Definition
• Pediatric embryonal tumor of either benign or malignant behavior arising from precursor cells of sympathetic nervous system origin
Pathogenesis
• Activating mutations in ALK gene and loss of function mutations in PHOX2B gene account for most familial cases of neuroblastomas, although they can also occur in sporadic cases
• Sporadic cases may result from the accumulation of multiple copy-number DNA variations in certain genes (such as FLJ22536, BARD1, NBPF23) that may, through modification of MDM2 and p53, act synergistically in tumorigenesis
• A number of other DNA changes can determine the phenotype of the tumor; whole chromosome gains may result in a good prognosis and a benign course whereas segmental chromosomal changes (such as MYCN amplification, seen in about 20% of tumors) may lead to a malignant phenotype
Clinical features
Epidemiology
• Incidence of all neuroblastomas is about 10 cases per million children younger than 15 years
• Most common cancer of children younger than 1 year
• Half of all cases are diagnosed by age 2, and about 90% are discovered by age 5; median age at diagnosis is 17 months
• Neuroblastomas more often arise from the adrenal medulla (about 40% of cases) and less commonly along the sympathetic ganglion chain; thoracic lesions account for about 20% of cases
Presentation
• Variable: some patients have an asymptomatic mass whereas others may have life-threatening compressive symptoms or widespread metastatic disease
• Those with tumors arising from the sympathetic chain may present with Horner’s syndrome (unilateral ptosis, miosis, and anhidrosis), pain, paresthesias, paralysis, or even respiratory distress as the mass may impinge on nearby thoracic structures
• Tumors are often metabolically active with production of catecholamines that can be detected in patients’ urine as homovanillic acid and vanillylmandelic acid and that may cause vasoactive symptoms such as flushing, hypertension, and watery diarrhea
• Radiographically, a large infiltrative paraspinal mass is present, often with skeletal erosion, calcification, and foci of hemorrhage and necrosis
• Radionuclide imaging using the epinephrine precursor iodine 123 metaiodobenzylguanidine can be used to detect metastatic lesions
• Patients may present with metastasis, most often to bone marrow, bone, liver, or lymph nodes
Prognosis and treatment
• Prognosis is determined based on various clinical, histological, and genetic factors
• Clinical factors: patient age (those younger than 1 year have favorable outcomes); the International Neuroblastoma Staging System: assessment of excision completeness, lymph node involvement, metastasis, and local tumor extension
• Genetic factors: DNA ploidy and MYCN status also prognostically important; hyperdiploid and MYCN nonamplification confer a better prognosis. Cytogenetics and molecular testing for the presence of MYCN amplification should be included in the pathology report
• Histological factors: International Neuroblastoma Pathologic Classification: modified version of Shimada classification; determines presence of Schwannian stroma (more stroma is prognostically favorable), cell differentiation, and mitosis–karyorrhexis index (MKI; count of combination of mitotic figures and apoptotic/karyorrhectic cells)
• Treatment for low stage tumors is complete surgical excision. Higher stage (III and IV) cases also receive chemotherapy with or without radiation therapy. Patients may also undergo myeloablative chemotherapy with hematopoietic stem cell rescue
Pathology
Gross
• Tumors are large and encapsulated; cut section demonstrates lobulated architecture and a soft to fleshy consistency. Hemorrhage is often present
Histology
• Proliferation of small, round blue cells with hyperchromatic nuclei, “salt and pepper” chromatin, and indistinct cytoplasm. Up to 50% of the stroma may resemble Schwannian stroma, but it may also be composed of neuropil (Schwannian stroma-poor)
• Divided into differentiating, poorly differentiated, and undifferentiated subtypes
• Differentiating tumors have ganglionic differentiation of 5% to 50% of neoplastic cells
• Poorly differentiated tumors have <5% ganglionic differentiation
• Undifferentiated tumors have no ganglionic differentiation and lack fibrillary network of neuropil
• Tumors are graded based on age, Schwannian stroma, differentiation, and MKI, which is a count of 5000 cells. If >200 mitotic or karyorrhectic cells are found, it is considered a high MKI. A count <100 is considered to be a low MKI
Immunopathology/special stains
• Immunohistochemical analysis will be positive against antibodies for chromogranin, synaptophysin, PGP 9.5, and NSE. ALK may be positive in some cases
• CD56 is positive, which may be helpful in cases where a known neuroblastoma has infiltrated the bone marrow, as it is a sensitive but not specific marker
Main differential diagnoses
• Rhabdomyosarcoma: may see rhabdomyoblasts; muscle markers (e.g., desmin, myogenin) positive
• Primitive neuroectodermal tumor/Ewing: chromogranin negative; CD99 and FLI-1 positive
• Lymphoma: no “salt and pepper” chromatin; may see atypical lymphoid cells; LCA and T-cell or B-cell markers positive
• Desmoplastic small round cell tumor: desmoplastic stroma, no rosettes, and WT1 (nuclear) positive
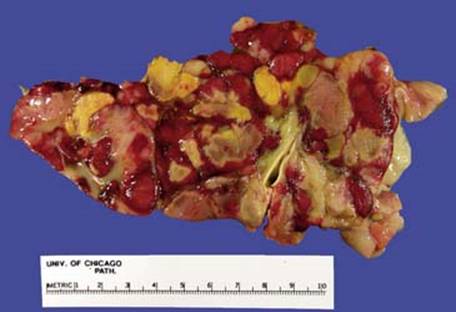
Fig 1 Mediastinal neuroblastoma. Gross photograph of a neuroblastoma showing nodular fleshy cut surface with areas of hemorrhage and necrosis.
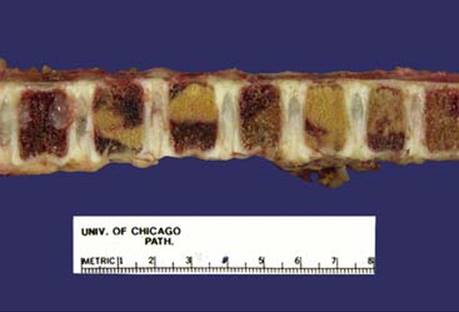
Fig 2 Mediastinal neuroblastoma. Metastatic neuroblastoma in vertebral column (yellow areas) from a 3-year-old who died of widely metastatic disease.
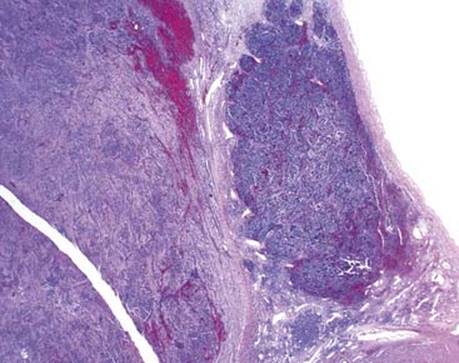
Fig 3 Mediastinal neuroblastoma. Low-power view demonstrates an encapsulated lesion with two areas: one with tightly packed dark cells and the other with apparently more voluminous cytoplasm.

Fig 4 Mediastinal neuroblastoma. Medium-power view of the darker area shows nests of monotonous, small, round blue cells in a pink, fibrillary stoma. Note small amount of Schwannian stroma with blood vessels separating the tumor cells into nests.
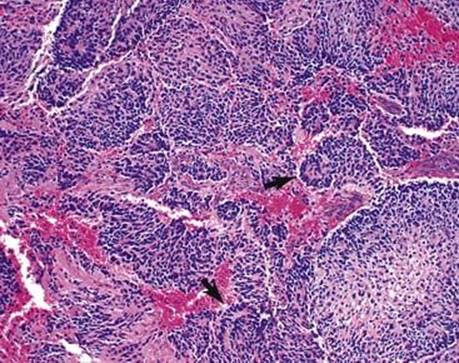
Fig 5 Mediastinal neuroblastoma. At higher power, small rosettes with central neuropil (arrows) are seen.
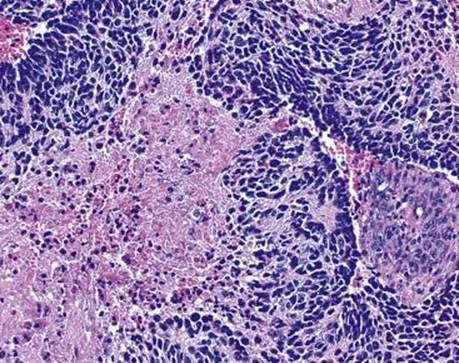
Fig 6 Mediastinal neuroblastoma. Highest power of same case as in Fig 5 shows low MKI.
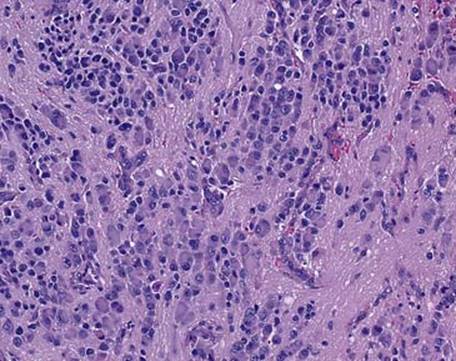
Fig 7 Mediastinal neuroblastoma. The second of the two areas from the same tumor shows cells with abundant pink cytoplasm and vesiculated, eccentric, large nuclei (ganglionic differentiation).
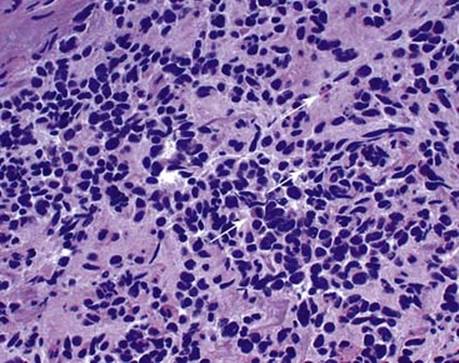
Fig 8 Mediastinal neuroblastoma. High power from a poorly differentiated neuroblastoma with a high MKI (arrows).
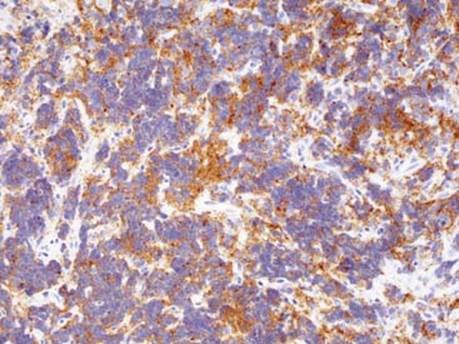
Fig 9 Mediastinal neuroblastoma. Synaptophysin staining by immunohistochemistry can be helpful in diagnosing undifferentiated neuroblastoma.
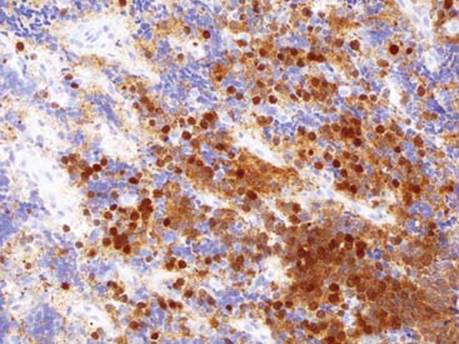
Fig 10 Mediastinal neuroblastoma. PGP 9.5 staining by immunohistochemistry can be helpful in undifferentiated neuroblastoma.