronically hypoxic rats, etomidate and to a lesser extent propofol showed vessel relaxation.93 The clinical significance of these contradictory results is unknown.
Etomidate
Etomidate is an imidazole that mediates its clinical actions primarily at GABA A receptors. As mentioned earlier, it appears to have vasorelaxant properties in isolated pulmonary arteries. Its major attribute as an induction agent is its stable hemodynamic profile. In patients with cardiac disease, an induction dose of etomidate increased mean arterial pressure (MAP), decreased SVR, and decreased PAP.94 In pediatric patients without PHTN presenting for cardiac catheterization, there was no significant change in any hemodynamic parameters after induction with etomidate.95
Opioids
Opioids seem to have little to no deleterious effects on the pulmonary vascular system. In anesthetized cats, administration of morphine, fentanyl, remifentanil, and sufentanil caused a vasodilatory response under elevated tone conditions in isolated lobar artery.96 The mechanism seems to involve histamine- and opioid-mediated receptor pathways. Clinical experience would echo the cardiostability of judicious narcotic administration in hemodynamically fragile patients.
Volatile Anesthetics
At clinically relevant concentrations, modern volatile anesthetics likely have little to no direct vasodilating effect on the pulmonary vasculature. In pigs, sevoflurane administration depressed right ventricular function with no change in PVR.97 This suggests that the decreases in PAP observed with volatile anesthetics may partially occur secondary to the decreases in CO seen with these agents. Nitrous oxide is typically avoided in patients with PHTN as it is believed to cause pulmonary vasoconstriction, perhaps via release of catecholamines from sympathetic nerves supplying the pulmonary vasculature. In patients with mitral stenosis and PHTN presenting for cardiac surgery, administration of nitrous oxide after fentanyl anesthesia (7.5 to 10 µg/kg) increased PVR, PAP, and cardiac index (CI).98 However, a subsequent study showed that in the presence of high-dose fentanyl (50 to 75 µg/kg), 70% nitrous oxide is actually was associated with a decrease in PAP and CO in patients with secondary PHTN, with no echocardiographic changes in right ventricular function.99
Neuromuscular Blockers
Pancuronium increases PAP in dogs with lung injury.100 It is theorized to do so indirectly by increases in CO and directly by increasing PVR, possibly by its antagonist actions at muscarinic receptors in the pulmonary vasculature. Rocuronium, cisatracurium, and vecuronium have little to no effect on most cardiac indices in patients undergoing coronary artery bypass graft (CABG).101
Magnesium
Magnesium is a vasodilator in both the systemic and pulmonary circulations. The mechanism of action of magnesium’s effects on vasodilation is likely through its effects on membrane channels involved in calcium flux and through its action in the synthesis of cAMP. It would appear to be an important cofactor for endothelial-dependent pulmonary vasodilation. It has been used successfully to wean NO in PHTN.102 Increasing doses of magnesium in piglets with acute embolic PHTN decreased mPAP, increased CO, and decreased PVR.103 Magnesium has been used to treat persistent PHTN of the newborn, but controversy surrounds its use.
Regional Analgesia
Pain can increase PVR.104 Perioperative thoracic epidural analgesia (TEA) is commonly used in abdominal and thoracic surgery. TEA may decrease PAP through decreases in CO or via attenuation of the pulmonary sympathetic outflow. In pigs, TEA depresses right ventricular function in acute PHTN.105 Unilateral thoracic paravertebral block with lidocaine has been shown to decrease myocardial contractility up to 30% and significantly decrease systemic pressure; an effect that may be attenuated by epinephrine. In general, the potential benefits of regional anesthesia in thoracoabdominal surgery typically outweigh the risks of hypotension and right ventricular dysfunction. As with most anesthetic interventions in patients with PHTN, careful titration and monitoring is paramount. A few reports illustrate successful use of epidural analgesia in this patient population.106
Vasopressors and Inotropes
Vasopressors and inotropes are commonly required during anesthesia to counteract the effects of cardiodepressant and vasodilating drugs. Treatment of hypotension in these patients can be difficult to manage given the typical cautious fluid administration most patient populations.
The innervation and receptor content of the pulmonary vasculature is complex. Neurotransmitter receptors in this system include those from the adrenergic, cholinergic, and dopaminergic families as well as histamine, serotonin, adenosine, purines, and peptides. The pulmonary vasculature’s response to sympathetic activation will generally result in an increase in PVR. In human pulmonary artery, administration of acetylcholine induces pulmonary relaxation.107
The response of the pulmonary system to exogenous vasopressor administration is dependent on the clinical situation. Consequently, results of studies are heterogeneous. In anesthetized dogs without PHTN, dopamine, epinephrine, norepinephrine, and phenylephrine all increase PAP to varying degrees by varying mechanisms but with no drug is there a significant increase in PVR.108 Dopamine does not increase PVR after lung transplantation in pigs.109 In anesthetized patients with chronic secondary PHTN undergoing cardiac surgery, both norepinephrine and phenylephrine increase PAP and PVRI with minimal change in CI.110 Within the clinically relevant MAP target in this study, norepinephrine decreased the mPAP to MAP ratio, but phenylephrine did not, suggesting it may be a better choice in this patient cohort. In a dog model of acute PHTN, however, phenylephrine restored perfusion to the ischemic right ventricle and therefore increased CO.111 This is a relevant observation, as it illustrates the importance of coronary artery perfusion in the setting of right ventricular strain and that maintenance of systemic pressure by whatever method may be the most important principle in this subset of patients.
Vasopressin has also been studied. In a chronic hypoxic rat model, vasopressin administration resulted in a V1 receptor–mediated pulmonary vasodilation.112 In an acute PHTN model in dogs, vasopressin increased PVR and resulted in a substantial decrease in right ventricular contractility.113 Human studies of effects of vasopressin on the pulmonary vasculature are limited. Vasopressin has been used successfully after cardiac surgery in patients with PHTN and resistant hypotension.114 The use of vasopressin to treat acute right ventricular failure in patients with IPPH has been described in obstetric anesthesia.115
Pulmonary Vasodilators
Pulmonary vasodilators are typically employed to improve right ventricular function in the setting of PHTN or in an effort to enhance regional pulmonary blood flow and improve intrapulmonary shunt. In the acute care setting, however, it is these agent’s pulmonary vasodilatory effects that are being exploited. In general, parenteral and oral vasodilators are hampered by their relatively nonselective actions in the pulmonary vascular bed. In addition to their hypotensive systemic hemodynamic effects, their use may also lead to perfusion of underventilated alveoli, worsen intrapulmonary shunt and, in turn, worsen oxygenation. The ideal pulmonary vasodilator should have a rapid onset of action, a short half-life, and produce regional pulmonary vasodilation. This would avoid systemic hypotension and the potential adverse effects on ventilation-perfusion matching that limit the use of systemic agents in critically ill patients. In this regard, inhaled vasodilators are attractive as they preferentially dilate ventilated alveoli and have less systemic effects.
Nitric Oxide
Inhaled nitric oxide (iNO) is preferentially delivered to ventilated lung units leading to improved perfusion to alveoli that are able to participate in gas exchange. This “selective effect” leads to a decrease in intrapulmonary shunt. Medical grade NO may be administered either noninvasively (via a face mask) or through a ventilator circuit. If administered through a circuit, a device is used that can regulate the concentration of NO and monitor levels of nitrogen dioxide—a by-product of NO when it combines with oxygen (Fig. 25-1). At present, iNO is only approved for infants with respiratory distress syndrome. This approval stems from large prospective placebo-controlled studies demonstrating that NO reduced the need for extracorporeal membrane oxygenation (ECMO) and reduced the requirement for oxygen therapy following intensive care unit (ICU) discharge.116 Although there is controversy about a dose-response relationship for NO and pulmonary vasodilation, the typical dose ranges from 10 to 40 ppm. Methemoglobin levels need to be monitored when NO is administered for more than 24 hours. Heart and lung transplantation represent two distinct areas where acute pulmonary vasodilation has strong theoretic benefit as it relates to improving acute right ventricular failure and attenuating reperfusion injury, respectively. The acute right ventricular failure complicating heart transplantation may be attenuated with the use of a pulmonary vasodilator. Although several studies suggest that NO may be useful preoperatively in risk-stratifying patients scheduled for cardiac transplant, only case series support the use of inhaled NO to reverse the right ventricular dysfunction following cardiac transplant. However, based on clinical experience, inhaled NO has become a standard of care in many transplant centers. The beneficial immune-modulating effects of inhaled NO in addition to its vasodilating properties were felt to be responsible for preliminary studies of using inhaled NO to prevent primary graft dysfunction (PGD) after lung transplantation.117 Although a randomized clinical trial failed to show benefit in preventing PGD, it is commonly used to treat the hypoxemia and PHTN seen in established, severe PGD.118Owing to the inherent cost of using inhaled NO, other pulmonary vasodilators have been evaluated.
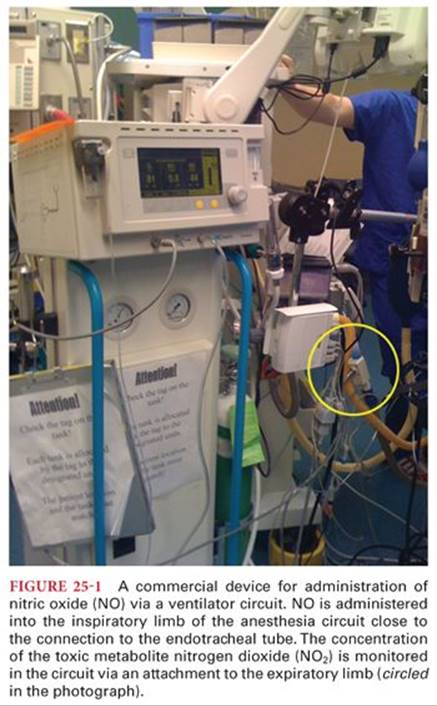
In nontransplant thoracic surgery, NO has been studied as a potential treatment for the gas exchange abnormalities associated with OLV. Its effects are controversial but it would appear that it exerts its maximal benefits in patients with elevated pulmonary vascular resistance index (PVRI) and the poorest gas exchange before administration.119 NO can be quickly delivered via the circuit of an anesthetic or intensive care ventilator; however, it is expensive and not widely available.
Prostaglandins
Prostanoids induce relaxation of vascular smooth muscle, inhibit growth of smooth muscle cells and are powerful inhibitors of platelet aggregation.120 Inhaled prostanoids involve an aerosol delivery mechanism that is attached by a nebulizer to the ventilator circuit (Fig. 25-2). Treatment may be limited by inefficiencies in aerosolization. Owing to the short half-life of epoprostenol, the drug must also be continuously nebulized.121 As a result, changes of dose delivery with alterations in ventilator volumes, FIO2, airway pressures, and solvent evaporation may be challenging. The synthetic prostanoids, treprostinil and iloprost, hold promise as inhaled vasodilators in that they may only require intermittent administration. When nebulized, prostanoids can lead to similar improvements in oxygenation and pulmonary pressures as compared to inhaled NO. A crossover study compared inhaled NO to inhaled prostaglandins in patients after lung (n = 19) or heart (n = 6) transplant. In this acute hemodynamic study, there was no significant difference in hemodynamics or oxygenation between agents.122 Prostacyclin can be delivered by nebulizer into a ventilator circuit at a starting dose of 50 ng/kg per minute and clinical effects should be evident within 10 minutes.123

Use of IV prostaglandins during OLV results in a decrease in both systemic and pulmonary pressures and either no change or a decrease in Pao2. Selective infusion of prostaglandin into the pulmonary artery of the ventilated lung in a human model during OLV resulted in stable systemic pressure and a reduction in PVR and increase in Pao2.124 However, this route of administration is not practical in routine thoracic anesthesia practice. Inhaled prostacyclin decreases PVRI and PAP with maintenance of favorable systemic pressures but does not change PaO2 during OLV.125
Both iNO and prostaglandins have been shown to affect platelet function. This could theoretically contribute to perioperative bleeding during large surgeries such as lung transplantation and is a concern in regard to neuraxial analgesia. The clinical relevance of platelet inhibition with these inhaled agents is unknown. In cardiac surgery patients, laboratory confirmation of platelet dysfunction with inhaled prostacyclin did not correlate with chest tube losses.126 Also, in an obstetrical patient with PHTN on IV prostacyclin, conversion to inhaled prostacyclin allowed for a successful labor epidural placement with no complications.127
Phosphodiesterase Inhibitors
Phosphodiesterase inhibitors prevent the degradation of cGMP and cAMP. cAMP and cGMP are activated by NO and are intermediaries in a pathway that leads to vasodilation via the activation of protein kinases and reduction in cytosolic calcium. Milrinone is an adenosine-3′, 5′-cAMP–selective phosphodiesterase enzyme (PDE) inhibitor. When nebulized, it has been shown to lead to a relative reduction in PVR compared to SVR.128 The inhalation of milrinone selectively dilated the pulmonary vasculature without systemic effects. When milrinone is combined with inhaled prostacyclin, there appears to be a potentiation and prolongation of the pulmonary vasodilatory effect.129
Owing to the relatively higher expression of phosphodiesterase 5 (PDE5) in the pulmonary circulation relative to the systemic circulation, PDE5 inhibitors have a relative selective effect on PVR as opposed to SVR. In addition to their relatively selective pulmonary vasodilatory effects, their effects on smooth muscle proliferation and cellular apoptosis may be responsible for benefit of these agents when administered chronically in patients with idiopathic pulmonary arterial hypertension (PAH). A direct effect on the right ventricle has been postulated; however, the clinical relevance of this finding is uncertain.
Although the benefits of oral sildenafil and tadalafil in chronic PAH have been evaluated in prospective controlled trials, most of the acute applications for these agents have been described in case reports or small cohort studies and as such have not been approved for these indications. In the acute setting, sildenafil has been demonstrated to enhance the effects of inhaled NO and may also be useful in blunting the rebound in pulmonary pressures that occurs during weaning of inhaled NO.130 The benefits of sildenafil in acute pulmonary embolism, cardiac transplantation, and in patients with PHTN being considered for pulmonary thromboendarterectomy have also been described.131
Hypoxic Pulmonary Vasoconstriction
IV anesthetic agents have no effect on hypoxic pulmonary vasoconstriction (HPV). All of the volatile anesthetics inhibit HPV in a dose-dependent fashion. Animal studies suggest that this inhibition is dependent on the agent: halothane > enflurane > isoflurane/desflurane/sevoflurane.132 The older agents were potent inhibitors of HPV and this may have contributed to the high incidence of hypoxemia reported during OLV in the 1960s and 1970s (see earlier); many of these studies used 2 to 3 minimum alveolar concentration (MAC) doses of halothane during anesthesia.
In doses of less than or equal to 1 MAC, the modern volatile anesthetics (isoflurane, sevoflurane,133 and desflurane134) are weak, and equipotent, inhibitors of HPV. The inhibition of the HPV response by 1 MAC of a volatile agent such as isoflurane is approximately 20% of the total HPV response, and this could account for only a net 4% increase in total arteriovenous shunt during OLV, which is a difference too small to be detected in most clinical studies.135 In addition, volatile anesthetics cause less inhibition of HPV when delivered to the active site of vasoconstriction via the pulmonary arterial blood than via the alveolus. This pattern is similar to the HPV stimulus characteristics of oxygen. During established OLV, the volatile agent only reaches the hypoxic lung pulmonary capillaries via the mixed venous blood. No clinical benefit in oxygenation during OLV has been shown for total IV anesthesia above that seen with 1 MAC of the modern volatile anesthetics.136 N2O inhibits HPV. N2O is usually avoided during thoracic anesthesia.
HPV is decreased by systemic vasodilators such as nitroglycerin and nitroprusside. In general, vasodilators can be expected to cause some deterioration in Pao2 during anesthesia. Thoracic epidural sympathetic blockade probably has little or no direct effect on HPV, which is a localized chemical response in the lung.137 However, thoracic epidural anesthesia can have an indirect effect on oxygenation if it is allowed to cause hypotension and a fall in CO, thus decreasing mixed venous oxygen saturation.
Intrinsic Pharmacologic Effects of the Lungs
The lungs receive essentially the entire CO and the surface area of their vascular bed is enormous (70 to 100 m2). The lungs contain nearly half of the body’s endothelium and have an extraordinarily high perfusion of 14 mL/min/g tissue (as opposed to the next-highest renal perfusion of 4 mL/min/g tissue). Thus, there is ample blood-endothelial interface for surface enzyme activity as well as uptake and secretion.138 The largest population of cells involved in pulmonary metabolism of blood-borne substances is, as might be expected, the pulmonary endothelium. Consistent with high metabolic activity, endothelial cells typically have both extensive cytoplasmic vesicles and prominent caveolae. The caveolae are tiny membrane invaginations and near-membrane vesicles similar to those found elsewhere in the body, measuring 50 to 100 nm, associated with caveolin proteins, and derived from lipid rafts within the membrane. The predominant activities of these caveolae, thought to include endocytosis and signal transduction, have not been fully delineated, and may be pleiotropic.139 The endothelial cells structurally have large luminal projections and invaginations, providing an even greater interface area at the microscopic level.
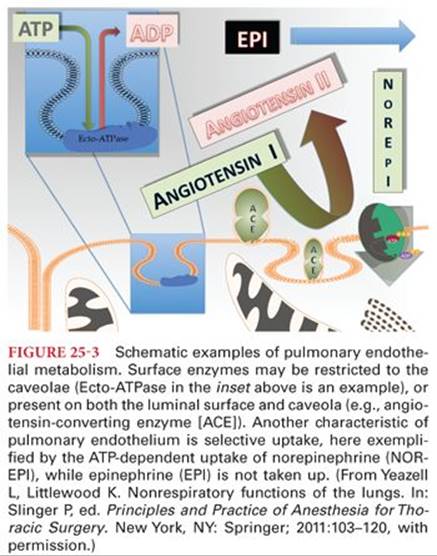
Metabolism by the endothelial cell occurs either on the surface of the cell via enzymes associated with the membrane (“ectoenzymes”) or by cytosolic processing after substances are taken up by the cell. Some surface enzymes are distributed along the luminal membrane, whereas others are associated exclusively with the caveolae. Figure 25-3 schematically depicts these processes with example substances and pathways. Metabolism may be further divided into exogenous versus endogenous substances as well as deactivated versus activated products. The terminology of pulmonary metabolism can be confusing and sometimes inconsistent. In general, “pulmonary uptake” (or “extraction”) is simply used to describe transfer from blood to lung. It does not indicate whether the substance of interest is subsequently metabolized or returned back into the blood (with or without alteration). “First-pass” uptake is used to describe the amount of substance removed from the blood on the first cycle through the lungs. “Extraction” is also sometimes misused synonymously with first-pass uptake. “Clearance” may be used to describe a substance undergoing actual elimination, either in terms similar to renal clearance as volume of blood from which the substance would be completely removed (milliliters per minute or milliliters per kilogram per minute), or as a comparison of pulmonary arterial concentration versus systemic arterial concentration.
The lung has a pronounced impact on the blood concentration of substances even when it does not ultimately break them down or secrete them. This is because of simple uptake and retention of substances, often followed by release back into the blood. This “capacitor effect”140 of the lungs in which any rapid rise or fall in concentration is attenuated is revisited in the following discussion regarding local anesthetic toxicity.
Exogenous Substances
Drugs
The cytochrome P450 monooxygenase enzyme systems are the most studied metabolic pathways for medications. The lungs have been found to have substantial concentrations of P450 isoenzymes, particularly within type II pneumocytes, Clara cells, and endothelial cells. While P450 and other enzyme systems have long been known to exist in the human lung, the actual activity of lung enzymes ranges from negligible to 33% of that of the liver.141
Opioids
Fentanyl has been shown to have a markedly variable first-pass uptake up to 90% in humans. The same investigators found that significant amounts of fentanyl then returned from the lungs into the blood with a biphasic pattern, equilibrating after about a minute in the fast phase and nearly 25 minutes for the slow phase. The uptake of fentanyl is higher than expected even for this basic and lipophilic drug. Active uptake of fentanyl has been demonstrated in human lung endothelial cells. Sufentanil demonstrates uptake that is a little more than half that of fentanyl. Morphine has a much lower uptake of about 10%.142
Local Anesthetics
For lidocaine, there is a first-pass uptake of approximately 50% with significant retention at 10 minutes.143 The uptake of lidocaine has also been examined in a variety of physiologic circumstances. Under extremes of metabolic acidosis and alkalosis, lidocaine demonstrates increased uptake with higher blood pH. It is postulated that this finding is the consequence of increased drug lipophilicity because, in a less acidic environment, more of the drug is in its nonionized form. Bupivacaine has been investigated less extensively than lidocaine and with less consistent results. In most animal species, peak extraction has been reported as high with variable first-pass retention. In humans, however, the effective first-pass extraction appears to be lower when studied by epidural dosing.144
Two areas of interest in the practice of clinical anesthesia are intimately linked with the pulmonary uptake of local anesthetics. The first is the relative safety of levobupivacaine and ropivacaine in comparison to bupivacaine. These drugs have, in fact, been the subject of several investigations. Early animal studies suggested decreased toxicity of these newer preparations. However, a review of the pharmacodynamics and pharmacokinetics of local anesthetics145 describes the challenges of comparing toxicities in clinical practice. A second area of interest is the treatment of local anesthetic toxicity with lipid emulsion. The issue of pulmonary uptake and delayed release of local anesthetics must be considered in the treatment of suspected local anesthetic toxicity with emulsified lipid.146
Hypnotics
Thiopental has been found to have nearly 15% first-pass uptake in humans147 with little or no metabolism. The pulmonary uptake of ketamine was found to be slightly less than 10% without subsequent metabolism.148 For propofol, most work shows about 30% first-pass uptake and negligible metabolism of propofol by the lungs.149
Endogenous Substances
Angiotensin-Converting Enzyme
The lung plays a critical role in the renin-angiotensin system because of the pulmonary endothelium’s high concentration of angiotensin-converting enzyme (ACE). When the kidney responds to changes in physiologic parameters such as vascular volume, blood pressure, and adrenergic stimulation by the cleaving of prorenin, the resultant renin catalyzes the formation of angiotensin I from angiotensinogen. It is ACE that then converts angiotensin I to the critically important vasoconstrictor, angiotensin II. Although ACE can be found on vascular endothelium throughout the body as well as in the plasma, the pulmonary endothelium has an abundance of ACE as a surface or ectoenzyme on the vascular membrane150 (Fig. 25-4). The newly formed angiotensin II is not taken up or further metabolized by the endothelial cell, but rather immediately returns to the blood. Clinically, ACE inhibitors have been useful drugs in the management of systemic hypertension.151
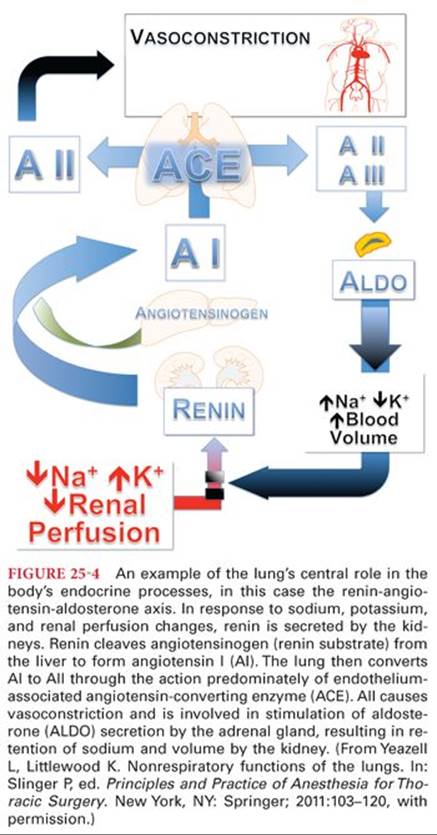
Bradykinin is a nine amino acid peptide produced in multiple sites throughout the body from kininogen through the action of plasma kallikrein. It is in turn metabolized by several peptidases. Bradykinin is degraded by ACE and more than 90% of bradykinin is eliminated on first-pass through the lungs.152 Bradykinin’s effects are wide-ranging, including antithrombotic and profibrinolytic activity in the coagulation system, as well as modulation of NO and prostacyclin release. Specific to the lung, bradykinin has vasodilating effects on normal pulmonary vessels but is vasoconstrictive when the pulmonary endothelium is destroyed in animal models.153 Bradykinin is a bronchoconstrictor.154 Some side effects of ACE inhibitors, such as angioedema and cough, and some of the beneficial impact, such as decreased myocardial infarctions and improved renal function, involve modification of bradykinin metabolism.
Biogenic Amines
Histamine; serotonin (5-hydroxytryptamine or 5-HT); and the three naturally occurring catecholamines dopamine, norepinephrine, and epinephrine comprise the group commonly termed biogenic amines. 5-HT is produced predominately by the gastrointestinal tract’s chromaffin cells. Ingested tryptophan undergoes a two-step conversion first by tryptophan-5-hydroxylase and then by L-amino acid decarboxylase to serotonin. Mast cells and neuroendocrine cells in the lung are also capable of producing serotonin by uptake of tryptophan along the same enzymatic pathway. Once released from the gastrointestinal tract, there is avid uptake of 5-HT, particularly by nerve endings and platelets. These cells do not metabolize 5-HT to any great extent. The remainder of 5-HT is extracted by the lung and, to a lesser degree, the liver. In the case of these organs, the 5-HT is metabolized to 5-hydroxyindoleacetic acid (5-HIAA) by cytosolic monoamine oxidase (MAO) and aldehyde dehydrogenase. 5-HIAA is a useful marker of carcinoid syndrome with increased histamine turnover. MAO inhibitors block the cytosolic metabolism of 5-HT but not its uptake, whereas several drugs, including volatile anesthetic agents, block uptake but not intracellular metabolisms.155
Because it is not lipophilic, the pulmonary uptake of 5-HT is an active process, predominately via endothelial cells and with some variability between species. The pulmonary uptake of 5-HT by the lung is typically reported to be 90% or greater, meaning that little 5-HT reaches the systemic vasculature under normal circumstances. This model of production and uptake of 5-HT plays a pivotal role in several pathologic processes relevant to clinical anesthesiology. In carcinoid syndrome, the right heart receives a high concentration of 5-HT before being extracted and metabolized by the pulmonary circulation. This is thought to be the reason that the right heart shows the greatest myocardial and valvular injury in this syndrome.156 The valvular injury of substances related to 5-HT such as methysergide and ergotamine, and those that increase 5-HT such as fenfluramine, and the recreational drug “ecstasy” (3,4-methylenedioxymethamphetamine), known to activate 5-HT receptors, are all similar to carcinoid cardiac disease. When an intracardiac right-to-left shunt is present in the carcinoid patient with a partial bypass of the pulmonary circulation, the left heart demonstrates valvular injury similar to that of the right heart.157
Pulmonary embolism presents another clinical situation pertinent to 5-HT activity. The mass effect of embolism does not, in itself, account for the typical cardiopulmonary consequences. The platelet aggregation and activation associated with acute pulmonary embolism results in degranulation with the release of 5-HT, well known to be a potent vasoconstrictor and to increase bronchial smooth muscle tone. This release of 5-HT and, perhaps, decreased local uptake of 5-HT are postulated to cause local and regional vascular changes. Other actions of elevated 5-HT, such as promotion of further platelet aggregation and inhibition of the vasodilating prostacyclin likely also play a role in the full response to pulmonary embolism.158 Histamine, in contrast to 5-HT, has almost no uptake in the pulmonary circulation.
Just as the lung has the enzymes to metabolize both histamine and serotonin but the ability to take up only serotonin, its uptake of catecholamines also demonstrates marked selectivity. Norepinephrine demonstrates a 35% to 50% first-pass uptake with subsequent metabolism by catechol-O-methyltransferase (COMT), MAO, aldehyde reductase, and aldehyde dehydrogenase.159 However, dopamine, isoproterenol, and epinephrine have essentially no uptake.
Arachidonic Acid Metabolites
Extensive production and metabolism of arachidonic acid derivatives occurs in the lung. The term eicosanoids refers to the 20-carbon carboxylic acids derived from the metabolism of the lipid membrane component icosatetraenoic acid, more commonly known as arachidonic acid. The action of phospholipase A2 converts the esterified form, as found in the membrane, and releases arachidonic acid from structural glycerol. Once free, arachidonic acid may follow three main metabolic pathways in the lung: the lipoxygenase pathway produces leukotrienes, lipoxins, and some of the hydroxyeicosatetraenoic acids (HETEs); the cyclooxygenase (COX) pathway produces prostaglandins, thromboxane, and prostacyclin; and the cytochrome P450 monooxygenase system produces cis-epoxyeicosatrienoic acids and HETEs that are different than the products of the lipoxygenase pathway.
The leukotrienes promote inflammatory responses in the lung. They are responsible for bronchoconstriction and increased pulmonary vascular permeability, are chemotactic and chemokinetic for neutrophils, and facilitate eosinophil degranulation.160 They are produced by activated inflammatory cells within the lung as well as those arriving in response to inflammation. The lipoxins have become identified as critical factors in the resolution of inflammation throughout the body.161 They inhibit eosinophil and neutrophil chemotaxis and adhesion, as well as natural killer cell activation. They are endothelium-dependent vasodilators of both pulmonary and systemic vasculature.
COX catalyzes the cyclization and oxygenation of arachidonic acid, producing prostaglandin PGG2 which is converted to PGH2. There are subtypes of the COX enzyme, most notably COX-1 and COX-2. There has been great interest in COX-2 since its discovery in the 1990s because its inhibition was hoped to be more specific in controlling pain and inflammation without injury to the gastroduodenal mucosa. Although effective, the emergence of a small but real increase in cardiovascular risk of COX-2 inhibitors has tempered their use.162 Complicating this issue further, many of the nonspecific COX inhibitors such as acetaminophen, salicylates, and the nonsteroidal antiinflammatory agents ibuprofen and naproxen show only slightly less COX-2 avidity than some of the newer COX-2–specific inhibitors. Following the production of PGH2, the metabolic pathway divides into branches producing the various bioactive prostanoids; the enzymes of particular interest here are PGD synthase, PGE synthase, prostacyclin synthase, and thromboxane synthase. The final products of these pathways typically have opposed or balancing effects locally and regionally. Prostaglandin E2 (PGE2) and PGI2 are bronchodilators, for example, whereas PGF2α, PGD2, and thromboxane A2 (TXA2) cause bronchoconstriction. Similarly, PGD2, PGE2, PGF2α, and TXA2 are potent vasoconstrictors, whereas PGE1 and PGF2 are vasodilators.
The cytochrome P450 monooxygenase system provides three pathways of arachidonic acid metabolism, which result in epoxyeicosatetraenoic acids (EETs), HETEs, or dihydroxyeicosatetraenoic acids (dHETEs). The HETEs and EETs have been shown experimentally to affect pulmonary vascular and bronchomotor tone. 20-HETE and 5-, 6-, 11-, and 12-EETs all have relaxing effects on both the lung vasculature and airways. They are further known to have general antiinflammatory effects, to modulate reperfusion injury, and to inhibit platelet aggregation. Within the lung, 15-HETE and 20-HETE may both modify hypoxic vasoconstriction.163
References
1. Wojciechowski P, Hurford W. Pharmacology of the airways. In: Slinger P, ed. Principles and Practice of Anesthesia for Thoracic Surgery. New York, NY: Springer; 2011:121–132.
2. Lewis MJ, Short AL, Lewis KE. Autonomic nervous system control of the cardiovascular and respiratory systems in asthma. Respir Med. 2006;100(10):1688–1705.
3. Burwell DR, Jones JG. The airways and anaesthesia—I. Anatomy, physiology and fluid mechanics. Anaesthesia. 1996;51(9):849–857.
4. Canning BJ, Fischer A. Neural regulation of airway smooth muscle tone. Respir Physiol. 2001;125(1–2):113–127.
5. Barnes PJ. Pharmacology of airway smooth muscle. Am J Respir Crit Care Med. 1998;158(5):S123–S132.
6. Lumb AB, Nunn JF. Nunn’s Applied Respiratory Physiology. 6th ed. Philadelphia, PA: Elsevier Butterworth Heinemann; 2005.
7. Johnson M. The beta-adrenoceptor. Am J Respir Crit Care Med. 1998;158(5, pt 3):S146–S153.
8. Widdicombe JG. Autonomic regulation. i-NANC/e-NANC. Am J Respir Crit Care Med. 1998;158(5, pt 3):S171–S175.
9. Drazen JM, Gaston B, Shore SA. Chemical regulation of pulmonary airway tone. Annu Rev Physiol. 1995;57:151–170.
10. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002–1014.
11. Nelson HS. Beta-adrenergic bronchodilators. N Engl J Med. 1995; 333(8):499–506.
12. Johnson M, Butchers PR, Coleman RA, et al. The pharmacology of salmeterol. Life Sci. 1993;52(26):2131–2143.
13. Drazen JM, Israel E, Boushey HA, et al. Comparison of regularly scheduled with as-needed use of albuterol in mild asthma. Asthma Clinical Research Network. N Engl J Med. 1996;335(12):841–847.
14. Israel E, Chinchilli VM, Ford JG, et al. Use of regularly scheduled albuterol treatment in asthma: genotype-stratified, randomised, placebo-controlled cross-over trial. Lancet. 2004;364(9444): 1505–1512.
15. Israel E, Drazen JM, Liggett SB, et al. The effect of polymorphisms of the beta(2)-adrenergic receptor on the response to regular use of albuterol in asthma. Am J Respir Crit Care Med. 2000;162(1): 75–80.
16. National Asthma Education and Prevention Program Coordinating Committee, National Heart, Lung, and Blood Institute, U.S. Department of Health and Human Services. Expert Panel Report 3 (EPR-3): Guidelines for the diagnosis and management of asthma—summary report 2007. http://www.nhlbi.nih.gov/guidelines/asthma/ asthsumm.pdf. Accessed January 7, 2010.
17. Gibson PG, Powell H, Ducharme FM. Differential effects of maintenance long-acting beta-agonist and inhaled corticosteroid on asthma control and asthma exacerbations. J Allergy Clin Immunol. 2007;119(2):344–350.
18. Bengtsson B. Plasma concentration and side-effects of terbutaline. Eur J Respir Dis Suppl. 1984;134:231–235.
19. Teule GJ, Majid PA. Haemodynamic effects of terbutaline in chronic obstructive airways disease. Thorax. 1980;35(7):536–542.
20. Wagner PD, Dantzker DR, Iacovoni VE, et al. Ventilation-perfusion inequality in asymptomatic asthma. Am Rev Respir Dis. 1978;118(3):511–524.
21. Repsher LH, Anderson JA, Bush RK, et al. Assessment of tachyphylaxis following prolonged therapy of asthma with inhaled albuterol aerosol. Chest. 1984;85(1):34–38.
22. Georgopoulos D, Wong D, Anthonisen NR. Tolerance to beta 2-agonists in patients with chronic obstructive pulmonary disease. Chest. 1990;97(2):280–284.
23. Nelson HS, Weiss ST, Bleecker ER, et al. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest. 2006; 129(1):15–26.
24. Williams SJ, Winner SJ, Clark TJ. Comparison of inhaled and intravenous terbutaline in acute severe asthma. Thorax. 1981;36(8): 629–631.
25. Pierce RJ, Payne CR, Williams SJ, et al. Comparison of intravenous and inhaled terbutaline in the treatment of asthma. Chest. 1981;79(5):506–511.
26. Spiteri MA, Millar AB, Pavia D, et al. Subcutaneous adrenaline versus terbutaline in the treatment of acute severe asthma. Thorax. 1988;43(1):19–23.
27. Flynn RA, Glynn DA, Kennedy MP. Anticholinergic treatment in airways diseases. Adv Ther. 2009;26(10):908–919.
28. Karpel JP, Schacter EN, Fanta C, et al. A comparison of ipratropium and albuterol vs albuterol alone for the treatment of acute asthma. Chest. 1996;110(3):611–616.
29. Restrepo RD. A stepwise approach to management of stable COPD with inhaled pharmacotherapy: a review. Respir Care. 2009; 54(8):1058–1081.
30. Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15): 1543–1554.
31. Gal TJ, Suratt PM. Atropine and glycopyrrolate effects on lung mechanics in normal man. Anesth Analg. 1981;60(2):85–90.
32. Gal TJ, Suratt PM, Lu JY. Glycopyrrolate and atropine inhalation: comparative effects on normal airway function. Am Rev Respir Dis. 1984;129(5):871–873.
33. Villetti G, Bergamaschi M, Bassani F, et al. Pharmacological assessment of the duration of action of glycopyrrolate vs tiotropium and ipratropium in guinea-pig and human airways. Br J Pharmacol. 2006;148(3):291–298.
34. Haddad EB, Patel H, Keeling JE, et al. Pharmacological characterization of the muscarinic receptor antagonist, glycopyrrolate, in human and guinea-pig airways. Br J Pharmacol. 1999;127(2):413–420.
35. Tzelepis G, Komanapolli S, Tyler D, et al. Comparison of nebulized glycopyrrolate and metaproterenol in chronic obstructive pulmonary disease. Eur Respir J. 1996;9(1):100–103.
36. Sutherland ER, Martin RJ. Airway inflammation in chronic obstructive pulmonary disease: comparisons with asthma. J Allergy Clin Immunol. 2003;112(5):819–827; quiz 828.
37. Fujimoto K, Kubo K, Yamamoto H, et al. Eosinophilic inflammation in the airway is related to glucocorticoid reversibility in patients with pulmonary emphysema. Chest. 1999;115(3):697–702.
38. Pizzichini E, Pizzichini MM, Gibson P, et al. Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitis. Am J Respir Crit Care Med. 1998;158(5, pt 1): 1511–1517.
39. Chanez P, Bourdin A, Vachier I, et al. Effects of inhaled corticosteroids on pathology in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1(3):184–190.
40. Suissa S, Ernst P, Benayoun S, et al. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med. 2000;343(5):332–336.
41. Donahue JG, Weiss ST, Livingston JM, et al. Inhaled steroids and the risk of hospitalization for asthma. JAMA. 1997;277(11):887–891.
42. Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789.
43. Barnes PJ. Molecular mechanisms of corticosteroids in allergic diseases. Allergy. 2001;56(10):928–936.
44. Pujols L, Mullol J, Torrego A, et al. Glucocorticoid receptors in human airways. Allergy. 2004;59(10):1042–1052.
45. Singh S, Amin AV, Loke YK. Long-term use of inhaled corticosteroids and the risk of pneumonia in chronic obstructive pulmonary disease: a meta-analysis. Arch Intern Med. 2009;169(3):219–229.
46. Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med. 1999;340(25):1941–1947.
47. McEvoy CE, Niewoehner DE. Adverse effects of corticosteroid therapy for COPD. A critical review. Chest. 1997;111(3):732–743.
48. Usery JB, Self TH, Muthiah MP, et al. Potential role of leukotriene modifiers in the treatment of chronic obstructive pulmonary disease. Pharmacotherapy. 2008;28(9):1183–1187.
49. Drazen JM, Israel E, O’Byrne PM. Treatment of asthma with drugs modifying the leukotriene pathway. N Engl J Med. 1999;340(3): 197–206.
50. Reiss TF, Chervinsky P, Dockhorn RJ, et al. Montelukast, a once-daily leukotriene receptor antagonist, in the treatment of chronic asthma: a multicenter, randomized, double-blind trial. Montelukast Clinical Research Study Group. Arch Intern Med. 1998;158(11):1213–1220.
51. Israel E, Rubin P, Kemp JP, et al. The effect of inhibition of 5-lipoxygenase by zileuton in mild-to-moderate asthma. Ann Intern Med. 1993;119(11):1059–1066.
52. Brabson JH, Clifford D, Kerwin E, et al. Efficacy and safety of low-dose fluticasone propionate compared with zafirlukast in patients with persistent asthma. Am J Med. 2002;113(1):15–21.
53. Malmstrom K, Rodriguez-Gomez G, Guerra J, et al. Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma. A randomized, controlled trial. Montelukast/Beclomethasone Study Group. Ann Intern Med. 1999;130(6):487–495.
54. Price DB, Hernandez D, Magyar P, et al. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3): 211–216.
55. Bernstein IL. Cromolyn sodium. Chest. 1985;87(1)(suppl):68S–73S.
56. Guevara JP, Ducharme FM, Keren R, et al. Inhaled corticosteroids versus sodium cromoglycate in children and adults with asthma. Cochrane Database Syst Rev. 2006;(2):CD003558.
57. Barnes PJ. Theophylline: new perspectives for an old drug. Am J Respir Crit Care Med. 2003;167(6):813–818.
58. Global Intiative for Asthma. www.ginasthma.com. Accessed January 7, 2010.
59. Global Initiative for Chronic Obstructive Lung Disease. www.gold copd.com. Accessed January 7, 2010.
60. Aubier M, De Troyer A, Sampson M, et al. Aminophylline improves diaphragmatic contractility. N Engl J Med. 1981;305(5):249–252.
61. Goff MJ, Arain SR, Ficke DJ, et al. Absence of bronchodilation during desflurane anesthesia: a comparison to sevoflurane and thiopental. Anesthesiology. 2000;93(2):404–408.
62. Rooke GA, Choi JH, Bishop MJ. The effect of isoflurane, halothane, sevoflurane, and thiopental/nitrous oxide on respiratory system resistance after tracheal intubation. Anesthesiology. 1997;86(6): 1294–1299.
63. Yamakage M. Direct inhibitory mechanisms of halothane on canine tracheal smooth muscle contraction. Anesthesiology. 1992; 77(3):546–553.
64. Yamakage M, Chen X, Tsujiguchi N, et al. Different inhibitory effects of volatile anesthetics on T- and L-type voltage-dependent Ca2+ channels in porcine tracheal and bronchial smooth muscles. Anesthesiology. 2001;94(4):683–693.
65. Gold MI, Helrich M. Pulmonary mechanics during general anesthesia: V. Status asthmaticus. Anesthesiology. 1970;32(5):422–428.
66. Parnass SM, Feld JM, Chamberlin WH, et al. Status asthmaticus treated with isoflurane and enflurane. Anesth Analg. 1987;66(2): 193–195.
67. Johnston RG, Noseworthy TW, Friesen EG, et al. Isoflurane therapy for status asthmaticus in children and adults. Chest. 1990;97(3): 698–701.
68. Schwartz SH. Treatment of status asthmaticus with halothane. JAMA. 1984;251(20):2688–2689.
69. Cheng EY, Mazzeo AJ, Bosnjak ZJ, et al. Direct relaxant effects of intravenous anesthetics on airway smooth muscle. Anesth Analg. 1996;83(1):162–168.
70. Eames WO, Rooke GA, Wu RS, et al. Comparison of the effects of etomidate, propofol, and thiopental on respiratory resistance after tracheal intubation. Anesthesiology. 1996;84(6):1307–1311.
71. Wanna HT, Gergis SD. Procaine, lidocaine, and ketamine inhibit histamine-induced contracture of guinea pig tracheal muscle in vitro. Anesth Analg. 1978;57(1):25–27.
72. Lin CC, Shyr MH, Tan PP, et al. Mechanisms underlying the inhibitory effect of propofol on the contraction of canine airway smooth muscle. Anesthesiology. 1999;91(3):750–759.
73. Brown RH, Wagner EM. Mechanisms of bronchoprotection by anesthetic induction agents: propofol versus ketamine. Anesthesiology. 1999;90(3):822–828.
74. Brown RH, Greenberg RS, Wagner EM. Efficacy of propofol to prevent bronchoconstriction: effects of preservative. Anesthesiology. 2001;94(5):851–855.
75. Yukioka H, Hayashi M, Terai T, et al. Intravenous lidocaine as a suppressant of coughing during tracheal intubation in elderly patients. Anesth Analg. 1993;77(2):309–312.
76. Hamill JF, Bedford RF, Weaver DC, et al. Lidocaine before endotracheal intubation: intravenous or laryngotracheal? Anesthesiology. 1981;55(5):578–581.
77. Maggiore SM, Richard JC, Abroug F, et al. A multicenter, randomized trial of noninvasive ventilation with helium-oxygen mixture in exacerbations of chronic obstructive lung disease. Crit Care Med. 2010;38(1):145–151.
78. Lordan JL, Holgate ST. H1-antihistamines in asthma. Clin Allergy Immunol. 2002;17:221–248.
79. Richter K, Gronke L, Janicki S, et al. Effect of azelastine, montelukast, and their combination on allergen-induced bronchoconstriction in asthma. Pulm Pharmacol Ther. 2008;21(1):61–66.
80. Rowe BH, Bretzlaff JA, Bourdon C, et al. Magnesium sulfate for treating exacerbations of acute asthma in the emergency department. Cochrane Database Syst Rev. 2000;(2):CD001490.
81. Blitz M, Blitz S, Hughes R, et al. Aerosolized magnesium sulfate for acute asthma: a systematic review. Chest. 2005;128(1):337–344.
82. Reimer C, Granton J. Pharmacology of the pulmonary circulation. In: Slinger P, ed. Principles and Practice of Anesthesia for Thoracic Surgery. New York, NY: Springer, 2011:133–142.
83. Lai HC, Wang KY, Lee WL, et al. Severe pulmonary hypertension complicates postoperative outcome of non-cardiac surgery. Br J Anaesth. 2007;99(2):184–190.
84. Strumpher J, Jacobsohn E. Pulmonary hypertension and right ventricular dysfunction: physiology and perioperative management. J Cardiothorac Vasc Anesth. 2011;25:687–704.
85. Hirota K, Lambert DG. Ketamine: its mechanism(s) of action and unusual clinical uses. Br J Anaesth. 1996;77(4):441–444.
86. Reich DL, Silvay G. Ketamine: an update on the first twenty-five years of clinical experience. Can J Anaesth. 1989;36(2):186–197.
87. Williams GD, Philip BM, Chu LF, et al. Ketamine does not increase pulmonary vascular resistance in children with pulmonary hypertension undergoing sevoflurane anesthesia and spontaneous ventilation. Anesth Analg. 2007;105(6):1578–1584.
88. Oklu E, Bulutcu FS, Yalcin Y, et al. Which anesthetic agent alters the hemodynamic status during pediatric catheterization? Comparison of propofol versus ketamine. J Cardiothorac Vasc Anesth. 2003;17(6):686–690.
89. Heller AR, Litz RJ, Koch T. A fine balance—one-lung ventilation in a patient with Eisenmenger syndrome. Br J Anaesth. 2004; 92(4):587–590.
90. Kopka A, McMenemin IM, Serpell MG, et al. Anaesthesia for cholecystectomy in two non-parturients with Eisenmenger’s syndrome. Acta Anaesthesiol Scand. 2004;48(6):782–786.
91. Kondo U, Kim SO, Nakayama M, et al. Pulmonary vascular effects of propofol at baseline, during elevated vasomotor tone, and in response to sympathetic alpha- and beta-adrenoreceptor activation. Anesthesiology. 2001;94(5):815–823.
92. Kondo U, Kim SO, Murray PA. Propofol selectively attenuates endothelium-dependent pulmonary vasodilation in chronically instrumented dogs. Anesthesiology. 2000;93(2):437–446.
93. Ouedraogo N, Mounkaila B, Crevel H, et al. Effect of propofol and etomidate on normoxic and chronically hypoxic pulmonary artery. BMC Anesthesiol. 2006;6:2.
94. Colvin MP, Savege TM, Newland PE, et al. Cardiorespiratory changes following induction of anaesthesia with etomidate in patients with cardiac disease. Br J Anaesth. 1979;51(6):551–556.
95. Sarkar M, Laussen PC, Zurakowski D, et al. Hemodynamic responses to etomidate on induction of anesthesia in pediatric patients. Anesth Analg. 2005;101(3):645–650.
96. Kaye AD, Hoover JM, Kaye AJ, et al. Morphine, opioids, and the feline pulmonary vascular bed. Acta Anaesthesiol Scand. 2008;52(7):931–937.
97. Kerbaul F, Bellezza M, Mekkaoui C, et al. Sevoflurane alters right ventricular performance but not pulmonary vascular resistance in acutely instrumented anesthetized pigs. J Cardiothorac Vasc Anesth. 2006;20(2):209–216.
98. Schulte-Sasse U, Hess W, Tarnow J. Pulmonary vascular responses to nitrous oxide in patients with normal and high pulmonary vascular resistance. Anesthesiology. 1982;57(1):9–13.
99. Konstadt SN, Reich DL, Thys DM. Nitrous oxide does not exacerbate pulmonary hypertension or ventricular dysfunction in patients with mitral valvular disease. Can J Anaesth. 1990;37(6): 613–617.
100. Hemmerling TM, Russo G, Bracco D. Neuromuscular blockade in cardiac surgery: an update for clinicians. Ann Card Anaesth. 2008;11(2):80–90.
101. McCoy EP, Maddieneri VR, Elliott P, et al. Hemodynamic effects of rocuronium during fentanyl anesthesia. Can J Anesth. 1993; 40:703–708.
102. al-Halees Z, Afrane B, el-Barbary M. Magnesium sulfate to facilitate weaning of nitric oxide in pulmonary hypertension. Ann Thorac Surg. 1997;63(1):298–299.
103. Haas NA, Kemke J, Schulze-Neick I, et al. Effect of increasing doses of magnesium in experimental pulmonary hypertension after acute pulmonary embolism. Intensive Care Med. 2004; 30(11):2102–2109.
104. Houfflin Debarge V, Sicot B, Jaillard S, et al. The mechanisms of pain-induced pulmonary vasoconstriction: an experimental study in fetal lambs. Anesth Analg. 2007;104(4):799–806.
105. Rex S, Missant C, Segers P, at al. Thoracic epidural anesthesia impairs the hemodynamic response to acute pulmonary hypertension by deteriorating right ventricular-pulmonary arterial coupling. Crit Care Med. 2007;35(1):222–229.
106. Armstrong P. Thoracic epidural anaesthesia and primary pulmonary hypertension. Anaesthesia. 1992;47(6):496–499.
107. Barnes PJ, Liu SF. Regulation of pulmonary vascular tone. Pharmacol Rev. 1995;47(1):87–131.
108. Pearl RG, Maze M, Rosenthal MH. Pulmonary and systemic hemodynamic effects of central venous and left atrial sympathomimetic drug administration in the dog. J Cardiothorac Anesth. 1987;1(1):29–35.
109. Roscher R, Ingemansson R, Algotsson L, et al. Effects of dopamine in lung-transplanted pigs at 32 degrees C. Acta Anaesthesiol Scand. 1999;43(7):715–721.
110. Kwak YL, Lee CS, Park YH, et al. The effect of phenylephrine and norepinephrine in patients with chronic pulmonary hypertension*. Anaesthesia. 2002;57(1):9–14.
111. Vlahakes GJ, Turley K, Hoffman JI. The pathophysiology of failure in acute right ventricular hypertension: hemodynamic and biochemical correlations. Circulation. 1981;63(1):87–95.
112. Jin HK, Yang RH, Chen YF, et al. Hemodynamic effects of arginine vasopressin in rats adapted to chronic hypoxia. J Appl Physiol. 1989;66(1):151–160.
113. Leather HA, Segers P, Berends N, et al. Effects of vasopressin on right ventricular function in an experimental model of acute pulmonary hypertension. Crit Care Med. 2002;30(11):2548–2552.
114. Tayama E, Ueda T, Shojima T, et al. Arginine vasopressin is an ideal drug after cardiac surgery for the management of low systemic vascular resistant hypotension concomitant with pulmonary hypertension. Interact Cardiovasc Thorac Surg. 2007;6(6):715–719.
115. Price LC, Forrest P, Sodhi V, et al. Use of vasopressin after Caesarean section in idiopathic pulmonary arterial hypertension. Br J Anaesth. 2007;99(4):552–555.
116. Roberts JD Jr, Fineman JR, Morin FC III, et al. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N Engl J Med. 1997;336(9): 605–610.
117. Mosquera I, Crespo-Leiro MG, Tabuyo T, et al. Pulmonary hypertension and right ventricular failure after heart transplantation: usefulness of nitric oxide. Transplant Proc. 2002;34(1): 166–167.
118. Meade MO, Granton JT, Matte-Martyn A, et al. A randomized trial of inhaled nitric oxide to prevent ischemia-reperfusion injury after lung transplantation. Am J Respir Crit Care Med. 2003; 167(11):1483–1489.
119. Rocca GD, Coccia C, Pompei L, et al. Hemodynamic and oxygenation changes of combined therapy with inhaled nitric oxide and inhaled aerosolized prostacyclin. J Cardiothorac Vasc Anesth. 2001;15(2):224–227.
120. Vane JR, Botting RM. Pharmacodynamic profile of prostacyclin. Am J Cardiol. 1995;75(3):3A–10A.
121. Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–329.
122. Khan TA, Schnickel G, Ross D, et al. A prospective, randomized, crossover pilot study of inhaled nitric oxide versus inhaled prostacyclin in heart transplant and lung transplant recipients. J Thorac Cardiovasc Surg. 2009;138(6):1417–1424.
123. Jerath A, Srinivas C, Vegas A, et al. The successful management of severe protamine-induced pulmonary hypertension using inhaled prostacyclin. Anesth Analg. 2010;110:365–369.
124. Chen TL, Lee YT, Wang MJ, et al. Endothelin-1 concentrations and optimization of arterial oxygenation and venous admixture by selective pulmonary artery infusion of prostaglandin E1 during thoracotomy. Anaesthesia. 1996;51:422–426.
125. Bund M, Henzler D, Walz R, et al. Aerosolized and intravenous prostacyclin during one-lung ventilation. Hemodynamic and pulmonary effects [in German]. Anaesthesist. 2004;53(7):612–620.
126. Haraldsson A, Kieler-Jensen N, Wadenvik H, et al. Inhaled prostacyclin and platelet function after cardiac surgery and cardiopulmonary bypass. Intensive Care Med. 2000;26(2):188–194.
127. Hill LL, De Wet CJ, Jacobsohn E, et al. Peripartum substitution of inhaled for intravenous prostacyclin in a patient with primary pulmonary hypertension. Anesthesiology. 2004;100(6):1603–1605.
128. Urdaneta F, Lobato EB, Beaver T, et al. Treating pulmonary hypertension post cardiopulmonary bypass in pigs: milrinone vs. sildenafil analog. Perfusion. 2008;23(2):117–125.
129. Lakshminrusimha S, Porta NF, Farrow KN, et al. Milrinone enhances relaxation to prostacyclin and iloprost in pulmonary arteries isolated from lambs with persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2009;10(1):106–112.
130. Atz AM, Wessel DL. Sildenafil ameliorates effects of inhaled nitric oxide withdrawal. Anesthesiology. 1999;91(1):307–310.
131. Dias-Junior CA, Vieira TF, Moreno H Jr, et al. Sildenafil selectively inhibits acute pulmonary embolism-induced pulmonary hypertension. Pulm Pharmacol Ther. 2005;18(3):181–186.
132. Lohser J. Evidence-based management of one-lung ventilation. Anesth Clinics. 2008;26:241–272.
133. Wang JY, Russel GN, Page RD, et al. A comparison of the effects of sevoflurane and isoflurane on arterial oxygenation during one-lung anesthesia. Br J Anesth. 2000;81:850–853.
134. Wang JY, Russel GN, Page RD, et al. A comparison of the effects of desflurane and isoflurane on arterial oxygenation during one-lung anesthesia. Anaesthesia. 2000;55:167–173.
135. Benumof J. Isoflurane anesthesia and arterial oxygenation during one-lung ventilation. Anesthesiology. 1986;64:419–422.
136. Reid CW, Slinger PD, Lewis S. Comparison of the effects of propofol-alfentanil versus isoflurane anesthesia on arterial oxygenation during one-lung anesthesia. J Cardiothorac Vasc Anesth. 1997;10:860–863.
137. Brimioulle S, Vachiery J-L, Brichant J-F, et al. Sympathetic modulation of hypoxic pulmonary vasoconstriction in intact dogs. Cardiovasc Res. 1997;34:384–392.
138. Yeazell L, Littlewood K. Nonrespiratory functions of the lungs. In: Slinger P, ed. Principles and Practice of Anesthesia for Thoracic Surgery. New York, NY: Springer; 2011:103–120.
139. Parat M, Kwang WJ. The biology of caveolae: achievements and perspectives. Int Rev Cell Mol Biol. 2009;273:117–162.
140. Upton RN, Doolette DJ. Kinetic aspects of drug disposition in the lungs. Clin Exp Pharmacol Physiol. 1999;26(5–6):381–391.
141. Pacifici GM, Franchi M, Bencini C, et al. Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica. 1988;18(7): 849–856.
142. Waters CM, Krejcie TC, Avram MJ. Facilitated uptake of fentanyl, but not alfentanil, by human pulmonary endothelial cells. Anesthesiology. 2000;93(3):825–831.
143. Krejcie TC, Avram MJ, Gentry WB, et al. A recirculatory model of the pulmonary uptake and pharmacokinetics of lidocaine based on analysis of arterial and mixed venous data from dogs. J Pharmacokinet Biopharm. 1997;25(2):169–190.
144. Sharrock NE, Mather LE, Go G, et al. Arterial and pulmonary arterial concentrations of the enantiomers of bupivacaine after epidural injection in elderly patients. Anesth Analg. 1998;86(4):812–817.
145. Mather LE, Copeland SE, Ladd LA. Acute toxicity of local anesthetics: underlying pharmacokinetic and pharmacodynamic concepts.[see comment]. Reg Anesth Pain Med. 2005;30(6):553–566.
146. Marwick PC, Levin AI, Coetzee AR. Recurrence of cardiotoxicity after lipid rescue from bupivacaine-induced cardiac arrest. Anesth Analg. 2009;108(4):1344–1346.
147. Roerig DL, Kotrly KJ, Dawson CA, et al. First-pass uptake of verapamil, diazepam, and thiopental in the human lung. Anesth Analg. 1989;69(4):461–466.
148. Henthorn TK, Krejcie TC, Niemann CU, et al. Ketamine distribution described by a recirculatory pharmacokinetic model is not stereoselective. Anesthesiology. 1999;91(6):1733–1743.
149. Upton RN, Ludbrook G. A physiologically based, recirculatory model of the kinetics and dynamics of propofol in man. Anesthesiology. 2005;103(2):344–352.
150. Orfanos SE, Langleben D, Khoury J, et al. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in humans. Circulation. 1999;99(12):1593–1599.
151. Muntner P, Krousel-Wood M, Hyre AD, et al. Antihypertensive prescriptions for newly treated patients before and after the main antihypertensive and lipid-lowering treatment to prevent heart attack trial results and seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure guidelines. Hypertension. 2009;53(4):617–623.
152. Skidgel RA. Bradykinin-degrading enzymes: structure, function, distribution, and potential roles in cardiovascular pharmacology. J Cardiovasc Pharmacol. 1992;20(suppl 9):S4–S9.
153. Skidgel RA, Erdos EG. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides. 2004;25(3):521–525.
154. Suguikawa TR, Garcia CA, Martinez EZ, et al. Cough and dyspnea during bronchoconstriction: comparison of different stimuli. Cough. 2009;5:6.
155. Cook DR, Brandom BW. Enflurane, halothane, and isoflurane inhibit removal of 5-hydroxytryptamine from the pulmonary circulation. Anesth Analg. 1982;61(8):671–675.
156. Shah PM, Raney AA. Tricuspid valve disease. Curr Probl Cardiol. 2008;33(2):47–84.
157. Mizuguchi KA, Fox AA, Burch TM, et al. Tricuspid and mitral valve carcinoid disease in the setting of a patent foramen ovale. Anesth Analg. 2008;107(6):1819–1821.
158. Stratmann G, Gregory GA. Neurogenic and humoral vasoconstriction in acute pulmonary thromboembolism [see comment]. Anesth Analg. 2003;97(2):341–354.
159. Philpot RM, Andersson TB, Eling TE. Uptake, accumulation, and metabolism of chemicals by the lung. In: Bakhle YS, Vane JR, eds. Metabolic Functions of the Lung. New York, NY: Marcel Dekker; 1977:123–171.
160. Haeggstrom JZ, Kull F, Rudberg PC, et al. Leukotriene A4 hydrolase. Prostaglandins Other Lipid Mediat. 2002;68–69:495–510.
161. Romano M, Recchia I, Recchiuti A. Lipoxin receptors. ScientificWorldJournal. 2007;7:1393–1412.
162. Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol. 2007;50(5):470–479.
163. Jacobs ER, Zeldin DC. The lung HETEs (and EETs) up. Am J Physiol Heart Circ Physiol. 2001;280(1):H1–H10.