Ilana Cass
Beth Y. Karlan
Abnormalities of the ovary or fallopian tubes may result from physiologic changes, infectious processes, or benign or malignant neoplasms. Patients may exhibit a pelvic mass with or without other signs or symptoms. These disorders may occur at any age from childhood to senescence. Depending on patient age, the physiologic and pathologic manifestations and implications may differ. For example, teenage patients with a pelvic mass most likely will have a benign germ cell or epithelial ovarian tumor, whereas postmenopausal women have a significantly greater risk of epithelial ovarian carcinoma. The differential diagnosis and the radiologic and tumor marker workups will vary, depending on patient age and the initial complaints. The clinician must be vigilant and willing to evaluate the vague symptoms that are frequently the hallmark of many ovarian and tubal pathologies, especially in women over 40.
In recent years, we have gained an increased understanding of ovarian and fallopian tube tumor biology and molecular genetics, but these advances can be translated only slowly to the clinic. This chapter will outline ovarian and fallopian tube neoplasms, including the common epithelial histologies as well as the rarer germ cell and stromal tumors. Epidemiology and risk factors for these cancers, including inherited cancer susceptibility due to mutations in BRCA1 and BRCA2 genes will be discussed, as well as preventive and screening strategies.
GERM CELL TUMORS OF THE OVARY
Germ cell tumors occur most frequently in young women and girls and account for 90% of prepubertal ovarian tumors and 60% of ovarian tumors in women younger than 20 years. These tumors arise from the germ cells originating in the embryonic yolk sac. Patients usually complain of abdominal pain and have an associated pelvic or abdominal mass. Others might have acute abdominal pain as a result of ovarian rupture, hemorrhage, or torsion. A misdiagnosis of acute appendicitis has been made in these circumstances.
Mature Cystic Teratoma
Mature cystic teratomas, also known as dermoid cysts, are the most common benign ovarian neoplasm, with a peak incidence from ages 20 to 40 years. However, they also can be seen in infancy, as well as in menopausal woman. Mature cystic teratomas originate from primordial germ cells and are composed of well-differentiated derivatives of any combination of the three germ layers: ectoderm, mesoderm, endoderm. Ectodermal elements usually predominate. Although these mature tissues are benign in the vast majority of cases, on rare occasion they may undergo malignant transformation, with squamous cell carcinoma being the most frequent malignant histology.
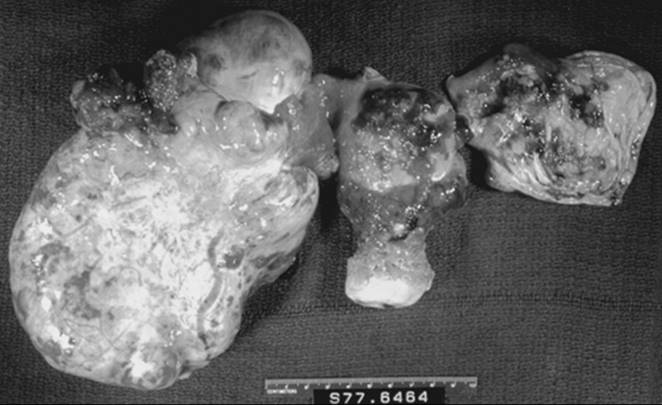
Grossly, the tumors are round or oval with a smooth, glistening, gray-white surface. Most tumors measure 5 to 10 cm in diameter and are bilateral in 8% to 15% of cases. The tumors are usually unilocular but occasionally multilocular, and often are filled with hair and a fatty material similar to sebum (Fig. 55.1). A solid portion located at one pole of the cyst that often projects into the cavity is known as a Rokitansky protuberance, where all three germ cell layers are found. Tissues also can be found at the protuberance, including hair, teeth, bone, glia, neural tissue, cartilage, retina, smooth muscle, fibrous and fatty tissue, gastrointestinal and bronchial mucosa, and thyroid and salivary gland tissue.
Microscopically, ovarian stroma is noted outside the cyst wall, with the cyst lined predominantly by squamous epithelium with underlying sebaceous and sweat glands (Fig. 55.2).
|
|
|
FIG. 55.1. Mature cystic teratoma. This mature cystic teratoma contains hair and teeth. |
|
|
|
FIG. 55.2. Mature cystic teratoma. Microscopically, there is a keratinizing squamous epithelium on the right and mature glial tissue on the left. |
In the past, benign cystic teratomas often were diagnosed on an abdominal radiograph in a young woman with acute abdominal pain, because osseous differentiation or teeth could be seen in the pelvis. Transvaginal sonography, however, has become the diagnostic tool of choice in most centers, although pelvic magnetic resonance imaging (MRI) can be helpful when the diagnosis is uncertain and may be especially useful when the mass is detected during pregnancy. Torsion is a frequent complication, because these are relatively buoyant and mobile tumors. Treatment usually involves ovarian cystectomy, with preservation of normal ovarian tissue and close inspection of the contralateral ovary. This often can be accomplished laparoscopically, when the dermoid cyst can be shelled out from the remaining normal ovary. Although concern for chemical peritonitis resulting from cyst rupture exists, with copious irrigation of the peritoneal cavity this complication can be avoided.
Struma Ovarii and Strumal Carcinoid
Rarely, ovarian teratomas will contain a single specialized tissue type. Struma ovarii is a subset of mature cystic teratomas in which the tumor is composed entirely or predominantly of thyroid tissue. Grossly, the cut surface shows gelatinous red to green-brown colloid, and microscopically mature thyroid tissue is seen (Fig. 55.3). Struma ovarii accounts for less than 3% of mature teratomas, with similar age distribution and similar clinical findings. However, thyroid gland enlargement is seen occasionally, and about 5% of patients with struma ovarii will experience signs and symptoms of thyrotoxicosis. Malignant changes in a struma ovarii are extremely rare, but can occur, and are most frequently of a follicular histology (unlike primary thyroid carcinomas). Approximately 30% of reported cases of malignant struma ovarii are associated with metastatic disease. Evaluation with iodine-131 scanning may be helpful, and treatment is often similar to that given for their thyroid carcinoma counterparts.
|
|
|
FIG. 55.3. Struma ovarii. Microscopically, the tumor is composed of normal thyroid tissue. |
Strumal carcinoids are rare teratomas characterized by a mixture of thyroid and trabecular carcinoid tissues. Primary carcinoid tumors of the ovary account for less than 5% of ovarian teratomas and most likely arise from argentaffin cells found in the gastrointestinal or bronchial tissues that make up the teratoma. These tumors often are hormonally active due to the secretion of serotonin, bradykinin, and other peptide hormones that are secreted directly into the systemic circulation and are not inactivated by a “first pass” through the liver. Approximately 50% of ovarian carcinoids over 4 cm in diameter will be associated with the carcinoid syndrome, including episodic cutaneous flushing, diarrhea, and bronchospasm.
Malignant Germ Cell Tumors
Malignant ovarian germ cell tumors account for less than 5% of all ovarian cancers. Table 55.1 provides a listing of the World Health Organization classification of germ cell tumors of the ovary. Their male counterpart, testicular cancer, is approximately 10 times more common than ovarian germ cell tumors, and many of the chemotherapy regimens that are used for treating ovarian germ cell tumors have been adapted from clinical trials in patients with the corresponding type of testicular cancer. These interesting tumors tend to be diagnosed at an earlier stage than the common epithelial ovarian carcinomas, and they are very sensitive to chemotherapy and, in the case of dysgerminoma, to radiotherapy, as well. Germ cell tumors have distinct histopathologic characteristics and tumor markers that are useful in diagnosis and follow-up. Table 55.2 outlines the serum tumor markers typically associated with germ cell tumors.
|
|
|
TABLE 55.1. World Health Organization classification of germ cell tumors |
|
|
|
TABLE 55.2. Tumor markers associated with germ cell neoplasms: likelihood of elevation |
Dysgerminoma
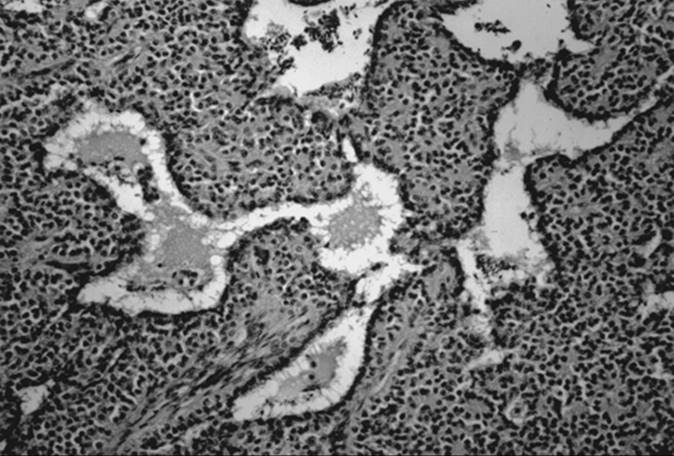
Dysgerminoma is the most common malignant germ cell tumor of the ovary, accounting for 2% of all ovarian malignancies. Approximately 50% of the patients with this tumor are younger than 20 years, and 80% are younger than 30 years. Children with dysgerminoma may exhibit precocious puberty or primary amenorrhea. The serum lactate dehydrogenase level frequently is elevated and may serve as a tumor marker during treatment and follow-up.
Although dysgerminomas tend to be unilateral, approximately 10% to 15% of these tumors are bilateral. They are usually gray-white, smooth, and fleshy in appearance, and on cut surface they are usually solid. Hemorrhage and necrosis may be seen. Cystic areas suggest mixed germ cell elements that would require further careful sampling of the specimen. The microscopic appearance of an ovarian dysgerminoma is similar in histology to its male counterpart, testicular seminoma, with aggregates of tumor cells surrounded by connective tissue stroma containing many lymphocytes and foreign body giant cells (Fig. 55.4). The tumor cells are usually large, with clear or lightly granular cytoplasm containing abundant glycogen. Nuclei are vesicular and large, occupying one half of the cells and containing one or more nucleoli. Mitotic activity is almost always present. In less than 10% of patients, syncytiotrophoblastic giant cells may be detected, producing human choriogonadotropin (hCG) that may be demonstrated in tissue section by immunohistochemical techniques and in the serum by a modestly elevated hCG level. Syncytiotrophoblastic cells in a dysgerminoma do not have prognostic significance; however, serum β-hCG levels can be used to monitor patients' responses to treatment and during follow-up.
|
|
|
FIG. 55.4. Dysgerminoma. Loose aggregates of tumor cells are separated by connective tissue containing abundant lymphocytes. |
Treatment is determined by patient age. Because most patients with dysgerminoma are of reproductive age, unilateral salpingo-oophorectomy and a full staging procedure with preservation of the contralateral ovary and the uterus are recommended, as long as there is no evidence of disease in the contralateral ovary. Table 55.3 is a listing of procedures required when a full staging procedure is performed. Table 55.4 illustrates the widely used International Federation of Gynecology and Obstetrics (FIGO) staging system for all ovarian cancers. Observational studies have revealed identical remission rates for conservative surgical treatment (e.g., unilateral salpingo-oophorectomy and staging) and complete extirpative surgery (e.g., bilateral salpingo-oophorectomy with or without a hysterectomy) in early-stage disease. Routine biopsy of the contralateral ovary is unnecessary if it is normal in appearance. If gross tumor is noted in both ovaries, a unilateral salpingo-oophorectomy of the larger ovary, a contralateral ovarian cystectomy, and a full staging procedure followed by chemotherapy may be appropriate in those patients who desire to retain fertility. Patients with advanced disease may require complete removal of the reproductive organs, which should be performed in consultation with a gynecologic oncologist.
|
|
|
TABLE 55.3. Complete staging procedure for ovarian malignancy |
|
|
|
TABLE 55.4. FIGO staging for ovarian cancer |
Dysgerminomas are very sensitive to both radiation and chemotherapy. Because they are found most frequently in young women, chemotherapy is the most appropriate postoperative treatment in these patients and should be offered to those with disease more advanced than stage Ia. Combination therapy with (a) vincristine sulfate (Oncovin), dactinomycin (Cosmegen), and cyclophosphamide (Cytoxan) (VAC) or (b) vinblastine sulfate (Velban), bleomycin sulfate (Blenoxane), and cisplatin (Platinol) (known as VBP) has been used with excellent results, especially in patients with stage I disease. The combination of bleomycin, etoposide, and cisplatin (BEP) has been even more effective in treating all stages of disease, with a sustained remission in essentially 100% of patients. The M.D. Anderson Cancer Center group and the Gynecologic Oncology Group (GOG) have used BEP in their treatment of dysgerminoma in all stages of disease and have reported a prolonged disease-free interval with three to six cycles of therapy, depending on disease status, residual disease, and tumor markers. In an effort to explore regimens with lower toxicity, the GOG reported a regimen of carboplatin and etoposide for completely staged and resected stage Ib to III dysgerminomas. Despite favorable results, BEP remains the recommended first line based on descriptive studies (type III evidence). In patients with pure dysgerminomas seemingly confined to the ovary (women who were staged inadequately), administration of three cycles of adjuvant BEP is recommended due to a recurrence rate of 20%. However, the scientific merit of this treatment plan requires further investigation. Another option is to offer a repeat surgery to fully stage the disease. There have been several reports of secondary neoplasm, especially hematologic malignancies, after the use of BEP and other etoposide-based regimens. Although these are rare occurrences, careful long-term follow-up and evaluation of risk–benefit ratio may be warranted to assess the efficacy of this regimen with and without etoposide.
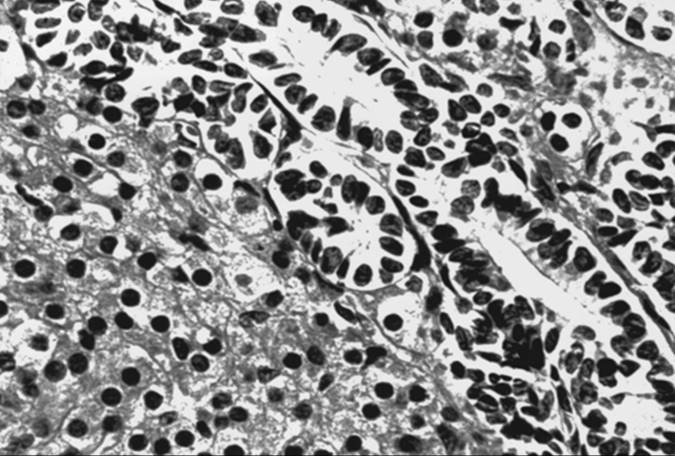
Endodermal Sinus Tumor
Endodermal sinus tumor, or yolk sac tumor, is the second most common germ cell tumor, representing 1% of all ovarian malignancies. It may be pure or part of a malignant mixed germ cell tumor. The reported age distribution ranges from 16 months to 46 years, but most patients are younger than 30 years. The serum α-fetoprotein (AFP) level frequently is elevated in these patients (see Table 55.2), making it a useful diagnostic test in the initial workup, in the assessment of response to therapy, and in follow-up for recurrence. Symptoms are typical of those observed with other germ cell tumors. Several cases have occurred during pregnancy. No endocrine manifestations have been seen with the pure form of endodermal sinus tumor. More than 70% of patients with endodermal sinus tumors are diagnosed in stage I, although they are biologically virulent.
Grossly, tumors are usually gray-yellow, large, and solid, ranging from 3 to 30 cm in diameter. Bilateral involvement has been noted only in patients with metastatic spread to other organs. Foci of hemorrhage, necrosis, and gelatinous changes are present. Microscopically, endodermal sinus tumors display a wide range of histologic patterns. The microcystic pattern is characterized by a loose network of channels and spaces forming a honeycomb lined by flat pleomorphic mesothelial-like cells with large hyperchromatic or vesicular nuclei. Hyaline globules or droplets, which are positive with periodic acid–Schiff stain, are found commonly. The endodermal sinus pattern is characterized by perivascular formations called Schiller-Duval bodies (Fig. 55.5). Other patterns include the alveolar–glandular pattern, composed of alveolar, gland-like, or cystic spaces lined by flat or cuboidal epithelium; the polyvesicular vitelline pattern, in which numerous small vesicles are surrounded by connective tissue; and the solid pattern, consisting of aggregates of small pleomorphic undifferentiated cells. These histologic patterns do not have prognostic significance.
|
|
|
FIG. 55.5. Yolk sac tumor. A characteristic finding in the endodermal sinus pattern is the Schiller-Duval body. |
In the past, patients with endodermal sinus tumors, even those with early-stage tumors, did poorly in spite of aggressive surgical and radiation treatments. With adjuvant chemotherapy after conservative pelvic surgery and staging laparotomy (see Table 55.3), there has been a marked improvement in the prognosis of patients with this malignancy. Adjuvant chemotherapeutic regimens for all nondysgerminomatous germ cell tumors (including endodermal sinus tumors, immature cystic teratoma, embryonal cell tumors, and others) are similar, because they are grouped together in treatment protocols in several major studies. The VAC regimen, first introduced in the 1970s, resulted in a successful cure rate of more than 80% in patients with stage I disease but less than 50% in patients with more advanced stages, as reported by a GOG study and the M.D. Anderson group experience. The VBP regimen has shown superior results, compared with VAC, but toxicity is increased. However, no randomized clinical trials have been performed to compare the two regimens due to the rarity of these malignancies. More recently, the BEP regimen has shown an excellent response rate of over 95% in patients with local or advanced disease and has become the primary therapeutic regimen. Further studies, however, are needed to explore alternative regimens to reduce the rate of toxicity while maintaining the same efficacy. Second-look laparotomy should not be a part of posttherapy surveillance for this tumor or any other germ cell tumors. Salvage therapies, such as surgery followed by chemotherapy, in patients who have failed primary chemotherapy have shown some success; however, they are anecdotal at best due to the limited number of cases.
Immature Cystic Teratoma
Like mature cystic teratoma, immature cystic teratoma is composed of tissues derived from all three germinal layers, except that they also contain embryonic tissue. This group of tumors is the third most common malignant germ cell tumor, representing about 25% of all such tumors in patients younger than 20 years. Unlike mature cystic teratoma, which occurs in all ages but more frequently during the reproductive years, immature cystic teratoma essentially is found during the first two decades of life. It usually grows rapidly through its capsule, forming adhesions to the surrounding structures, and implants in the peritoneal cavity.
Tumors are usually smooth and unilateral, ranging from 9 to 28 cm. A mature cystic teratoma may be present in the other ovary. Tumors are predominantly solid, with some cystic areas filled with serous or mucinous fluid or fatty material (Fig. 55.6). The cut surface is soft and usually gray to pink to brown. Microscopically, the most common immature tissue present is neural, derived from the ectoderm (Fig. 55.7). A histologic grading system was, therefore, proposed based on the relative amount of mature and immature neuroepithelial tissues, mitotic activity, and degree of differentiation:
|
|
|
FIG. 55.6. Immature teratoma. This bulky neoplasm, removed from a 20-year-old woman, weighed 1,800 g. The neuroectodermal elements appeared opaque and friable (arrow). |
|
|
|
FIG. 55.7. Immature teratoma. These tumors are graded based on the presence and extent of neuroepithelial elements. Note the rosettes. |
· Grade 0-Mature tissue only
· Grade 1-Limited immature neuroepithelial tissue and mitotic activity
· Grade 2-Moderate amount of immature tissue and mitotic activity
· Grade 3-Large quantities of immature tissue and mitotic activity
Prognosis correlates with histologic grade of tumors, because the presence of immature elements worsens the prognosis. In patients with stage Ia grade 1 disease, surgery alone, consisting of an exploratory laparotomy, unilateral salpingo-oophorectomy, and a complete staging procedure (see Table 55.3), is sufficient. For more advanced disease and high-grade tumors, postoperative adjuvant chemotherapy is necessary. Depending on disease extent, preservation of the contralateral ovary and the uterus is usually feasible. Serum AFP levels may be elevated (see Table 55.2). Chemotherapy is discussed in detail above.
Embryonal Carcinoma
Embryonal carcinoma is rare, accounting for less than 5% of all germ cell tumors. It usually occurs in children, with a median age of 15 years. Like its testicular counterpart, embryonal carcinoma is a highly malignant neoplasm. Because it often is seen as part of mixed germ cell tumors, serum AFP and hCG levels often are elevated. Clinically, it may be associated with precocious puberty, as well as abnormal vaginal bleeding in adults.
The gross pathologic picture is variable, but the tumor is usually large and soft, with a cut surface that is solid and gray-white with areas of hemorrhage and necrosis. Microscopically, embryonal carcinoma consists of primitive, undifferentiated sheets of variably sized epithelial cells (Fig. 55.8). Nuclei are vesicular, and mitoses are frequent. Syncytiotrophoblastic giant cells frequently are seen in the stroma or directly adjacent to clusters of embryonal carcinoma cells.
|
|
|
FIG. 55.8. Embryonal carcinoma. Tumor cells form syncytial aggregates that surround cleft-like spaces. |
Polyembryoma
Polyembryoma is a rare germ cell tumor, characterized by numerous embryoid bodies that morphologically resemble normal presomite embryos. It is usually part of a mixed germ cell neoplasm and causes similar symptoms. The median age of patients with polyembryoma is 15 years. The tumor is usually unilateral, ranging from 10 cm to a mass that fills the entire abdominal cavity; cut section reveals mostly solid areas with hemorrhage and necrosis. Microscopically, embryonic bodies of varying degrees of differentiation are noted, including an embryonic disk, amniotic cavity, yolk sac, and extraembryonic mesenchyme (Fig. 55.9). Syncytiotrophoblastic cells have been detected, also. AFP, hCG, and sometimes human placental lactogen can be demonstrated in the serum and in cells by immunohistochemical staining.
|
|
|
FIG. 55.9. Polyembryoma. In less differentiated forms, embryoid bodies may have a bizarre appearance. |
Choriocarcinoma
Choriocarcinoma is a rare germ cell tumor of either pure or mixed form. Pure choriocarcinoma usually is found in prepubertal children. Isosexual precocious puberty is a common clinical finding in premenarcheal patients. In postmenarcheal patients, the presence of other germ cell components is helpful in distinguishing ovarian germ cell tumors from a gestation choriocarcinoma. A diagnosis of ectopic pregnancy often is entertained in postmenarcheal patients due to the shared signs and symptoms. The gross appearance of choriocarcinoma depends on the composition of the germ cell elements. The tumor is usually large, unilateral, and solid, with areas of necrosis and hemorrhage. Microscopically, both cytotrophoblasts and syncytiotrophoblasts are present (Fig. 55.10). Treatment is as described previously for the other germ cell tumors.
|
|
|
FIG. 55.10. Choriocarcinoma. There is a dimorphic population of malignant cytotrophoblasts and syncytiotrophoblasts. Associated hemorrhage is a common finding. |
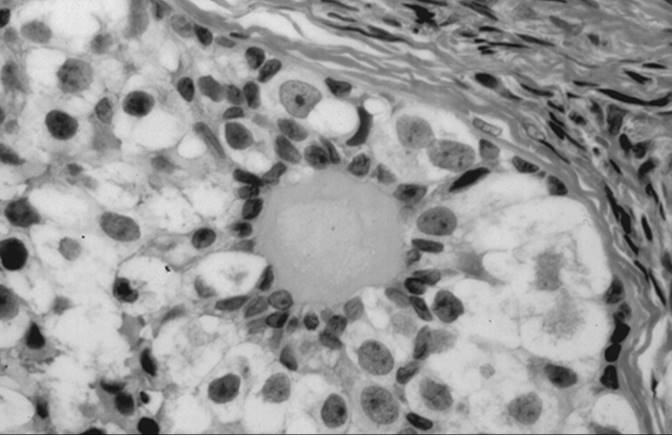
Gonadoblastoma
Gonadoblastoma almost always arises in a congenitally abnormal gonad, with associated sexual maldevelopment. Patients with gonadoblastoma most often have pure or mixed gonadal dysgenesis or are male pseudohermaphrodites.
Predominantly, karyotypes 46,XY, 45,X/46,XY mosaicism and, rarely, 46,XX or 45,X have been associated with this tumor. Gonadoblastoma is much more common in phenotypic females than in phenotypic males, with a ratio of 4:1. It frequently is associated with dysgerminoma and occasionally with other germ cell neoplasms, including yolk sac tumor, embryonal carcinoma, and choriocarcinoma.
Patients with gonadoblastoma usually complain of primary amenorrhea, virilization, or developmental abnormalities of the genitalia. A karyotype should be included in the evaluation of the young women with a pelvic mass and any symptoms of abnormal sexual development. It is during the workup of these conditions that gonadoblastoma is usually diagnosed. It is detected most frequently during the second decade. Hot flushes and other menopausal symptoms have been noted after tumor excision, suggesting estrogen-secreting cells are in these tumors. However, the exact source of the androgen or estrogen production is unknown, because the steroid production was noted in the absence of Leydig or lutein cells.
Gonadoblastoma is more common in the right gonad than in the left and is bilateral in 38% of patients. The tumor can range from a microscopic lesion to a mass, measuring up to 8 cm in diameter, that is soft and fleshy to firm and hard, depending on the degree of calcification. When mixed with other malignant germ cell elements, it can grow even larger. Microscopically, gonadoblastoma is composed of cellular nests containing a mixture of germ cells and immature sex cord–stromal cells, such as Sertoli and granulosa cells (Fig. 55.11). Hyaline bodies and calcification are often present in the nests. In 50% of patients, a supervening dysgerminoma displaces most of the gonadoblastoma, pushing the cell nests to the periphery.
|
|
|
FIG. 55.11. Gonadoblastoma. Large germ cells are admixed with smaller sex cord derivatives that surround rounded hyaline material resembling a Call-Exner body. |
Treatment for patients with gonadal dysgenesis is a bilateral gonadectomy, because they have an increased risk for a germ cell tumor, especially gonadoblastoma. If the gonads are not removed and tumors develop, the prognosis of patients with pure gonadoblastoma is excellent, as long as the tumor and the other ovary are both removed. The prognosis in patients with gonadoblastoma associated with dysgerminomatous elements remains very good, even with metastasis. If the gonadoblastoma is associated with other germ cell tumors, such as endodermal sinus tumor, embryonal carcinoma, or choriocarcinoma, the prognosis is poor if untreated. However, combination chemotherapy (BEP) results in significant improvement, as described previously.
SEX CORD–STROMAL TUMORS OF THE OVARY
Sex cord–stromal tumor accounts for approximately 8% of all ovarian tumors. The origin of tumor cells may be the coelomic and mesonephric epithelium or the mesenchymal stroma of the genital ridge. This category includes an array of tumors derived from the sex cords (granulosa and Sertoli cells) and from the gonadal stroma (theca and Leydig cells). The most common types of sex cord–stromal tumor are granulosa cell tumors and fibrothecomas, whose peak age of incidence is approximately 50 years. These tumors have the potential for steroid hormone secretion, with estrogen being the predominant hormone. They usually are not seen in women younger than 20 years, except for juvenile granulosa cell tumors. The classification of sex cord–stromal cell tumors is outlined in Table 55.5.
|
|
|
TABLE 55.5. World Health Organization classification of sex cord–stromal tumors |
Thecoma
Thecoma is a benign tumor affecting all ages, with a predominance in the postmenopausal group; it is rare in patients younger than 35 years. It accounts for 2% of all ovarian tumors. Many women with a thecoma have abnormal or postmenopausal uterine bleeding; some have an endometrial adenocarcinoma as a result of unopposed estrogen produced by the tumor. Thecoma is composed of lipid-laden stromal cells resembling theca cells. Rather than arising as a de novo neoplasm, it may represent changes occurring in background cortical stromal hyperplasia.
Tumor size ranges from a nonpalpable incidental finding to a large solid mass with a diameter of 15 to 20 cm. It is usually unilateral and almost never malignant. Its outer surface is smooth, and its cut surface is typically solid, lobulated, and yellow. Cystic change may be seen. Microscopic evaluation reveals masses of oval or round cells with abundant, pale, lipid-containing cytoplasm. Hyaline plaques are noted often. A luteinized thecoma sometimes occurs, usually in younger women.
Treatment for thecoma is tailored to patient age and ranges from a total abdominal hysterectomy and bilateral salpingo-oophorectomy for menopausal or postmenopausal women to a salpingo-oophorectomy or ovarian cystectomy, if possible, in patients who desire to retain fertility.
Fibroma
Like thecoma, fibroma is a benign tumor affecting all ages, although most occur in women aged 40 to 60 years; less than 10% of patients are 30 years or younger. Fibroma is not associated with hormone production. In some cases, hydrothorax and ascites are found in association with a pelvic mass—a constellation of findings known as Meigs syndrome. In other cases, fibroma is seen in patients with a hereditary basal cell nevus syndrome, characterized by early-appearing basal cell carcinomas, keratocysts of the jaw, calcification of the dura, and mesenteric cysts.
Fibroma, like thecoma, ranges in size from a nonpalpable incidental finding to larger than 20 cm; it is usually unilateral but multinodular. Cut surface is firm and hard and has a whorled appearance. Microscopically, fibroma is composed of bundles of collagen-producing spindle cells arranged in a storiform pattern (Fig. 55.12). Hyalinization and intercellular edema are characteristic. The cytoplasm of tumor cells may contain small quantities of lipid, making distinction from thecoma difficult.
|
|
|
FIG. 55.12. Fibroma. The tumor is composed of intersecting bundles of spindled cells. |
Treatment is similar to that for thecoma. In patients with Meigs syndrome, the hydrothorax and ascites usually resolve after resection of the pelvic tumor.
Juvenile Granulosa Cell Tumor
Approximately 44% of all juvenile granulosa cell tumors occur during the first decade of life and 97% during the first three decades. Isosexual pseudoprecocious puberty commonly is associated with this tumor, along with Ollier disease (enchondromatosis), Maffucci syndrome (enchondromatosis and hemangiomatosis), and abnormal karyotypes with ambiguous genitalia. Symptoms usually include abdominal pain, increasing abdominal girth, and hemoperitoneum due to rupture. Occasionally, it may be associated with pregnancy.
The gross appearance of juvenile granulosa cell tumor is commonly a large yellow-to-gray, solid and cystic mass, similar to the adult form. A mixture of granulosa and theca cells may be present. Microscopic features that distinguish juvenile granulosa cell tumor from the adult form are hyperchromatism of the tumor cells, which generally lack grooves, and the frequent luteinization of both the granulosa and theca cells (Fig. 55.13). The follicles are usually immature. Call-Exner bodies, which are pathognomonic of adult granulosa cell tumor, are rarely present in the juvenile form.
|
|
|
FIG. 55.13. Juvenile granulosa cell tumor. Tumor cells from follicles of varying sizes and shapes that contain weakly basophilic secretions. |
Treatment in young women usually consists of a unilateral salpingo-oophorectomy with a complete staging (see Table 55.3), especially if preservation of reproductive function is desired and the tumor appears to be grossly confined to one ovary. Although the survival rate for patients with stage I disease is greater than 90%, juvenile granulosa cell tumor appears to behave more aggressively in advanced disease, with a survival rate of less than 50%. Isolated reports of successful treatment with various combination chemotherapies have been recorded. Due to the rarity of these tumors, a standard chemotherapeutic regimen has not emerged.
Adult Granulosa Cell Tumor
The adult form of granulosa cell tumor accounts for 1% to 2% of all ovarian tumors and 95% of all granulosa cell tumors. Although these tumors average 10 to 12 cm in diameter, a pelvic mass is not always detectable. Peak age of occurrence is 50 to 55 years, and most granulosa cell tumors produce estrogen, which in premenopausal women is manifested by menstrual irregularities such as menorrhagia, amenorrhea due to anovulation, or metrorrhagia. In postmenopausal women, vaginal bleeding results from endometrial stimulation. Endometrial hyperplasia or carcinoma is a relatively common occurrence, approximately twice that seen in premenopausal women. The best estimate for the frequency of associated endometrial carcinoma is 5%.
Grossly, the tumors are solid and gray-white or yellow, with cystic areas and hemorrhage. Microscopically, they are composed of granulosa cells and theca cells or fibroblasts, or both. Theca cells and fibroblasts are most likely a response of the ovarian stroma to granulosa cell proliferation, because only granulosa cells are found in metastatic sites. Granulosa cells may be round, polygonal, or spindle shaped, with scant cytoplasm and round or ovoid nuclei. Many different histologic patterns may be seen separately or together; these include the microfollicular pattern with its distinctive Call-Exner bodies (Fig. 55.14), macrofollicular, insular, trabecular, solid-tubular and, rarely, hollow-tubular patterns. Less differentiated forms include the watered silk or diffuse pattern. Rarely, a granulosa cell tumor undergoes sarcomatous transformation, producing the most aggressive form of the disease. Inhibin may be a useful immunohistochemical marker and serum inhibin levels can be used to monitor the clinical course.
|
|
|
FIG. 55.14. Granulosa cell tumor. The microfollicular pattern is characterized by granulosa cells with angulated nuclei surrounding small cavities that simulate the Call-Exner bodies of the developing follicle. |
Although most granulosa cell tumors have a very low potential for malignant behavior, with 90% of tumors being stage Ia, they do have a propensity for late recurrence up to 10 to 20 years or more after initial diagnosis. In addition to full surgical staging (see Table 55.3), surgery should include a total abdominal hysterectomy and bilateral salpingo-oophorectomy in postmenopausal patients. Conservative surgery is indicated in younger patients who wish to maintain fertility, if the tumor is confined to one ovary. Stage appears to be the most important prognostic factor. Although others include capsular rupture, tumor size, nuclear atypia, mitotic activity, and histologic pattern, studies of these factors have not been conclusive. It is difficult to evaluate the efficacy of chemotherapy in this group of tumors. Most retrospective series have been limited by their small sample size and short follow-up. Platinum-based therapy including the PAC, VBP, and BEP regimens have been the most successful, with a 5-year survival of 50%. Radiotherapy also has been used with varying success in studies limited by small numbers. In recurrent disease, surgery may offer the best mode of therapy. Chemotherapy and radiation therapy have been used.
Androblastoma and Sertoli-Leydig Cell Tumor
Hilus Cell Tumor
Hilus cell tumor is a subtype of Leydig cell tumor, originating from the ovarian hilus. The other subtype, which is very rare, is a nonhilar-type Leydig cell tumor, derived from ovarian stromal cells and having similar clinical and pathologic features. The average age of patients with a hilus cell tumor is 58 years, and many show symptoms of abnormal menstruation, hirsutism, and virilization. Some may exhibit estrogenic manifestations. Hilus cell tumors are almost always benign.
Grossly, this tumor is circumscribed, lobulated, solid, soft, and red to yellow. Tumor enlargement is usually minimal, and the tumor often is physically undetectable. Sometimes, it is discovered incidentally when the ovaries are sectioned for pathology examination for another purpose. Microscopic evaluation reveals circumscribed masses of steroid cells with abundant eosinophilic cytoplasm. Lipochrome pigment may be present, also. The diagnostic elongate eosinophilic crystalloids of Reinke must be found for the tumor to be definitively classified a Leydig cell neoplasm. Treatment of this benign tumor is unilateral salpingo-oophorectomy or ovarian cystectomy if preservation of fertility is desiredSK.
Sertoli-Leydig Cell Tumors
Sertoli-Leydig cell tumors are also termed Sertoli–stromal cell tumors and can be divided further into several subtypes (see Table 55.5). Stage and tumor differentiation seem to be important prognostic factors. Despite the name of these tumors, hormone production is not always associated with them. Tumors that produce hormones may result in masculine or feminine phenotypes, because few of the tumors actually produce estrogen or progesterone.
Sertoli Cell Tumor
Sertoli cell tumors are very rare. Patients, ranging in age from 7 to 79 years (median, 33 years), usually exhibit a pelvic or abdominal mass. If the mass is functional, patients may experience some form of estrogenic effect, such as endometrial hyperplasia or isosexual precocious pseudopuberty. Androgenic and progestogenic effects may also occur. Most tumors are stage I unilateral masses that are well circumscribed, averaging about 9 cm in diameter. Cut surface reveals solid, yellow-to-brown, lobulated masses. Microscopic evaluation discloses closely packed hollow or solid tubules lined by Sertoli cells that can also contain abundant cytoplasmic lipid. An association between Sertoli cell tumors and Peutz-Jeghers syndrome has been reported in the literature.
Most Sertoli cell tumors are benign or early-stage malignant neoplasms that are cured by surgery. Conservative surgery with a unilateral salpingo-oophorectomy is indicated in many of these patients who are young and desire preservation of ovarian function. Only rarely are these tumors poorly differentiated and aggressive. Experience with chemotherapy in these tumors is limited due to their rarity.
Sertoli-Leydig Cell Tumor
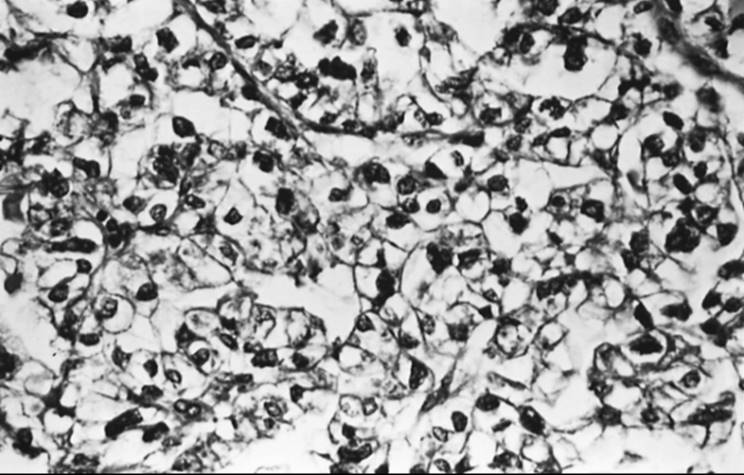
Sertoli-Leydig cell tumor accounts for less than 0.5% of all ovarian tumors. It is seen most often in young women, with a mean age of occurrence of 25 years. Less than 10% of these tumors occur in women older than 50 years, and fewer than 5% occur in prepubertal girls. Sertoli-Leydig cell tumor often is associated with androgen production; however, virilization develops in only 50% of patients. This may be due to a lack of hormone production or insufficient androgen production. Typically, patients complain of oligomenorrhea followed by amenorrhea, breast atrophy, acne, hirsutism, temporal balding, deepening of the voice, and enlargement of the clitoris. The latter two symptoms may not resolve after tumor removal. Patients without endocrine manifestations may complain of abdominal swelling or pain. Occasionally, symptoms of estrogen production such as menorrhagia or menometrorrhagia may be seen. They are a result of the Sertoli cell component of the tumor or peripheral androgenic conversion. Sertoli-Leydig cell tumor must be distinguished from other virilizing tumors, such as adrenal tumors, which often are associated with an elevated urinary level of 17-ketosteroids; the urinary level of 17-ketosteroids in Sertoli-Leydig cell tumor is usually normal or only slightly elevated. Serum AFP level may be increased and may be useful as a tumor marker.
The gross appearance of Sertoli-Leydig cell tumor is highly variable. Overall, it has an average diameter of 12 to 15 cm, and its cut surface is usually tan or yellow and may be cystic. Hemorrhage and necrosis frequently are seen in the poorly differentiated tumors. On microscopic examination, the tumor is composed of a mixture of Sertoli, Leydig, and undifferentiated gonadal stromal cells, with or without heterologous components, in varying proportions and degrees of differentiation. In well-differentiated lesions, Sertoli cells form tubules and Leydig cells are found in the intervening stroma (Fig. 55.15). Sertoli cells are cytologically bland, and mitotic figures are rare. Leydig cells may contain abundant lipochrome pigment or crystalloids of Reinke. Intermediate and poorly differentiated tumors are characterized by more immature components of the Sertoli and Leydig cells. Cartilage, mucinous epithelium, skeletal muscle, and other heterologous elements are found in 20% to 25% of these tumors, most of which are of intermediate differentiation. When heterologous elements have been found in poorly differentiated neoplasms, the tumors are clinically malignant. Sertoli-Leydig cell tumor with a retiform pattern may be seen with prominent hyalinized cores and papillae lined by stratified epithelial cells.
|
|
|
FIG. 55.15. Sertoli-Leydig cell tumor. This well-differentiated tumor is composed of hollow tubules lined by Sertoli cells and adjacent sheets of Leydig cells. |
The treatment for Sertoli-Leydig cell tumor usually depends on patient age and tumor stage, degree of differentiation, and presence of heterologous elements in the tumor. The most important prognostic factor is stage. In young women with a stage Ia well-differentiated tumor and who desire future pregnancy, a unilateral salpingo-oophorectomy and a staging procedure are adequate treatment (see Table 55.3). However, more aggressive cytoreductive surgery, including a hysterectomy and bilateral salpingo-oophorectomy, tumor resection, and staging procedure may be indicated in postmenopausal patients or those with more advanced disease.
Adjuvant therapy is recommended for patients who have stage Ia lesions with poorly differentiated elements or heterologous components or for those who have metastatic disease. However, due to the limited number of cases, no standard adjuvant therapy has been accepted for these patients. Most of the information available comes from small series and case reports. Treatment of advanced sex cord–stromal cell tumors, unlike that used for germ cell tumors, has not met with much success. Platinum-based therapies have yielded the best results, with an overall survival of approximately 50%. These include the PAC, VBP, and BEP regimens. As with chemotherapy, radiotherapy has been used successfully in a limited number of cases.
Steroid Cell Tumors Not Otherwise Specified
Termed lipid cell or lipoid tumors in the past, these are tumors composed entirely of cells resembling typical steroid hormone–secreting cells (e.g., lutein cells, Leydig cells, and adrenal cortical cells), except that specific features such as location of origin in the hilus or crystalloids of Reinke are not identified. Steroid cell tumors not otherwise specified (NOS) account for approximately 0.1% of all ovarian tumors, with a mean age of occurrence of 43 to 60 years. Androgenic changes, occurring in 75% to 90% of patients, may be of many years' duration. Estrogenic and progestogenic changes are noted occasionally. Although the estrogenic manifestations may be a result of estrogen production by the tumors, the aromatization of androgen to estradiol in adipose tissue may be more plausible. Cushing syndrome may be found in some patients, accompanied by elevated serum cortisol levels. Diagnosis often depends on the clinical manifestation of virilization or the rare occasion of isosexual pseudoprecocity. Tumor removal results in rapid resolution of most of the hormonal effects, except for deepening of the voice and clitoromegaly.
Grossly, steroid cell tumors NOS are solid, well circumscribed, and yellow to orange-tan, measuring 5 to 8 cm in diameter. Hemorrhage, necrosis, and cystic degeneration occasionally are observed. The cut surface of the tumor is soft and lobulated. Microscopically, tumor cells may resemble Leydig or hilar cells (Fig. 55.16). In other instances, cells have abundant pale cytoplasm, resembling adrenocortical cells. These cells are polygonal to round and larger than Leydig cells, with central nuclei and lipid-rich cytoplasm. The striking resemblance of many of these tumors to adrenocortical tumors has led some to speculate that they may arise from the adrenocortical nests. The association with manifestations of Cushing syndrome would seem to support this theory, and detailed examination has revealed the presence of these nests in the broad ligament and the ovarian hilus. Alternatively, given that steroid cell tumors NOS often are confined to the ovary may simply mean that adrenocortical hormones are being produced by cells of ovarian origin rather than by an ectopic adrenal tumor. Steroid cell tumors NOS are rarely malignant; approximately 10% to 15% of them recur or metastasize.
|
|
|
FIG. 55.16. Steroid cell tumor not otherwise specified. If crystalloids of Reinke were identified in these cells, the tumor would be classified a Leydig cell tumor or possibly a hilus cell tumor, depending on the its location. The granular appearance of the cytoplasm in these polygonal-to-rounded tumor cells suggests that they contain little lipid. |
A unilateral salpingo-oophorectomy is adequate for stage Ia disease in young, reproductive age women. An abdominal hysterectomy and bilateral salpingo-oophorectomy with staging and resection of all extraovarian disease are indicated in women with advanced disease or in those beyond reproductive age.
EPITHELIAL OVARIAN TUMORS
Benign Neoplasms
Serous Cystadenoma
Serous and mucinous cystadenomas are the most common benign epithelial ovarian neoplasms. Serous tumors account for approximately 25% of all benign ovarian neoplasms, with an age range of 20 to 50 years; they are bilateral in 12% to 20% of patients. Symptoms are variable. For many patients, the diagnosis is made during routine pelvic examination.
Grossly, serous cystadenoma is a cystic, usually unilocular lesion, ranging from 5 to 15 cm in diameter. The inner lining of the cyst wall may be flat or partially covered by papillary projections. Microscopically, the epithelial lining can range from simple cuboidal cells, resembling the ovarian surface epithelium, to tall columnar cells, resembling the fallopian tube (Fig. 55.17). Ciliated and secretory cells may be present. Mitoses are rare, and nuclear atypia is absent. Psammoma bodies, which are concentric calcifications, are seen in 15% of tumors. Multiple, large, calcified deposits may be visible on radiologic examination of the abdomen. The stroma may vary from fibrous to cellular to hyalinized, with marked stromal edema. The papillary processes are fibrous and lined by a single layer of epithelial cells.
|
|
|
FIG. 55.17. Serous cystadenoma. The lining epithelium commonly appears tubal. |
The preoperative workup depends on patient age and the degree of suspicion of malignancy. A unilateral salpingo-oophorectomy usually is performed in patients who have completed childbearing. In those desiring future reproduction, an ovarian cystectomy usually is performed. A bilateral salpingo-oophorectomy may be performed in selected individuals, particularly in those with bilateral tumors. In such cases, discussion of a hysterectomy is reasonable but not necessary.
Mucinous Cystadenoma
Mucinous cystadenoma accounts for approximately 25% of all benign ovarian neoplasms, with an age range of 20 to 50; in 2% to 3% of patients it is bilateral. The tumor arises from the surface epithelium of the ovary, resembling müllerian-type epithelium of the endocervix, intestinal-type epithelium, or both these types. Because of the large size that this tumor frequently attains, patients usually have a palpable pelvic or abdominal mass and may have associated pain.
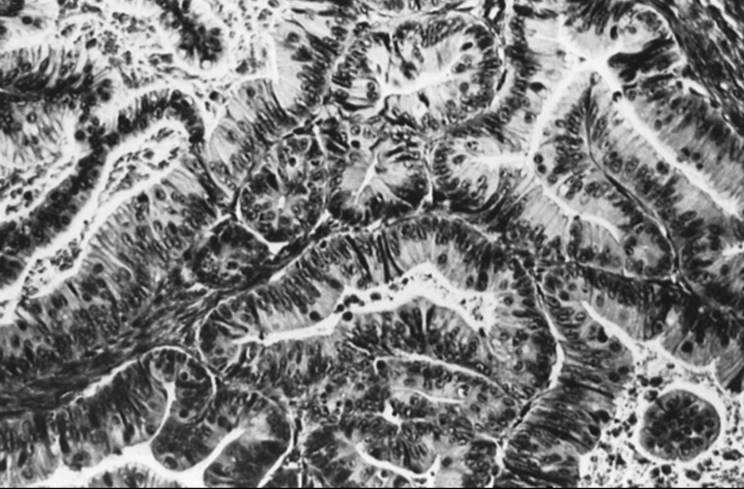
Gross evaluation of a mucinous cystadenoma reveals that it is usually multilocular and larger than its serous counterpart, ranging up to 50 cm or more in diameter. The external surface is usually smooth, pinkish gray, and sometimes lobulated. Inside, locules tend to be small and contain thick, sticky, tenacious mucinous material (Fig. 55.18). Microscopically, the epithelium consists of a single layer of uniform tall columnar cells that resemble a picket fence in the endocervical type or may contain goblet, argentaffin, and Paneth cells in the gastrointestinal type (Fig. 55.19). Treatment is similar to that for serous cystadenoma. The appendix should be removed in any suspected ovarian mucinous tumor because of frequent synchronous appendiceal mucinous tumors (mucocele), even in a clinically normal-appearing appendix.
|
|
|
FIG. 55.18. Mucinous cystadenoma. The tumor is multilocular. |
|
|
|
FIG. 55.19. Microscopic appearance of the lining of a mucinous cystadenoma. The “picket fence” epithelium is characteristic of this tumor. |
Cystadenofibroma
As the name suggests, cystadenofibroma is a variant of serous cystadenoma, containing both cystic and solid components. This benign tumor is usually unilateral and has a similar age distribution to that of serous cystadenoma. It is variably papillary to solid. Papillae are broad, firm, and nonfriable.
Microscopically, solid areas are found to contain small cystic structures that are histologically identical to serous cysts. As with other benign epithelial ovarian tumors, treatment is individualized based on patient age and reproductive desire.
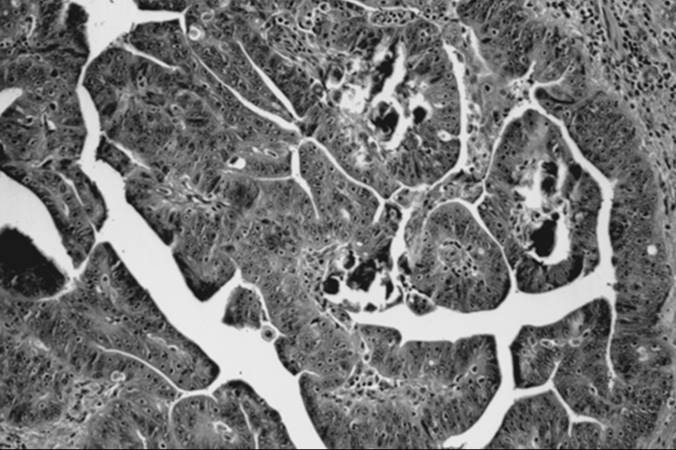
Brenner Tumor
Also known as transitional cell tumor, Brenner tumor constitutes 2% of all primary ovarian tumors. Patients with this tumor range in age from 30 to 70 years, with a mean age of 50 years. Brenner tumor is thought to be derived from ovarian surface epithelium that undergoes a metaplastic transformation to cells resembling urothelium. It may occur synchronously with mucinous cystadenoma. Most transitional cell tumors are benign, but transformation to a malignant form has been observed. Clinical picture may be variable, because the patient may be asymptomatic or may have a palpable mass or pain. Occasionally, patients may experience vaginal bleeding, probably due to hormonal activity in the stroma.
Grossly, the tumor is solid—less commonly cystic—and usually unilateral, although 6% to 7% of tumors are bilateral. Ranging in size from microscopic to as large as 30 cm in diameter, it is usually gray, white, or yellow, with a faintly lobulated cut surface. Microscopic review depicts a characteristic pattern of circumscribed epithelial nests of cells embedded in an abundant fibromatous stroma (Fig. 55.20). Epithelial cells may be round to polygonal, with eosinophilic or clear cytoplasm. When longitudinal grooves of the nuclei are present, the cells are described as having a “coffee bean” appearance. Often, these nests of epithelial cells undergo benign cystic changes lined by either transitional cells or mucinous cells. Treatment is usually resection of the tumor, and this may involve a cystectomy or salpingo-oophorectomy with or without a hysterectomy, depending on patient age and reproductive desires.
|
|
|
FIG. 55.20. Brenner tumor. Epithelial nests embedded in a fibromatous stroma may show cystic change. Longitudinal grooves in the cells can impart a “coffee bean” appearance. |
Tumors of Low Malignant Potential
Epidemiology
Ovarian tumors of low malignant potential (LMP), also known as atypical proliferating tumors, comprise a group of tumors showing greater epithelial proliferation than that seen in their benign counterparts although, by definition, they are still noninvasive. Recognized by FIGO in 1971, LMP ovarian tumors account for approximately 15% of all epithelial ovarian cancers; mean age of occurrence is 40 years. A meta-analysis performed by the Collaborative Ovarian Cancer Group found that, as with malignant epithelial ovarian cancer, parity, multiple births, history of breast-feeding, and oral contraceptive use are protective against LMP tumors. A history of infertility and use of fertility drugs may increase the risk of developing an LMP tumor, although these data are weak and controversial. Prospective trials are needed to resolve the controversy surrounding the use of fertility drugs and these tumors.
Clinical Features
Patients are usually asymptomatic, but they may come to medical attention with a pelvic mass and complaints of abdominal or pelvic pain or both, increasing abdominal girth, or abnormal bleeding. Ultrasonography or a computed tomography (CT) scan may be helpful in making the diagnosis of an ovarian mass. Serum CA-125 levels are not always elevated. When they are, the tumor is usually of serous histology. LMP tumors usually have an indolent course. Many biomarkers, such as DNA ploidy, tumor markers, oncogenes, and tumor suppressor genes, have been studied in an attempt to define a high-risk group or predict tumor recurrence and thus indicate which patients might benefit from adjuvant treatment. To date, no such marker has been identified.
Pathologic Classification
LMP ovarian tumors have been described for all epithelial subtypes; the most common types are serous and mucinous tumors. The absence of stromal invasion is an absolute criterion for making the diagnosis. Careful examination of the tissue blocks is necessary to minimize the potential for “sampling error” or omitting an area of invasive carcinoma in LMP tumors. Approximately 20% to 30% of ovarian tumors diagnosed as borderline by frozen section prove to be carcinomas with review of the permanent sections. The mean diameter of serous LMP tumors is 12 cm, and bilateral tumors are reported in 33% to 75% of patients. These tumors are usually cystic, with mural clusters of papillary projections (Fig. 55.21 and Fig. 55.22).
|
|
|
FIG. 55.21. Serous tumor of low malignant potential (atypical proliferating tumor). On the right is the external surface of the ovary; on the left the cut surface. The tumor is multicystic, and within some cysts are complex papillary projections. |
|
|
|
FIG. 55.22. Serous tumor of low malignant potential (atypical proliferating tumor). Ramifying papillae are lined by a relatively bland serous epithelium that forms tufts and seemingly free-floating clusters. |
Mucinous LMP tumors are larger than their serous counterparts, with an average diameter of 17 to 20 cm; they are infrequently bilateral. They are characterized by multiloculated cystic masses, with smooth outer surfaces and areas of papillations and solid thickening on the inner surface. Microscopically, the epithelial lining of the cysts consists of tall, columnar, mucin-secreting cells, resembling the epithelium of the endocervix or intestine. Stratified epithelial cells may be atypical with hyperchromatic nuclei and mitotic figures, but without stromal invasion (Fig. 55.23).
|
|
|
FIG. 55.23. Mucinous tumor of low malignant potential (atypical proliferating tumor). Epithelial proliferation results in pseudostratification and tufting. Mild cytologic atypia is present. |
Stage for stage, the 5-year survival rate for patients with LMP epithelial ovarian tumors is far better than that for patients with malignant epithelial ovarian cancer. A review of the literature by several investigators revealed a survival rate of greater than 95% in patients with stage I LMP ovarian tumors. Furthermore, Kurman and Trimble found that a majority of patients with LMP tumors actually died with the disease, not from it, because invasive carcinoma developed in only 8 (0.8%) of 953 patients with a mean follow-up of 7 years. The other patients died from radiation- or chemotherapy-associated complications. Recognizing that a minority of LMP tumors behave aggressively like invasive ovarian carcinomas, Seidman and Kurman have challenged the current classification of LMP tumors in an attempt to develop a nomenclature that better reflects prognosis.
These investigators believe that LMP tumors are actually a heterogeneous group of tumors, both histologically and clinically, that can be divided into malignant or benign phenotypes based upon certain features.
For serous tumors, they propose that patient outcome depends upon the presence or absence of micropapillary features within the ovarian tumor and invasive versus noninvasive peritoneal implants. In a large meta-analysis of serous LMP tumors, they report that patients with micropapillary serous carcinoma have 5- and 10-year survival rates of 81% and 71%, respectively. Patients with a serous borderline tumor without invasive implants (atypical proliferative serous tumor) had 5- and 10-year survival rates of greater than 98%. However, in serous borderline tumor with invasive implants, survival rates drop to 60% to 70% after 5 and 10 years. The vast majority of patients with invasive implants, or with those which progressed to invasive carcinoma, were found to have micropapillary features within the primary ovarian tumor upon careful sectioning. Thus, atypical proliferative serous tumor and serous borderline tumor without invasive implants follow in a very benign clinical course, while micropapillary serous LMP tumors behave much like invasive ovarian carcinomas.
Micropapillary serous carcinoma is characterized by thin, elongated micropapillae with minimal or no fibrovascular support arising directly from thick, more centrally located papillary structures (Fig. 55.24). Mitotic activity may be seen in some of the cases, ranging from one to three figures per 10 high-power fields. The distinction between invasive and noninvasive implants may be difficult. Noninvasive implants usually have a scant epithelial component surrounded by reactive spindle cells with imperceptibly meshed epithelial and stromal cells (Fig. 55.25). On the other hand, invasive implants usually have a more cellular epithelial component, with complex epithelial proliferation composed of multiple micropapillae and small round nests that display a destructive infiltrative growth (Fig. 55.26).
|
|
|
FIG. 55.24. Micropapillary serous carcinoma. In this fully malignant serous carcinoma, strands of neoplastic cells stream from thick, fibrous cores, stimulating a Medusa head. |
|
|
|
FIG. 55.25. Noninvasive implant in a serous borderline tumor. A circumscribed focus of tumor is surround by fibroadipose tissue that shows no stromal reaction. Cell nests are heavily calcified. |
|
|
|
FIG. 55.26. Invasive implant in a serous borderline tumor. The tumor here is more cellular. The invasive implant extends below the peritoneal surface and is associated with reactive desmoplasia. |
Mucinous LMP tumors tend to be confined to the ovary. Advanced stage tumors are typically associated with pseudomyxoma peritonei, and frequently represent metastatic disease from the gastrointestinal tract. In such cases, careful histopathological examination of the appendix is necessary to rule out an occult appendiceal primary. Primary mucinous carcinomas of the upper gastrointestinal tract and appendix can present with small primary lesions and large bilateral ovarian masses that stimulate primary ovarian carcinoma. Mucinous ovarian LMP confined to the ovary have an excellent prognosis, while the survival of patients with advanced stage disease is approxmiately 50%.
Pseudomyxoma Peritonei
Pseudomyxoma peritonei may accompany mucinous neoplasms of the ovary and is characterized by extracellular gelatinous material in the pelvic and abdominal cavity. The primary site of origin of pseudomyxoma peritonei is debatable. Some series cite that the majority begin in the appendix, while others have described a synchronous origin in the ovary and appendix. Pseudomyxoma is associated most frequently with borderline or well-differentiated ovarian mucinous tumors in approximately 8% of cases and only rarely is seen in mucinous cystadenomas. The mainstay of treatment is cytoreductive surgery; multiple treatment modalities with systemic and intraperitoneal chemotherapies have not demonstrated survival benefit. Patients have a high recurrence rate, with variable times to recurrence. Most series agree that when patients have recurrent disease, repeat laparotomy is required to relieve symptoms. The overall 5-year survival rate is 50%, and most patients die from bowel obstruction.
Treatment
The primary surgical treatment for patients with LMP tumors who have completed childbearing is identical to the recommendation for invasive ovarian disease, including a total abdominal hysterectomy, bilateral salpingo-oophorectomy, tumor debulking, and full staging (see Table 55.3). An appendectomy should be performed in patients with a mucinous LMP tumor because of the association with a synchronous primary appendiceal tumor.
In younger patients with early-stage diagnosis and a desire for childbearing, conservative surgery with preservation of the uterus, the contralateral ovary, and in some cases the ipsilateral ovary (i.e., cystectomy) may be the appropriate treatment. Consultation with a gynecologic oncologist and pathologist can identify those patients who are candidates for conservative management. Several studies, both cohort and observational, have reported excellent outcome with conservative management of such patients. One of the largest studies reports a 12% recurrence rate for patients treated conservatively with either unilateral salpingo-oophorectomy (n = 110) or ovarian cystectomy (n = 74), versus 2.5% for patients treated with definitive hysterectomy and bilateral salpingo-oophorectomy. Recurrences or progression to carcinoma (1.5%) were more common among patients with invasive implants or advanced stage disease. The feasibility of doing a cystectomy for an LMP ovarian tumor and conserving the rest of the ovarian tissue in early-stage disease has been described but requires further study.
LMP ovarian tumors have been diagnosed during pregnancy. Conservative surgery usually is performed, and pregnancy does not appear to be deleterious in regard to the prognosis for these patients. Most patients went on to deliver at full term without any complications.
Postoperative Therapy
There is no evidence to suggest that adjuvant chemotherapy for patients with early-stage LMP tumors or with optimal cytoreduction of advanced LMP tumors improves survival. In fact, patients may be more likely to die from the side effects of adjuvant therapy than from the disease itself. Platinum-based therapy may be appropriate for a select group of patients with micropapillary serous tumors and invasive serous implants, based upon the high rates of recurrence in these patients. However, patients must be counseled that available literature does not demonstrate improved survival with chemotherapy. An ongoing prospective GOG trial will address these issues. Second-look laparotomy should not be a part of the standard therapy in treating these patients.
Malignant Neoplasms
Epidemiology
Epithelial ovarian carcinomas account for 80% to 90% of all ovarian malignancies. Incidence rates in the United States are about 12 to 15 per 100,000 for white women, compared with about 8 to 10 per 100,000 for the U.S. residents of non-European heritage. The incidence rate continues to increase to about 40 cases per 100,000 women by age 50, and then continues to increase slowly to about 50 cases per 100,000 by age 65. Although the incidence rates in Asian nations are lower, studies have indicated increasing incidence rates in these countries. Other than race, risk factors for ovarian cancer identified by epidemiologic studies include age over 60 years, early menarche, late menopause, nulliparity, infertility, personal history of breast or colon cancer, and family history of ovarian, breast, or colon cancer. In some inherited cases, the increased risk of ovarian cancer may be more than 50%, depending on the type and number of family members affected and age of onset. Other factors that have been implicated but lack adequate epidemiologic evidence include high-fat diets, talcum powder, and infertility drugs. Use of an oral contraceptive, bilateral tubal ligation, and hysterectomy are protective. Epithelial tumors are derived from the ovarian surface epithelium, which is the site of ovulation. One hypothesis suggests that incessant ovulation contributes to ovarian carcinogenesis because of the repeated injury and repair of the ovarian epithelium secondary to monthly ovulation. Repair of the ovarian surface promotes proliferation-associated DNA damage. Data from the United States Collaborative Analysis support the suppression of ovarian activity as a means of preventing ovarian cancer. A study found that a high number of ovulatory cycles was associated with more genetic alterations in patients with ovarian cancer. Oral contraceptives appear to protect against ovarian cancer through suppression of ovulation and a direct antiproliferative effect of the progestins on ovarian epithelium.
Clinical Picture
Many patients with epithelial ovarian cancer come to medical attention with advanced stage disease, with symptoms of abdominal fullness, pain, distension, early satiety, and weight loss. Other gastrointestinal symptoms include nausea, dyspepsia, constipation, or diarrhea. Abdominal distension can be due to ascites or a large abdominopelvic mass. Abnormal vaginal bleeding is seen in 30% of the cases. The most common physical signs are ascites, manifested by a fluid wave and the absence of shifting dullness, and a pelvic mass. The mass is frequently firm, hard, and fixed, with multiple nodularities. Small tumors may not be palpated easily in the presence of ascites. Possible sites of spread within the abdomen are multiple (Fig. 55.27).
|
|
|
FIG. 55.27. Possible sites of spread of epithelial ovarian cancer. |
Preoperative Evaluation
All patients should have a complete blood cell count and serum chemistry analysis, including liver function tests, performed. These tests, although routinely done preoperatively, will select patients who may be at higher anesthetic risk. For example, any liver or kidney dysfunction may require further investigation, or anemia may justify a workup and transfusion prior to surgery. Serum tumor markers should be assayed. Although CA-125 is the best tumor marker for women with epithelial ovarian carcinoma, it can be elevated in a variety of benign gynecologic conditions and other nongynecologic malignancies. CA-125 will be elevated in more than 80% of patients with serous epithelial ovarian cancer. Although it is not useful for screening, CA-125 has higher sensitivity and specificity for detecting ovarian cancer in the postmenopausal patient. Preoperatively, CA-125 may be useful in helping to predict the potential for malignancy and, if elevated, it can be used to follow response to therapy and detect an early recurrence.
Chest radiographs are performed routinely to look for malignant pleural effusions, which occur in 10% of patients, and metastatic pulmonary disease, which is very rare. Barium enema examination is not performed routinely but may be helpful in patients with a left lower quadrant mass, blood in the stool, obstipation, or anemia, or in patients with carcinomatosis in whom a primary gastrointestinal tract malignancy must be ruled out. Barium enema examination is not very useful in predicting the need for colon resection. Mammography may be performed to rule out a possible metastatic or synchronous breast carcinoma.
Sonography is the most useful diagnostic examination in the evaluation of a pelvic mass. It is an easily accessible and inexpensive imaging modality that provides an accurate description of ovarian morphology. Some characteristics associated with ovarian cancer include irregular ovarian cyst borders, solid elements, papillary projections, bilateral ovarian pathology, and ascites. Color Doppler imaging evaluates blood flow to an ovarian mass and can suggest a malignant process based upon abnormal neovascularization. Several studies suggest that color Doppler imaging improves detection of ovarian cancers, although it lacks the ability to definitively identify malignancy. Three-dimensional ultrasonography has the potential benefit of enhanced imaging of the ovarian architecture and measuring the ovarian cyst volume; however, its application in evaluation of an adnexal mass has not been fully studied.
CT scan may be helpful in characterizing the liver, lymphatic spread, the omentum, and the mesentery. CT scan may be helpful in distinguishing a gynecologic malignancy from a metastatic pancreatic neoplasm for which surgery may not be warranted. MRI in patients with an ovarian mass has not been shown to have any clear advantage over CT scanning, except for the evaluation of pregnant patients whose ultrasonographic examination was inconclusive and CT scan would result in undesirable ionizing irradiation.
Other studies such as bone and liver scintigraphy do not add any useful information. Intravenous pyelograms or renal scans may be helpful in patients with abnormal renal function or abnormal findings on ultrasonography, but they rarely are used. Immunoscintigraphy using CYT-103 or OC-125 that detects occult extraabdominal or miliary metastasis is experimental but may be helpful in patients prior to second-look laparotomy or in those with recurrent disease. Positron emission tomography (PET) is a form of computer-assisted imaging that uses radionuclide-labeled analogues of glucose labeled with positron-emitting isotopes. Cancer cells are detected by their increased glucose metabolism compared with noncancerous cells. PET has been examined in several preliminary studies involving the preoperative evaluation for suspected recurrent or metastatic ovarian tumors. Although the results are promising, PET's role in the preoperative evaluation of such patients awaits further study.
Staging
Staging is a critical step in the treatment of ovarian cancer. It is done at the time of surgical exploration in accordance with the FIGO system revised in 1987 (see Table 55.4). Proper staging is absolutely necessary, because it impacts both prognosis and subsequent treatment method (see Table 55.3). Observational studies have shown that 30% of patients who were thought to have stage I or stage II disease found during initial surgery had more advanced disease with more comprehensive restaging laparotomy. A midline vertical incision is recommended, and it should be extended to above the umbilicus, if necessary, to allow proper exposure of the upper abdominal cavity for tumor debulking. If an ovarian malignancy is discovered unexpectedly through a low transverse incision, the rectus muscle can be either severed or detached from the pubic symphysis to enhance surgical exposure.
Pathology
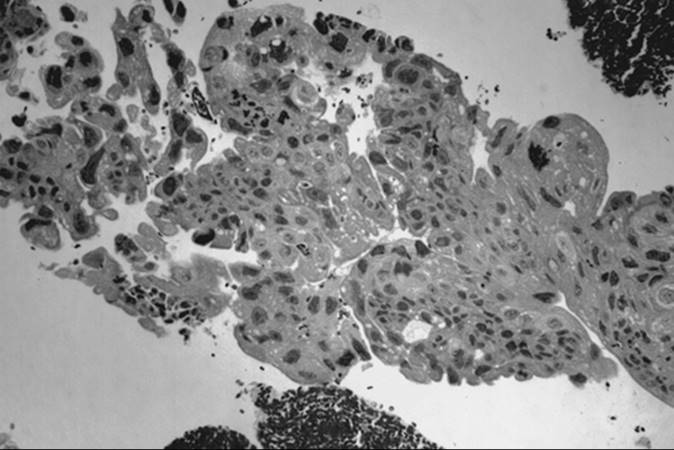
Serous Carcinoma
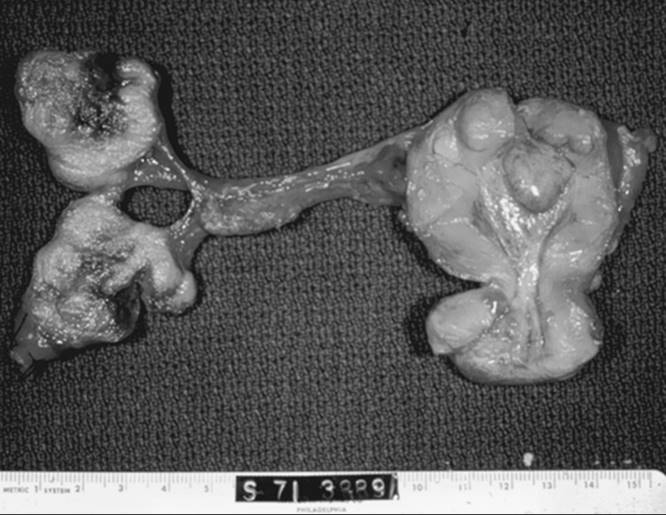
Serous tumor is the most common epithelial ovarian carcinoma, accounting for 40% to 50% of all such tumors. In most patients, serous tumors are bilateral and are disseminated at the time of diagnosis. They are soft, friable, mostly cystic, containing turbid or bloody fluid and having extensive papillary projections. Papillary excrescences also may be seen on the external surface or attached to adjacent structures (Fig. 55.28).
|
|
|
FIG. 55.28. Serous carcinoma. The tumor forms large, bilateral, solid, and cystic masses that have grown through the capsule. Tumor also involves the uterine serosa. |
Microscopic evaluation in well-differentiated serous carcinoma reveals well-formed papillary structures that grow into cystic spaces or on the peritoneal surface of the tumor. Psammoma bodies are present in most cases (Fig. 55.29). With less differentiated tumors, the papillary pattern becomes less visible, the mitotic activity more brisk, and tumors become solid sheets of uniform, dark cells that are often slightly spindled and have a high nucleus-to-cytoplasm ratio.
|
|
|
FIG. 55.29. Serous carcinoma. In this tumor, papillae are lined by high-grade tumor cells with prominent nucleoli. Psammoma bodies are present. |
Mucinous Carcinoma
Mucinous carcinoma represents 5% to 10% of all epithelial ovarian malignancies. Bilaterality is less common, present in less than 15% of cases. Disseminated spread is seen much less frequently than in patients with serous carcinoma. Mucinous carcinoma is usually larger than its serous counterpart, measuring approximately 15 to 30 cm in diameter. The tumor is multiloculated, solid and cystic, and filled with thick, viscous mucin.
Microscopically, invasive, well-differentiated mucinous carcinoma typically is composed of more than three layers of epithelium, with prominent mucin production (Fig. 55.30). It may show only subtle irregularities in gland contour and irregular budding, with no other stromal signs of invasion. In moderately differentiated carcinoma, the glands are more back-to-back, with obvious stromal invasion, cellular stratification, and nuclear atypia. In poorly differentiated mucinous carcinoma, cells are disorganized, embedded in a dense reactive ovarian stroma. Signet-ring cells may be present, as in a Krukenberg tumor. Mucinous tumor often contains a wide range of histologic differentiation; therefore, extensive sampling must be performed to obtain the correct diagnosis.
|
|
|
FIG. 55.30. Mucinous carcinoma. Infiltrating, crowded glands are lined by tall columnar cells with pale cytoplasm. Note the atypical, stratified nuclei (original magnification × 160). |
Endometrioid Tumor
Endometrioid carcinoma accounts for 20% of all epithelial ovarian malignancies. It is the second most common type of epithelial ovarian carcinoma and is bilateral in 30% of cases. In up to one third of patients, endometriosis has been noted in the same ovary or elsewhere in the pelvis. In addition, a synchronous endometrial adenocarcinoma has been seen in 20% of patients; these tumors are superficial and are associated with endometrial hyperplasia. Well-differentiated ovarian endometrioid adenocarcinoma is characterized by well-developed glands lined by tall, columnar, pseudostratified or multilayered epithelium like its uterine counterpart (Fig. 55.31). Squamous differentiation may be seen in about 30% of tumors and is usually benign. As tumors become less differentiated, the glandular pattern becomes less organized, and a solid growth pattern becomes predominant. Mitoses are more frequent, and nuclei are more high grade.
|
|
|
FIG. 55.31. Endometrioid carcinoma. In a well-differentiated endometrioid carcinoma, the glands are smoothly rounded, closely packed, and lined by an endometrioid type of columnar epithelium. |
Clear Cell Tumor
Clear cell carcinoma constitutes 5% to 10% of malignant epithelial ovarian tumors. It is bilateral in 15% to 20% of patients and confined to the ovary in 60%. Despite the limited spread of disease at the time of diagnosis, clear cell carcinoma is a notoriously virulent tumor. It frequently is associated with endometriosis, with 25% of tumors arising from the lining of the endometriotic cysts. A mixed form of epithelial ovarian carcinoma with clear cell tumor and endometrioid or serous carcinoma can occur, suggesting a similar histogenesis.
Microscopically, clear cell tumor may be characterized by sheets of polyhedral clear cells divided by fine connective tissue septa. Cells usually have abundant glycogen in the cytoplasm that is responsible for the clear appearance of the cells. The other characteristic feature is the tubulopapillary pattern formed by the columnar secretory cells with nuclei that bulge into the lumina of the glands, giving a hobnail appearance (Fig. 55.32).
Eosinophilic cells instead of secretory columnar cells may surround tubules or papillae, or a mixture of these cells may be present.
|
|
|
FIG. 55.32. Clear cell carcinoma. In clear cell carcinoma, tumor cells are polygonal, with central nuclei and abundant clear cytoplasm (original magnification × 250). |
Undifferentiated Tumor
Accounting for 5% to 10% of all ovarian malignancies, undifferentiated tumor exhibits such poor differentiation that it cannot be classified into any of the categories previously described. There is a variable histologic pattern, ranging from sheets of large anaplastic cells, to undifferentiated small cells, to large pleomorphic giant cells with eosinophilic cytoplasm. The prognosis is usually very poor because patients often seek treatment in advanced stages with large tumor burdens.
Brenner Tumor
Malignant Brenner tumor is very rare. It ranges from 10 to 30 cm in diameter and is usually unilateral. Microscopically, malignant Brenner tumor exhibits solid sheets of heterogeneous epithelial cells with minimal stroma. Nuclear grade and mitotic activity are high. Histologically, these cells resemble transitional cells, like those in the urologic tract. Differentiation from a carcinoma of urinary origin is based on clinical picture and surgical findings. Origin in the ovary is supported by finding a transition from a benign or proliferating Brenner tumor.
Müllerian Mesenchymal and Mixed Tumors
These rare ovarian tumors are divided into subtypes, including adenosarcoma and malignant mixed mesodermal tumor; they are associated with a poor prognosis. Histologically, they resemble their uterine counterparts. Adenosarcoma contains a benign epithelial component and a sarcomatous mesenchymal component. Malignant mixed mesodermal tumor has both malignant epithelial and malignant mesenchymal components (i.e., carcinosarcoma).
Similar to epithelial ovarian tumors, these tumors usually are surgically staged. However, optimal cytoreduction and adjuvant chemotherapy regimens for these patients may not be as effective as they are for patients with the other types of epithelial tumors. The chemotherapeutic regimens are also different, but studies are limited by the rarity of these tumors. Agents that have been tried include doxorubicin (Adriamycin), cisplatin, and ifosfamide (IFEX), and paclitaxel (Taxol).
Treatment
Surgery
Surgery is the most important aspect in the initial care of patients with epithelial ovarian cancer. Surgery alone is curative for many patients with early-stage ovarian carcinoma. For more advanced disease, surgery establishes the diagnosis and allows appropriate staging and cytoreduction, optimally to less than 1 cm of residual disease. Conservative surgical management of young women who desire to preserve fertility with a unilateral salpingo-oophorectomy with a full staging procedure may be appropriate for stage Ia disease. The contralateral ovary should be evaluated carefully, because there is a 5% chance of occult metastasis or a separate primary carcinoma. Biopsy of the contralateral ovary is no longer recommended unless there are grossly apparent abnormalities. Such patients also must have strict follow-up. Although a hysterectomy and removal of remaining adnexae is recommended for women who have completed childbearing, this recommendation is not based on randomized clinical trials.
Primary cytoreduction of ovarian carcinoma has been recommended for almost 30 years. The theory is that removal of large tumor nodules allows better penetration of chemotherapeutic agents and removes potential foci of chemoresistance. Optimal cytoreduction is defined as residual disease smaller than 1 cm. Several reports over the last 25 years have shown that the diameter of the largest residual disease correlates with response rate, progression-free interval, and overall survival. The GOG evaluated survival by maximal diameter of residual disease (data from protocols 52 and 97) and found that the survival rate for patients with residual disease of 2 cm or greater is about 20% after 4 years, compared with a 60% survival rate after 4 years in those with only microscopic residual disease. For patients with residual disease less than 2 cm, the survival rate was 40% after 4 years (Fig. 55.33). Median survival of patients who have received optimal cytoreduction was approximately twice that of patients with suboptimal cytoreduction.
|
|
|
FIG. 55.33. Survival by residual disease: Gynecologic Oncology Group, protocols 52 and 97. (From Hoskins WJ, McGuire WP, Brady MF, et al. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am J Obstet Gynecol 1994;170:974–980, with permission.) |
There has been great debate in the literature regarding the relative importance of surgical cytoreduction as compared with the inherent tumor biology of the cancer as determinants of patient survival. Patient age, the number and size of tumor nodules at time of diagnosis, and tumor grade have been cited as significant predictors of outcome. Other series have found that optimal surgical cytoreduction did not equate the survival benefit experienced by patients who initially had small-volume disease. Some investigators have suggested that platinum-based therapy has had a larger impact on survival than surgical debulking. Data, including a large meta-analysis, have demonstrated that maximal initial surgical cytoreduction was the most powerful predictor of patient survival. Patients with ovarian cancer are more likely to have optimal cytoreduction under the care of a gynecologic oncologist than with general surgeons or general gynecologists. Despite the benefit of improved survival, most patients with ovarian cancer in the United States never see a gynecologic oncologist. Ongoing efforts to educate physicians and patients regarding appropriate referral to specialized centers are needed.
At the end of chemotherapy, patients who are clinically in complete remission may be offered second-look evaluation to determine the success of therapy, as measured by the presence of any residual tumor cells. The reasons for second-look evaluation are as follows: (a) to determine disease presence, thus allowing continued therapy in an attempt to improve survival, (b) to debulk any residual carcinoma, and (c) a prognostic tool for patient survival. Most second-look evaluations can be completed successfully via the laparoscope among surgeons with adequate expertise, and second-look laparoscopy has equivalent rates of detection of residual disease when compared with laparotomy. A laparoscopic approach reduces the length of hospital stay (frequently done as outpatient surgery) and patient recovery time. Older studies have shown that survival rates for those who underwent second-look surgery, as compared with those who did not, were essentially the same, suggesting no benefit for the procedure. However, these studies have not evaluated the effect of newer chemotherapy and more effective salvage therapies. Although second-look surgery should not be considered standard of care, it should be performed in patients enrolled in clinical trials to evaluate the efficacy of newer first-line therapies and the efficacy of better salvage therapies in improving survival.
Secondary cytoreduction at the time of second-look laparotomy also has been examined in several studies that revealed a benefit in a select group of patients with gross disease that is reduced successfully to microscopic disease. However, only a minority of patients will have gross residual disease after clinical response to platinum-based chemotherapy. More recent data suggest that the presence or absence of residual disease at second-look surgery is one of the most significant prognostic factors for patient survival. Therefore, until more accurate diagnostic tests can be identified to determine residual disease after standard treatment, second-look surgery may be offered to patients with advanced stage disease in clinical remission to help determine prognosis, with the caveat that the procedure has prognostic value but no impact on survival.
Interval debulking surgery is performed after neoadjuvant chemotherapy in patients with advanced ovarian cancer. Although it remains a controversial subject, the Gynecological Cancer Cooperative Group of the European Organization for Research and Treatment of Cancer conducted a large randomized study documenting the benefit of interval debulking surgery. All patients received three cycles of cyclophosphamide and cisplatin followed by randomization to debulking versus no debulking surgery, and all patients then received three more cycles of chemotherapy. The study revealed that interval debulking surgery resulted in significantly better progression-free and overall survival (Fig. 55.34). Although the 6-month improvement in median overall survival is relatively small, interval debulking surgery may be considered in patients in whom cytoreduction was not optimal during initial surgery, yet who have responded to neoadjuvant chemotherapy.
|
|
|
FIG. 55.34. Survival of patients with advanced epithelial ovarian cancer according to whether or not they underwent debulking surgery. (From van der Burg MEL, van Lent M, Buyse M, et al. The effect of debulking surgery after induction chemotherapy on the prognosis in advanced epithelial ovarian cancer. N Engl J Med 1995;332:629–634, with permission.) |
Secondary cytoreductive surgery in patients with recurrent epithelial ovarian carcinoma has also been evaluated in several studies. Most of these reports show a statistically significant survival benefit for patients whose cytoreduction was optimal during secondary surgery. The likelihood of successful secondary cytoreduction and improved patient survival increases for patients with a longer disease-free interval (>12 months), unifocal disease, and initial optimal cytoreduction.
Chemotherapy
Following surgery, most patients will require chemotherapy. However, for patients with completely staged Ia or Ib grade 1 or 2 ovarian cancers, no survival benefit has been demonstrated with adjuvant chemotherapy. The studies have shown clear benefit to adjuvant chemotherapy in select patients with stage Ia or Ib grade 3 and stage Ic disease compared with observation. The GOG and the Gruppo Italiano Collaborativo in Oncologia Ginecologica studies found that patients with stage Ia or Ib grade 1 or 2 disease had comparable survival with observation and melphalan. In contrast, high-risk patients with stages Ia, Ib, Ic and II disease had improved disease-free intervals with cisplatin-containing regimens compared with observation, intraperitoneal phosphorus-32, or melphalan (Alkeran). The optimal number of cycles of chemotherapy for early-stage disease remains unclear, and it is the objective of a current prospective trial to compare the efficacy of three versus six cycles of paclitaxel and cisplatin in patients with early-stage ovarian cancer.
Various chemotherapeutic agents are active against ovarian cancer. Platinum-based therapy has been the mainstay of treatment for ovarian cancer for over 25 years. Combination platinum-based regimens have demonstrated superior response rates to those obtained with single-agent platinum, although there is insufficient evidence to suggest longer overall survival in these patients. The lack of survival benefit in some trials may relate to the high crossover treatment of patients who failed on one treatment arm and received other study treatments, as well as the varied length of patient follow-up in these studies.
Paclitaxel, a microtubule stabilizer, was found to have significant activity in many cancers, including epithelial ovarian carcinoma. In 1996, the GOG published a study comparing the efficacy of paclitaxel and cisplatin with cyclophosphamide and cisplatin as first-line adjuvant therapy in patients with suboptimally debulked epithelial ovarian carcinoma (stages III or IV). Disease-free survival and overall survival for the paclitaxel-cisplatin group was statistically longer than for the cyclophosphamide-cisplatin group (median survival 38 to 24 months). Paclitaxel-cisplatin was also more effective in patients with advanced stage ovarian cancer after optimal cytoreduction.
Substitution of carboplatin for cisplatin and decreased paclitaxel infusion times have reduced cost and patient side effects compared with the paclitaxel-cisplatin regimen. A subsequent prospective randomized trial replaced paclitaxel with docetaxel (Taxotere), another microtubule stabilizing agent, and found comparable response rates with less peripheral neuropathy. During the last 5 years, many new drugs have been found to be active second-line agents for epithelial ovarian cancer including topotecan, etoposide, altretamine (Hexalen), docetaxel (Taxotere), gemcitabine hydrochloride (Gemzar), vinorelbine tartrate (Navelbine), and doxorubicin hydrochloride liposomal injection (Doxil). Both topotecan, a topoisomerase inhibitor, and doxorubicin have shown promising activity in patients with recurrent, platinum-resistant disease with response rates of 20% to 30%. A large, prospective trial is randomizing patients to one of several combination multiagent chemotherapy regimens using gemcitabine, doxorubicin, or topotecan with paclitaxel-carboplatin as the control arm, to better define the best first-line regimen for patients with advanced stage ovarian cancer.
Neoadjuvant chemotherapy to reduce the tumor load before performing a laparotomy is being investigated. Based on available noninvasive testing, it is difficult to predict accurately which patients will not be candidates for a successful cytoreduction. Furthermore, neoadjuvant therapy has been employed only in small retrospective observational series. Although neoadjuvant chemotherapy has resulted in less morbidity from subsequent cytoreductive surgery, this approach has not translated into higher survival rates in the limited number of patients studied.
Intraperitoneal therapy allows direct exposure of a tumor to a drug concentration that may be 10-fold to 1,000-fold higher than that achieved with systemic therapy. In theory, this approach may have value for epithelial ovarian cancer, but its superiority has not yet been demonstrated despite extensive study. The clinical situations in which intraperitoneal therapy may be considered include salvage therapy for patients with disease of 5 mm or less, consolidation therapy for patients with surgically documented complete chemotherapeutic response, initial therapy for patients with high-grade early-stage tumors, or combination intraperitoneal therapy with systemic infusion. A randomized trial of intraperitoneal cisplatin and intravenous cyclophosphamide chemotherapy as first-line therapy showed promising survival rates with much less toxicity than older standard intravenous cisplatin-cyclophosphamide regimens. This study suggests that combination intraperitoneal cisplatin with an intravenous agent as primary therapy may be advantageous. However, optimal intraperitoneal regimens remain to be defined and will require randomized trials comparing intraperitoneal infusion with other approaches.
Radiation Therapy
Abdominopelvic radiation therapy has been used postoperatively in selected patients who have microscopic or small-volume residual disease; however, with more effective chemotherapy, radiation therapy is an infrequently used adjuvant therapy for ovarian cancer. One of the largest series, compiled by investigators in Toronto, treated patients with stage II and III disease with abdominopelvic radiation and showed a 10-year disease-free survival of 38% (n = 91) for patients with residual smaller than 2 cm and 6% (n = 91) for patients with residual larger than 2 cm. Further studies on the usefulness of radiation therapy for ovarian cancer are warranted.
Prognosis
With the addition of improved chemotherapeutic regimens and aggressive surgical cytoreduction, survival has improved in patients with advanced stage ovarian cancer. The overall 5-year survival is approximately 50%. Further strides in the management of ovarian cancer will require more knowledge of the molecular alterations that cause it. Advances in molecular biology indicate that ovarian tumorigenesis results from an accumulation of sequential genetic mutations. With a better understanding of the neoplastic transformation of ovarian epithelium, targeted therapies may be directed against the specific genetic aberrations in an individual cancer to improve patient outcome. Several innovative trials have combined gene therapy or immunotherapy and standard chemotherapy with promising early results. Ongoing efforts in both basic and clinical research are critical for the development of prevention strategies, screening for early detection, and better therapies.
FALLOPIAN TUBE CARCINOMA
Primary Fallopian Tube Epithelial Carcinoma
Fallopian tube carcinoma accounts for approximately 0.1% to 0.5% of all gynecologic malignancies. Peak incidence occurs at ages 60 to 64 years. Epidemiologic data suggest that age and nulliparity are associated with fallopian tube carcinoma, similar to endometrial and ovarian carcinomas. The constellation of symptoms including pelvic pain, a pelvic mass, and serosanguinous vaginal discharge (Latzko's triad) occur in the minority of cases. The most common symptom is vaginal staining or bleeding. Hydrops tubae profluens is characterized by colicky lower abdominal pain relieved by a profuse, serous, watery, yellow, intermittent vaginal discharge. The most common physical sign is a pelvic or abdominal mass. This mass often is thought to be an ovarian mass, with the correct diagnosis made only during surgery. Occasionally, a Pap smear revealing abnormal glandular cells with negative cervical or endometrial findings may lead the clinician to the diagnosis of fallopian tube cancer. Serum CA-125 levels often are elevated in advanced disease, as is noted in patients with ovarian carcinoma. Fallopian tube carcinoma is staged according to the FIGO. In contrast to ovarian cancer, most patients are diagnosed with early-stage disease: stage I, 37%; stage II, 21%; stage III, 31%; stage IV, 10%. (Table 55.6).
|
|
|
TABLE 55.6. FIGO staging for fallopian tube cancer |
Fallopian tube carcinoma usually is characterized by swollen tubes secondary to intraluminal growth. Most fallopian tube adenocarcinomas are serous and histologically identical to ovarian serous carcinoma (Fig. 55.35). Other types of epithelial fallopian tube carcinoma have been reported but are very rare. Several diagnostic criteria have been established to differentiate fallopian tube malignancy from ovarian and other primary tumors. Microscopically, the tumor must arise from the endosalpinx, with a histologic pattern consistent with tubal mucosal epithelium. The transition from benign to malignant epithelium is helpful for diagnosis. The ovaries and endometrium are either normal or have tumor smaller than that in the tube. As in epithelial ovarian carcinoma, fallopian tube epithelial tumors commonly spread by exfoliation and implantation throughout the peritoneal cavity. The extensive lymphatic channels within the fallopian tube facilitate lymphatic dissemination of tumor, and paraaortic lymph node metastases are common.
|
|
|
FIG. 55.35. Fallopian tube carcinoma. The fallopian tube lumen and its wall have been replaced by tumor. |
Surgery remains the principal treatment, including a total abdominal hysterectomy, bilateral salpingo-oophorectomy, tumor debulking, and a full staging, as with ovarian cancer (see Table 55.3). Treatment of primary fallopian tube epithelial tumors is based on the standard management for ovarian carcinoma, due to the lack of large-scale studies for this rare tumor. Platinum-based therapies are recommended following cytoreductive surgery, based on several series showing superior response rates and survival compared with those obtained with other agents.
Due to the rarity of fallopian tube cancer, paclitaxel has not yet been widely studied in this disease. However, based on data from ovarian cancer, platinum-taxane chemotherapy is used for patients with fallopian tube cancer. As in ovarian carcinoma, the prognosis is dependent upon the extent of disease and the amount of residual tumor at the end of surgery. Second-look surgery may be helpful in determining treatment efficacy and persistent disease but, as in ovarian carcinoma, it is not considered standard therapy.
Fallopian Tube Sarcoma
Sarcomas of the fallopian tubes may be classified as pure or mixed. They are exceedingly rare—fewer than 50 cases have been reported—and usually are found in postmenopausal women. Abdominal pain and watery or bloody vaginal discharge with signs of peritoneal spread are common findings. An ovarian origin must be ruled out by clearly identifying residual normal ovarian structure. The life expectancy of patients with these sarcomas usually is measured in months. As described in case reports, radiation and chemotherapy have not been effective.
Metastatic Disease
Metastatic fallopian tube disease is more common than primary tumors of the fallopian tubes. It often originates from a primary tumor of the ovary or endometrium and rarely from a primary cancer of the breast, gastrointestinal tract, or uterine cervix. Lymphatic involvement and an intact tubal epithelium distinguish a metastatic tumor from a primary fallopian tube tumor. The diagnostic criteria outlined above assist in differentiating between primary and metastatic fallopian tube tumors.
PRIMARY PERITONEAL CARCINOMA
Carcinoma may arise from the nonovarian epithelium of the peritoneal cavity and can involve diffusely all peritoneal surfaces, with minimal or absent involvement of the ovaries. These tumors share many of the clinical and histologic features of ovarian carcinoma. Most of these tumors are of serous histology. Clinical criteria have been established to distinguish peritoneal serous papillary carcinoma (PSPC) from its ovarian papillary serous counterpart, and molecular analyses of PSPC confirm that it is a distinct entity from ovarian carcinoma. Based upon these criteria, an estimated 7% to 15% of all cases of papillary serous ovarian cancer could be reclassified as PSPC.
The etiology of PSPC remains unknown. Considering the common embryologic origin of ovarian and peritoneal epithelium, a “field effect” of malignant epithelial transformation could involve multiple sites in the müllerian tract. Cases of PSPC have been described following prophylactic oophorectomy in patients with strong family histories of ovarian cancer or with documented BRCA mutations. PSPC appears to be another phenotypic variant of the BRCA-associated cancer syndromes. PSPC is staged and treated like ovarian carcinoma, with comparable response rates and survival.
HEREDITARY OVARIAN CANCER
Hereditary Forms of Ovarian Cancer
Approximately 5% to 10% of patients with ovarian cancer are thought to have an inherited genetic predisposition. Although this represents a small percentage of patients with ovarian cancer, great emphasis has been placed on these genetic mutations, given their clinical, social, and ethical implications.
Hereditary ovarian cancer syndromes are linked to dominantly inherited genetic mutations. A detailed family history from both maternal and paternal sides of the family must be obtained to determine the genetic susceptibility for the women at risk. Average age at diagnosis for this group of patients is often younger than for the general population, and most tumors are of serous histology.
The breast-ovarian cancer syndrome accounts for up to 85% of all hereditary ovarian cancer cases and is associated most frequently with mutations in the BRCA1 or BRCA2 genes. The BRCA1 and BRCA2 genes were identified and linked to hereditary breast and ovarian cancer in the 1990s. BRCA1 is located on chromosome 17q12-21, and BRCA2 is located on chromosome 13q12-13. Although the precise function of the BRCA genes is unknown, they appear to be involved in the recognition and repair of DNA damage. A sample pedigree of a breast-ovarian cancer syndrome family is illustrated in Fig. 55.36. The presence of a BRCA-associated syndrome is suspected whenever a pedigree reveals three or more affected relatives in two generations, bilateral or premenopausal breast cancers, a relative with breast and ovarian cancer, or a male with breast cancer in the pedigree (Table 55.7).
|
|
|
FIG. 55.36. Sample family pedigree. |
|
|
|
TABLE 55.7. Pertinent family history—risk factors for genetic mutations |
Many mutations have been described, located throughout the BRCA1 and BRCA2 genes, with nonsense mutations or frameshift mutations being predominant. Nonsense mutation occurs when a single nucleotide substitution results in a stop codon, and frameshift mutation occurs when one or more nucleotides are deleted to produce a downstream stop codon. Certain ethnic groups have higher carrier frequencies of founder BRCA mutations. Three specific founder mutations, 185delAG and 5382insC on BRCA1 and 6174delT on BRCA2, have been seen in approximately 2% to 2.4% of the Ashkenazi Jewish population. Studies have found that 40% to 60% of Jewish patients with ovarian cancer have a mutation in BRCA1 or BRCA2, in contrast to 5% of non-Jewish women with ovarian cancer.
The lifetime risk of ovarian cancer for patients with BRCA1 mutations is 20% to 60%, and the risk for a BRCA2 mutation carrier is 10% to 35%. Patients with BRCA1-associated ovarian cancer appear to be diagnosed at a younger age than patients with sporadic ovarian cancer or BRCA2-associated ovarian cancer. Studies show that BRCA2 mutations also are associated with other malignancies, including cancer of the pancreas, biliary tract, and prostate and melanoma (Table 55.8).
|
|
|
TABLE 55.8. Cancer risks by site for gene mutation carriers |
Data have also demonstrated that fallopian tube carcinoma can occur in patients with an inherited genetic predisposition to ovarian cancer by virtue of germline mutations in the BRCA genes. A large population-based series of patients with fallopian tube cancer found that 16% of patients had inherited mutations in BRCA1 or BRCA2. Based upon these data, fallopian tube carcinoma should be considered a clinical component of the BRCA-associated breast-ovary cancer syndrome. Patients with germline BRCA mutations who elect to have prophylactic surgery should have both ovaries and fallopian tubes removed, with careful histologic evaluation to eliminate occult fallopian tube carcinoma. It is unknown whether hysterectomy is necessary, in addition to salpingo-oophorectomy, in order to remove the cornual portion of the fallopian tube as surgical prophylaxis for these patients.
Hereditary nonpolyposis colorectal cancer (HNPCC) syndrome accounts for approximately 10% of all hereditary ovarian cancer cases. It is an autosomal dominant genetic syndrome characterized by three or more first-degree relatives with colon cancer (over 70% in the proximal colon) or endometrial cancer, where one is a first-degree relative of the other two, and two of them must be diagnosed with cancer before age 50 years. Four genes that are part of the DNA mismatch repair (MMR) pathway have been identified as being responsible for the HNPCC phenotype: hMSH2 (chromosome 2p), hMLH1 (chromosome 3p), hPMS1 (chromosome 2q), and hPMS2 (chromosome 7p). Most affected patients are found to have defects in either hMSH2 or hMLH1. An inherited defect in any one of these genes increases an individual's risk of developing cancer because of an impaired ability to repair somatic genetic mutations. Family members with HNPCC syndrome are at risk for cancer at other gastrointestinal sites, the urologic tract, and the ovary. The risk of endometrial cancer among women in the HNPCC syndrome is estimated to be 40% to 60% by the age of 70 compared with 1.5% in the general population. Limited studies have reported a 3.5-fold increase in the risk of ovarian cancer in members of these families (see Table 55.8).
Clinical Implications
Genetic Testing
Although hereditary cancers account for only 10% of ovarian malignancies, individual risk assessment and appropriate genetic testing can identify those individuals with significant lifetime risk.
The first concern in genetic testing is the scientific and technical utility of the tests: the reliability of tests, their predictive value, their interpretation, and ultimately their ability to prevent cancer in patients who have positive test results. The second major concern relates to the ethical, legal, and social implications of gene testing. The proper selection of patients for testing is very important. Genetic counseling is imperative. As testing for these genes becomes more available to the public in both commercial and academic settings, clinicians and patients must work together to ensure that the results are used in a fashion that will consider carefully the ethical, legal, and psychosocial issues that may arise from genetic testing. Multidisciplinary services that include pretest and posttest counseling, screening, treatment, and psychosocial sessions have been established in the major referral centers. Patients must be counseled regarding the potential advantages and disadvantages of genetic testing, as well as the utility of screening and prophylactic measures that are available.
American Society of Clinical Oncology guidelines recommend offering testing to anyone with a greater than 10% risk of carrying a mutation based on pedigree analysis, and only if the test result will influence medical management. First- and second-degree relatives of an affected individual from a family with breast-ovarian cancer syndrome should be offered genetic testing if they carry a mutation of the BRCA1 or BRCA2 gene. Certain ethnic groups, such as Ashkenazi Jews, are at increased risk for carrying a BRCA mutation, and a lower threshold for genetic testing may be appropriate in these individuals. Genetic testing for the MMR genes should be considered when there is a first-degree relative with a known mutation and when the patient meets the Amsterdam or the Bethesda Criteria for HNPCC syndrome (see Table 55.7).
A comprehensive family history can provide an estimate of an individual's risk of carrying a genetic mutation; however, genetic testing provides more accurate information regarding the chance that an individual patient will develop cancer. The best way to determine if a cancer-associated mutation is present in a family is to test an affected family member, who is usually the most likely proband to carry a deleterious mutation. The first family member often will need comprehensive gene sequencing, and subsequent individuals can then be tested for the identified mutation, which may be unique to the particular family. In the Ashkenazi Jewish population, genetic testing for the three founder mutations is required because of the high carrier frequency in this population and occasional reports of individuals carrying both BRCA1 and BRCA2 mutations.
Test results may come back positive or negative for an identifiable mutation or may report a mutation of indeterminate clinical significance. When a test result returns negative, interpretation will depend on the patient's family history. If an affected family member has tested positive for a mutation, then the patient has likely not inherited the deleterious mutation, and her cancer risk approximates that of the general population. If there is no documented positive mutation in the family, it is still possible that the patient has a cancer-associated mutation that is not detectable with testing. Approximately 12% of testing results are genetic variants or polymorphisms, reported as indeterminate clinical significance. Further study of these genetic variants and associated cancer risks in large populations will help reduce the number of indeterminate reports.
Follow-up and Management
Screening
Follow-up and treatment of patients with an inherited genetic predisposition to ovarian cancer is complex due to the variable penetrance of genetic alterations and the lack of effective early detection methods. Serum CA-125 levels and transvaginal ultrasonography with color Doppler flow are the two modalities that have shown potential usefulness in surveying these patients. CA-125 levels may be elevated in other benign diseases and may not be elevated in early stage I ovarian cancer; thus, it lacks sufficient sensitivity. Transvaginal ultrasonography is very sensitive in detecting adnexal masses; however, it cannot differentiate between malignant and benign masses, resulting in a high false-positive result rate and unnecessary surgery, especially in premenopausal women. Consequently, screening of the general population is not warranted with the currently available technology.
There is no conclusive evidence to support screening patients with higher risk of ovarian cancer. Ovarian surveillance modalities have not improved significantly the detection of early-stage invasive epithelial ovarian cancers, with minimal impact on the survival among the screened patients. One prospective study of high-risk women demonstrated enhanced survival among the screened population versus historic controls; however, conclusions are limited by small patient numbers. Despite the available data, the National Institute of Health Consensus Statement on Ovarian Cancer and the Cancer Genetic Studies Consortium recommend screening starting at ages 25 to 35 years as part of the annual or semiannual routine examination for women with germline BRCA1 or BRCA2 mutations. The benefit, however, is not proven, because the evidence is based on expert opinion only. Table 55.9 lists the provisional recommendations for cancer surveillance for the carriers of BRCA1 and BRCA2 mutations. Screening and prophylactic surgery for patients with HNPCC-associated mutations is also based on expert opinion, although colonoscopy and stool occult blood testing have been shown in randomized clinical trials to reduce the incidence of and mortality from colon cancer (see Table 55.9).
|
|
|
TABLE 55.9. Recommendations for treatment of women at high genetic risk for gynecologic malignancies |
Prevention
Chemoprophylaxis with oral contraceptive pills (OCP) for 5 years decreases ovarian cancer risk by 50% in the both general population and in high-risk women. A case-controlled study of 207 known BRCA mutation carriers and their sister controls found a 60% reduction of ovarian cancer risk with OCP use. Other risk-reducing strategies such as tubal ligation and hysterectomy have also demonstrated reduced incidence of ovarian cancer among many high-risk women.
Prophylactic Salpingo-oophorectomy
Prophylactic bilateral salpingo-oophorectomy (PBSO) is another preventive strategy for patients with BRCA mutations to reduce cancer risk. Given that most hereditary ovarian cancer occurs after the age of 35 to 40, prophylactic oophorectomy is an option in carefully selected patients at unequivocally high risk for ovarian cancer who have completed their childbearing. This procedure can be performed laparoscopically, and the decision for concomitant hysterectomy is individualized. The surgical pathologist must be alerted to perform a careful examination of the high-risk patient's ovaries and fallopian tubes given the reports of occult ovarian and tubal carcinoma in these specimens ranging in frequency from 2% to 15%.
The efficacy of PBSO in hereditary breast-ovarian cancer syndrome has been evaluated in several studies. Prospective studies have documented a significant reduction in both ovarian, peritoneal, and breast carcinoma among BRCA mutation carriers following PBSO, one study of 248 patients found a 98% decrease of ovarian and primary peritoneal cancers, and a 50% reduction in subsequent breast cancer compared with age-matched controls. This supports the findings of another prospective study that documented a 50% reduction of breast cancer with PBSO among premenopausal and postmenopausal BRCA mutation carriers that persisted irrespective of the use of hormone replacement therapy. Despite oophorectomies, primary peritoneal cancer has been reported in 0.4% to 10.7% of high-risk patients, and patients must be counseled regarding this disease. It is believed that that all coelomic epithelium, including the peritoneal covering of the entire abdominal pelvic cavity, is prone to malignant transformation in BRCA mutation carriers. Several other significant issues remain unresolved regarding the physiologic adjustments to premature surgical menopause and the safety of hormone replacement therapy in this group, especially in those at high risk for breast cancer.
Recommendations from other agencies or groups are as follows:
· American College of Obstetricians and Gynecologists (1992). Women with familial ovarian or hereditary breast-ovarian cancer syndromes who do not wish to maintain their reproductive capacity may be offered PBSO. Such women should have a documented familial syndrome, preferably established via a full pedigree analysis by a geneticist. These women should be informed that removal of the tubes and ovaries does not provide 100% protection; primary peritoneal carcinoma has been reported after bilateral salpingo-oophorectomy in some cases.
· National Institutes of Health Consensus Statement on Ovarian Cancer (1994). The probability of a hereditary ovarian cancer syndrome in a family pedigree increases with the number of affected relatives, with the number of affected generations, and with young age of onset of disease. Therefore, prophylactic oophorectomy should be considered in these patients after careful weighing of the risks and potential benefits. The risk of ovarian cancer in women from families with hereditary ovarian cancer syndrome is sufficiently high to recommend prophylactic oophorectomy at age 35 years or after completion of childbearing.
SUMMARY POINTS
· Germ cell tumors are the most common gynecologic malignancy in young women. The cure rate is 90% due to the significant advancement of chemotherapeutic regimens. The most commonly used regimen is BEP. The risk of secondary malignancy, especially hematologic, with the use of etoposide must always be considered when this regimen is administered.
· Sex cord–stromal tumors, such as granulosa cell and Sertoli-Leydig cell tumors, have the potential for steroid hormone secretion, with estrogen being the most frequently produced hormone. Aside from juvenile granulosa cell tumors, sex cord–stromal tumors rarely occur in women under 20 years. Primary treatment usually entails primary tumor resection and staging. When the pathology is malignant, the need for chemotherapy must be individualized, because no standard treatment regimen has been established.
· Tumors of LMP account for approximately 15% of epithelial ovarian cancers and have an earlier age of onset, around 40 years. All epithelial histologic subtypes of LMP tumors exist, with serous being the most common. Pseudomyxoma peritonei, characterized by extracellular gelatinous material in the peritoneal cavity, may accompany mucinous LMP tumors. Therapy for LMP tumors is surgical excision and appropriate tumor staging and, when necessary, debulking.
· Although significant strides have been made in the treatment of epithelial ovarian cancer, the 5-year survival rate remains approximately 50%. Continued efforts are needed to understand the etiology of these tumors, which ultimately will result in the development of prevention strategies, early detection, and improved therapies. Optimal cytoreductive surgery followed by platinum-taxane combination chemotherapy is the current standard treatment for advanced stage disease.
· Identification of the genes that are responsible for several familial ovarian cancer syndromes, especially BRCA1 and BRCA2, has opened up new areas of investigation and provided valuable information for understanding the development of both hereditary and sporadic ovarian carcinomas. Although there is no level I evidence to support the use of screening and prophylactic surgery in patients who are at high risk for ovarian cancer, these modalities may be considered to reduce the chance of disease development. Oral contraceptive pill use may also decrease ovarian cancer incidence in these high-risk women.
SUGGESTED READINGS
Germ Cell and Stromal Tumors
Gershenson DM. Chemotherapy of ovarian germ cell tumors and sex cord–stromal tumors. Semin Surg Oncol 1994;10:290–298.
Gershenson DM. Management of early ovarian cancer: germ cell and sex cord–stromal tumors. Gynecol Oncol 1994;55:S62–S72.
Kurman RJ. Blaustein's pathology of the female genital tract, fourth ed. New York: Springer-Verlag, 1994.
Powell JL, Johnson NA, Bailey CL, et al. Management of advanced juvenile granulosa cell tumor of the ovary. Gynecol Oncol 1993;48:119–123.
Williams SD. Ovarian germ cell tumors: an update. Semin Oncol 1998;25:407–413.
Low Malignant Potential Tumors
Gershenson DM, Silva EG, Levy L, et al. Ovarian serous borderline tumors with invasive peritoneal implants. Cancer 1998;82:1096–1103.
Trimble C, Trimble EL. Management of epithelial ovarian tumors of low malignant potential. Gynecol Oncol 1994;55:S52–S61.
Seidman JD, Kurman RJ. Ovarian serous borderline tumors: a clinical review of the literature with emphasis on prognostic indicators. Hum Pathol2000;31:539–557.
Young RH, Gilks CB, Sculy RE. Mucinous tumors of the appendix associated with mucinous tumors of the ovary and pseudomyxoma peritonei: a clinicopathologic analysis of 22 cases supporting an origin in the appendix. Am J Surg Pathol 1991;15:415–429.
Zanetta G, Rota S, Chairi S, et al. Behavior of borderline tumors with particular interest to persistence, recurrence, and progression to invasive carcinoma: a prospective study. J Clin Oncol 2001;19:2658–2664.
Epithelial Ovarian Tumors
Bristow RE, Lagasse LD, Karlan BY. Secondary surgical cytoreduction for advanced epithelial ovarian cancer: patient selection and review of the literature. Cancer 1996;78:2049–2062.
Bristow RE, Tomacruz RS, Armstrong DK, et al. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol 2002;20:1248–1259.
Colombo N, Chiari S, Maggioni A, et al. Controversial issues in the management of early epithelial ovarian cancer: conservative surgery and role of adjuvant therapy. Gynecol Oncol 1994;55:S47–S51.
Hoskins WJ, Bundy BN, Thigpen JT, et al. The influence of cytoreductive surgery on recurrence-free interval and survival in small-volume stage III epithelial ovarian cancer: a Gynecologic Oncology Group Study. Gynecol Oncol 1992;47:159–166.
McGuire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med 1996;334:1–6.
Podratz KC, Malkasian GD Jr, Hilton JF, et al. Second-look laparotomy in ovarian cancer: evaluation of pathologic variables. Am J Obstet Gynecol1985;152:230–238.
Rubin SC, Randall TC, Armstrong KA, et al. Ten-year follow-up of ovarian cancer patients after second-look laparotomy with negative findings. Obstet Gynecol 1999;93:21–24.
Thomas GM, Dembo AJ. Integrating radiation therapy into the management of ovarian cancer. Cancer 1993;71:1710–1718.
van der Burg MEL, van Lent M, Buyse M, et al. The effect of debulking surgery after induction chemotherapy on the prognosis in advanced epithelial ovarian cancer. N Engl J Med 1995;332:629–634.
Williams L, Bruetto VL, Yordan E, et al. Secondary cytoreductive surgery at second-look laparotomy in advanced ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol 1997;66:171–178.
Young RC, Walton LA, Ellenberg SS, et al. Adjuvant therapy in stage I and stage II epithelial ovarian cancer. N Engl J Med 1990;322:1021–1027.
Fallopian Tube Carcinomas
Aziz S, Kuperstein G, Rosen B, et al. A genetic epidemiological study of carcinoma of the fallopian tube. Gynecol Oncol 2001;80:341–345.
Nordin AJ. Primary carcinoma of the fallopian tube: a 20-year literature review. Obstet Gynecol Surv 1994;49:349–361.
Pecorelli S, Odicino F, Maisonneuve P, et al. Carcinoma of the fallopian tube. J Epidemiol Biostat 1998;3:63–74.
Peters WZ III, Andersen WA, Hopkins MP. Results of chemotherapy in advanced carcinoma of the fallopian tube. Cancer 1989;63:836–838.
Primary Peritoneal Carcinomas
Blouses JD, Lao YS, Buller RE, et al. Extraovarian peritoneal serous papillary carcinoma: a case-control retrospective comparison to papillary adenocarcinoma of the ovary. Gynecol Oncol 1993;50:347–351.
Chu CS, Menzin AW, Leonard DGB, et al. Primary peritoneal carcinoma: a review of the literature. Obstet Gynecol Surv 1999;54:323–335.
Karlan BY, Baldwin RL, Lopez-Luevanos E, et al. Peritoneal serous papillary carcinoma, a phenotypic variant of familial ovarian cancer: implications for ovarian cancer screening. Am J Obstet Gynecol 1999;180:917–928.
Hereditary Ovarian Cancer
American College of Obstetrics and Gynecology Genetic risk and screening techniques for epithelial ovarian cancer. ACOG Committee Opinion No. 117L, 1992.
Boyd J, Rubin SC. Hereditary ovarian cancer: molecular genetics and clinical implications. Gynecol Oncol 1997;64:196–206.
Burke W, Daly M, Garber J, et al. For the Cancer Genetic Studies Consortium. Recommendations for follow-up care of individuals with an inherited predisposition to cancer II: BRCA1 and BRCA2. JAMA 1997;227:997–1003.
Colgan TJ, Murphy J, Cole DE, et al. Occult carcinoma in prophylactic oophorectomy specimens: prevalence and association with BRCA germline mutation status. Am J Surg Pathol 2001;25:1283–1289.
Karlan BY, Platt LD. The current status of ultrasound and color Doppler imaging in screening for ovarian cancer. Gynecol Oncol 1994;55:S28–S33.
Kauf ND, Satagopan JM, Robson ME, et al. Risk-reducing salpingo-oophorectomy in women with BRCA1 or BRCA2 mutation. N Engl J Med 2002;346:1609–1615.
Narod SA, Risch H, Moslehi R, et al. Oral contraceptives and the risk of hereditary ovarian cancer. N Engl J Med 1998;339:424–428.
NIH Consensus Conference. Ovarian cancer screening, treatment and follow-up. JAMA 1995;273:491–497.
Rebbeck TR, Levin AM, Eisen A, et al. Breast cancer risk after bilateral prophylactic oophorectomy in BRCA1 mutation carriers. JNCI 1999;19:1475–1479.
Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med1997;336:1401–1408.
Weber BL, Punzalan C, Eisen A, et al. Ovarian cancer risk reduction after bilateral prophylactic oophorectomy (BPO) in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet 2000;67:59.