Bruce A. Chabner
INTRODUCTION
In the past decade, the taxanes have emerged as one of the most powerful classes of anticancer drugs (1). Two unmodified taxanes, paclitaxel and docetaxel, are approved for clinical use in multiple tumors. An albumin-stabilized paclitaxel (abraxane) is also available for treatment of breast cancer (2), and a new analogue, cabazitaxel, is approved for hormone refractory prostate cancer. Despite their similar structures and a common mechanism of action (disruption of microtubule function), the taxanes differ in their pharmacological profiles, toxicity, and their patterns of clinical activity. Taxanes are predominantly employed in solid tumor chemotherapy in combination with platinum derivatives, with other cytotoxics, or with monoclonal antibodies such as Herceptin (trastuzumab). Both unmodified taxanes act synergistically with trastuzumab against HER2/neu overexpressed breast cancer cells in vitro and in vivo, and the combination of taxane and trastuzumab improves survival against HER2/neu amplified breast cancer in the adjuvant setting. The two original taxanes differ in their interaction with doxorubicin, paclitaxel potentiating the anthracycline’s cardiac toxicity, while docetaxel and doxorubicin are well tolerated and highly active in combination (3). The taxanes are also primary agents for treating other malignancies, including ovarian, lung, and bladder cancer.
A closely related antimitotic agent, ixabepilone, is approved for second-line breast cancer treatment after taxanes, and differs from taxanes in its greater neurotoxicity and its lack of cross-resistance in MDR-positive tumors.
STRUCTURE
Paclitaxel was first isolated from the bark of the Pacific yew, Taxus brevifolia. Paclitaxel and its analogue, docetaxel, are now synthesized from 10-deacetylbaccatin III, a precursor found in the leaves of the European yew, Taxus baccata (4). Both molecules are composed of a 15-member taxane ring system linked to a 4-member oxetan ring at the C-4 and C-5 positions of the molecule. The structures of paclitaxel and docetaxel differ in substitutions at the C-10 ring position and in the configuration of an ester side chain attached at C-13. Docetaxel is slightly more water soluble than paclitaxel and a more potent inhibitor of tubulin in cell-free systems. The side chain substitutions at C-13 position are essential for antimicrotubule activity. The chemical structures of paclitaxel and docetaxel are shown in Figure 3–1. Abraxane is identical to paclitaxel, but is formulated within a microalbumin particle that eliminates the hypersensitivity caused for the lipid excipient used to deliver paclitaxel. Cabazitaxel retains the taxane nuclear ring system but has multiple side chain modifications to increase its solubility and decrease susceptibility to multidrug resistance.


FIGURE 3-1 The chemical structure of antimitotics: (A–C) taxanes, (D) ixabepilone, and (E) eribulin.
MECHANISM OF ACTION
The taxanes stabilize microtubules. They bind to the interior surface of the β-microtubule chain and enhance microtubule assembly by promoting the nucleation and elongation phases of tubulin polymerization. In solution they reduce the critical tubulin subunit concentration required for microtubule assembly. Unlike the vinca alkaloids, which prevent microtubule assembly, the taxanes decrease the lag time to assembly and dramatically shift the dynamic equilibrium from tubulin dimers to microtubule polymers (5).
The taxane binding site on β-tubulin is distinct from those of vinca alkaloids, podophyllotoxin, colchicines, maytansine, and the maytansine-like antimitotic attached to anti CD30 antibody (brevituximab vendotin). Paclitaxel and docetaxel bind reversibly to both the N-terminal residues and the internal amino acid residues at 217–233 positions. This binding increases the rate of tubulin polymerization, disrupts the orderly formation of mitotic spindles and segregation of chromosomes, halts progression through mitosis, and promotes apoptosis. Taxanes block the anti-apoptotic effects of the BCL-2 gene family, and induce p53 gene activation with consequent mitotic arrest, formation of multinucleated cells, and cell death.
In addition to their direct cytotoxic effects, taxanes potently inhibit vascular endothelial cell proliferation, and enhance the cytotoxic effects of radiation at clinically achievable concentrations.
DRUG RESISTANCE
Two major mechanisms of taxane resistance have been characterized in cells selected in vitro (6). Taxanes are one of many natural product drugs affected by multidrug resistance (MDR) as mediated through increased expression of the 170-kD p-glycoprotein, an efflux pump encoded by the MDR-1 gene. The p-glycoprotein promotes rapid efflux of taxanes, anthracyclines, and vinca alkaloids, as well as other natural products. MDR resistance can be reversed in vitro and in animal test systems by calcium channel blockers, tamoxifen, hormones, cyclosporine A, and even cremaphor, the principal lipid used to formulate paclitaxel. The precise role of MDR-1 in conferring resistance to the taxanes in the clinical setting is not firmly established. For example, clinical observations to date suggest that in breast cancer, there is incomplete cross-resistance between taxanes and anthracyclines, implying that MDR-1 expression is not responsible for drug resistance in all cases. A second form of resistance to taxanes is seen in cells that express an altered β-tubulin phenotype, either through mutations or due to minor polymorphisms that modify taxane binding. Paclitaxel-resistant, β-tubulin mutant cells have an impaired ability to polymerize tubulin dimers into microtubules. Amplification of β-tubulin encoding genes, mutation of the β-tubulin binding sites, and isotype switching of β-tubulin all have been reported in taxane-resistant cell lines.
An additional mechanism responsible for taxane resistance has been attributed to increased expression of MCL-1 and BCL-2, both of which inhibit apoptosis. Ixabepilone and cabazitaxel are less susceptible to MDR-mediated resistance, as compared to the original taxanes. β-Tubulin mutations have been linked to ixabepilone resistance in preclinical experiments.
CLINICAL PHARMACOLOGY AND METABOLISM
The taxanes are active only in their parent form, and all are administered intravenously. Oral bioavailability of either paclitaxel or docetaxel is poor due to high-level expression of p-glycoprotein and other ATP-binding cassette (ABC) transporters in intestinal epithelium, and first-pass drug metabolism in the liver.
The metabolism of all clinically approved taxanes is mediated through hepatic cytochrome p450 mixed-function oxidases. Paclitaxel is inactivated to hydroxylated metabolites through stepwise catalysis by cytochrome 2C8, producing 6α-OH, and CYP3A4, producing 6α-OH-3′OH, and finally to the dihydroxyl product (7). Docetaxel is oxidized at C13 by CYP3A4. The involvement of cytochrome enzymes in taxane biotransformation has two important implications: first, co-medications capable of inducing or inhibiting cytochromes influence the rate of inactivation and the metabolic fate of taxanes (8). Second, polymorphisms affecting enzymatic function have been described for both CYP2C8 and CYP3A4, thereby leading to interpatient variability of pharmacokinetics. Pharmacokinetic data for paclitaxel and docetaxel are shown in Table 3-1.
TABLE 3-1 COMPARATIVE PHARMACOKINETIC CHARACTERISTICS OF TAXANES

![]() PACLITAXEL
PACLITAXEL
Pharmacokinetic studies have disclosed substantial interpatient variability and nonlinearity of the relationship between paclitaxel dose and drug concentration in plasma (Table 3-2). Nonlinearity is particularly prominent with shorter (1- to 3-h) drug infusion schedules, and may indicate variability of tissue binding and clearance mechanisms.
TABLE 3-2 PHARMACOKINETIC PARAMETERS FOR PACLITAXEL
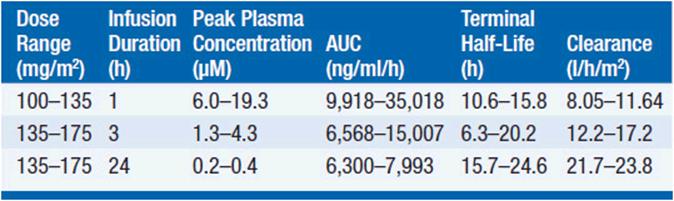
The pharmacokinetics of this drug have been evaluated in doses ranging from 100 to 300 mg/m2 infused in time periods of 1, 3, and 24 h. Following intravenous administration, the drug exhibits a biphasic decline in plasma concentration, reaching peak concentrations between 5 and 10 μM for 1-3 h infusions, and remaining in the inhibitory range for myelopoiesis (above 50 nM) for 12–24 h. Both the terminal half-life of 10-24 h and the mean clearance of paclitaxel appear to either remain unchanged or slightly increase as the infusion time is increased. Approximately 80% of paclitaxel is excreted in feces in the form of CYP2B8 and 3A4 metabolites, the 6α-hydroxy-paclitaxel, the C3′-hydroxy paclitaxel, and the dihydroxy products accounting for the bulk of the dose. Renal clearance of paclitaxel and its metabolites is minor, accounting for about 15% of administered dose and only 5% is excreted unchanged. The dose should be reduced by 50% in patients with a bilirubin greater than 1.5 mg/dl, and the drug should be withheld in patients with severe hepatic dysfunction.
![]() DOCETAXEL
DOCETAXEL
The pharmacokinetic behavior of docetaxel on a 1-h schedule at doses of 75–115 mg/m2 or less displays a linear relationship between dose and drug concentrations in plasma. The terminal half-life is about 17 h. As with paclitaxel, docetaxel is cleared by CYP-mediated metabolism and is widely distributed among tissues except for central nervous system.
DRUG INTERACTIONS
Because of its reliance for clearance upon the cytochrome system, taxanes have pharmacokinetic interactions with other cancer drugs (Table 3-3).
TABLE 3-3 CLINICALLY SIGNIFICANT TAXANE-DRUG INTERACTION
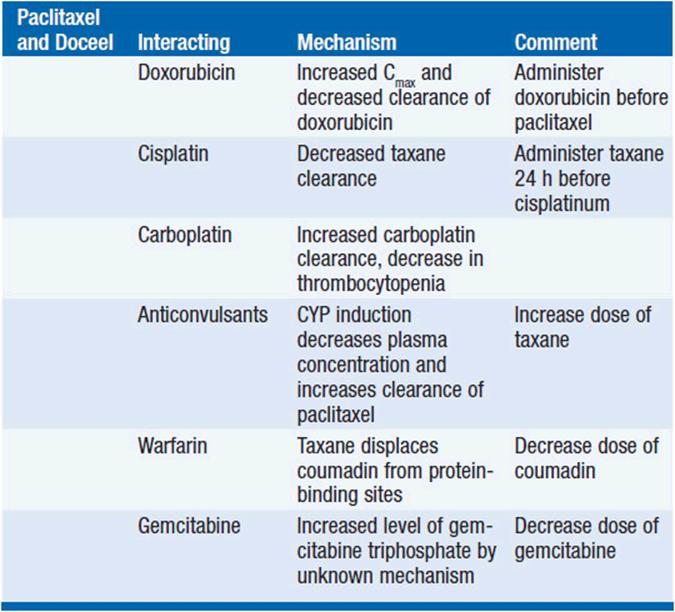
Paclitaxel preceding doxorubicin increases the frequency of mucositis, cardiotoxicity, and neutropenia than would be anticipated from additive effects of the two drugs. Pharmacokinetic studies indicate that paclitaxel decreases doxorubicin clearance. Administration of these drugs 24 h apart may ameliorate this effect.
Alternating sequences of paclitaxel and cyclophosphamide revealed that cytopenias were profound when paclitaxel was infused prior to, but not after, cyclophosphamide. Docetaxel given before ifosfamide increases the clearance of the alkylator, and dose-limiting toxicity occurred at lower doses of docetaxel when ifosfamide preceded the taxane (9).
Enzyme-inducing anticonvulsants increase CYP3A4 activity, accelerate taxane clearance, and markedly increase the dose of paclitaxel required to reach a maximum tolerated dose. CYP3A inhibitors, such as ketoconazole, profoundly slow taxane clearance.
TOXICITY
Neutropenia is the principal and dose-limiting toxicity of both taxanes, docetaxel being the most myelosuppressive. The severity and frequency of paclitaxel-induced neutropenia increases when infusion is prolonged from 3 to 24 h and at doses above 175 mg/m2. However, neutropenia is non-cumulative, and its duration even in heavily pretreated patients is usually brief. Weekly treatments with lower doses of 80–100 mg/m2 of paclitaxel yield antitumor activity at least equivalent to higher doses given every 3 weeks in breast cancer, with less acute toxicity. Severe thrombocytopenia and anemia are uncommon except in heavily pretreated patients.
In the absence of antihistamines and glucocorticoids, paclitaxel administration causes a high incidence of acute hypersensitivity reactions, primarily related to the cremaphor solvent. The incidence and the intensity of hypersensitivity to paclitaxel formulations are significantly diminished by pretreatment with dexamethasone, diphenhydramine, and ranitidine.
Cardiac arrhythmias, especially asymptomatic bradycardias, are seen after paclitaxel. Paclitaxel should be used with EKG monitoring in patients with a history of cardiac conduction disturbances. As discussed previously, paclitaxel increases the incidence of anthracycline-induced congestive heart failure (CHF), when the two drugs are used together. There is no conclusive evidence for an increased rate of CHF in patients receiving docetaxel/anthracycline combinations. Neither taxane increases the cardiac toxicity of trastuzumab. Dose-related myalgia and neuropathy, especially an increase in neurosensory symptoms (numbness in a symmetrical glove and stocking distribution), may become significant complaints with paclitaxel, particularly with higher doses, after multiple cycles, and in combination with cisplatin.
Neurotoxicity is more frequently associated with shorter (1–3 h) infusion schedules and with weekly, “dose-dense” schedules commonly used in ovarian and breast cancer, indicating that peak plasma concentrations and dose density are principal determinants. Mild to moderate peripheral neurotoxicity occurs in approximately 40% of patients receiving every 3 week paclitaxel, especially with those who have previously received cisplatin, but neurotoxicity, asthenia, and muscular weakness become prominent complaints from patients who have received large cumulative doses and those treated on a long-term weekly schedule (10).
The toxicity of docetaxel closely mimics that of paclitaxel with several important exceptions. Docetaxel is more myelosuppressive and stomatitis is more frequent. Nausea, vomiting, and diarrhea have been observed with both taxanes, but severe gastrointestinal toxicity is uncommon.
During its early phases of development, docetaxel treatment led to a cumulative fluid retention syndrome in approximately 50% of patients after three to five cycles of therapy. Ankle edema, pleural effusions, and even ascites may become dose limiting. Premedication with dexamethasone, 8 mg twice daily for 3–5 days, beginning 1 day before drug administration, significantly decreases the incidence and severity of the fluid retention syndrome.
FORMULATION AND ADMINISTRATION
![]() PACLITAXEL
PACLITAXEL
Taxanes are insoluble in water. Paclitaxel is formulated in 50% alcohol and 50% polyoxyethylated castor oil derivative (cremaphor). An initial dose of 135 mg/m2 of paclitaxel on a 24-h schedule was approved for patients with refractory or recurrent ovarian cancer, but later regulatory approval was obtained for a dose of 175 mg/m2 infused over 3 h every 3 weeks in ovarian cancer as well as for other indications. In ovarian and breast cancer patients treated on different schedules of 3-, 24-, and 96-h infusion every 3 weeks, the various schedules may be equally effective (11), although the 96-h infusion produces less myelosuppression and systemic complaints. In children, higher doses may be well tolerated.
In ovarian cancer treatment, a “dose-dense” weekly schedule of 70-90 mg/m2 with carboplatin, AUC 3, was more effective than every 3-week infusions of paclitaxel with carboplatin, AUC 5 (12). Paclitaxel, even in low doses of 20 mg/m2 weekly, reduces the thrombocytopenic effect of carboplatin. An alternative regimen in ovarian cancer employs a combination of intravenous, day 1, and intraperitoneal, day 8, paclitaxel with intraperitoneal cisplatin (see Ovarian Cancer, Chapter 54), but is associated with catheter-related toxicity and significant systemic toxicity.
![]() DOCETAXEL
DOCETAXEL
Docetaxel is formulated in polysorbate 80, and it can be administered after dilution in 0.9% saline, or 5% dextrose solution to a concentration of between 0.3 and 0.9 mg/ml. It is administered in doses of 60-100 mg/m2 over 1 h every 3 weeks. Weekly schedules of 30-40 mg/m2 cause a higher incidence of cumulative muscular weakness and neurotoxicity. This toxicity was especially noticeable with docetaxel doses exceeding 36 mg/m2 weekly.
![]() ABRAXANE
ABRAXANE
The newest approved taxane is abraxane, a formulation of paclitaxel in 3%-4% human albumin. This formulation of paclitaxel causes markedly less hypersensitivity than either paclitaxel or docetaxel, and is administered without premedication. The presence of the albumin particle surrounding the taxane enhances uptake in tumor cells that have an albumin receptor complex (SPARC) (13), which is found on many breast cancers and some normal tissues. The abraxane formulation leads to greater tissue penetration of paclitaxel (a much larger volume of distribution), a longer plasma half-life (27 h), and a greater free drug concentration in plasma. In clinical trials, the drug has at least equivalent activity to paclitaxel as second-line therapy in metastatic breast cancer, but produces greater myelosuppression, sensory neuropathy, and asthenia. The recommended dose and schedule are 260 mg/m2 infused over 30 min every 3 weeks.
![]() CABAZITAXEL
CABAZITAXEL
Cabazitaxel (a dimethyloxy derivative of docetaxel, see Figure 3-1) was selected for clinical development because of its lack of substrate affinity for the p-glycoprotein export pump. It is approved for treatment of prostate cancer refractory to docetaxel and hormonal inhibitors, based on an improvement of 2.1 months in overall survival, compared to survival in patients receiving mitoxantrone (14). The drug is administered in doses of 25 mg/m2 as a 60-min infusion every 3 weeks, in combination with prednisone 10 mg daily throughout treatment. It has a prolonged terminal half-life of 92 h and is primarily cleared by hepatic CYP3A4 metabolism. At this dose, its primary toxicities are grade 3 or greater neutropenia in 20% of patients. G-CSF use in prostate patients is recommended in high-risk patients (age greater than 65 years, comorbidities), and after the first cycle of therapy if patients develop severe neutropenia. It also causes a mild neuropathy in 10% of patients. Ongoing studies are evaluating a lower and potentially less toxic dose (20 mg/m2), and are comparing cabazitaxel to docetaxel as initial therapy for castration-resistant prostate cancer (13).
![]() CLINICAL PHARMACOLOGY OF OTHER ANTIMITOTICS
CLINICAL PHARMACOLOGY OF OTHER ANTIMITOTICS
Ixabepilone
The epothilones, an entirely new class of antimitotics with action similar to the taxanes (stabilization of tubulin polymers, mitotic arrest), were isolated from a fungal fermentation broth. Ixabepilone (a derivative of epothilone B, see Figure 3-1), in combination with capecitabine, is approved for treating breast cancer patients whose tumor is resistant to taxanes or anthracyclines. The drug has a large volume of distribution, limited protein binding, a long terminal half-life in plasma of 52 h, and is cleared predominantly by CYP3A4 metabolism, a pathway inhibited by imidazole antifungals. The recommended dose is 40 mg/m2 in a 3-h infusion every 3 weeks.
Ixabepilone, like other epothilones, produces a potent but usually reversible sensory neurotoxicity (burning, hypesthesias, neuropathic pain), which becomes increasingly prominent with successive cycles of therapy and may lead to drug delay or discontinuation. Patients with hepatic dysfunction or preexisting neuropathy should be treated with extreme caution. Dose reduction is recommended for patients with hepatic enzyme or bilirubin elevation. It also causes myelosuppression, and with its castor oil formulation, hypersensitivity reactions. Histamine antagonists (benadryl, 50 mg, and ranitidine, 150–300 mg) are recommended for premedication, and for patients experiencing hypersensitivity symptom, glucocorticoids should be given with succeeding doses (15).
Eribulin
The newest antimitotic to receive FDA approval is eribulin (Figure 3-1), a macrocyclic ketone derivative of halichondrin B, a natural product of a marine sponge. The halichondrins are extremely potent mitotic inhibitors in a manner distinct from the taxanes, but competitive with vinca alkaloids. They bind to the plus end position of β-tubulin and prevent microtubule extension, with no effect on shortening at the minus end of the polymer. The result is an aggregation of tubulin dimers, and arrest in G2-M. Eribulin is a weak substrate for the p-glycoprotein transporter. It was approved for taxane and anthracycline-resistant metastatic breast cancer, based on a positive phase III study showing an overall survival advantage of 2.5 months in comparison to physician’s choice of standard treatment. The primary toxicities were grade 3/4 neutropenia in 52% and peripheral neuropathy in fewer than 10%. In this heavily pretreated population, pharmacokinetic analysis revealed a long terminal half-life of 36–48 h, and slow clearance by hepatic metabolism, with dose adjustment of 50% indicated in patients with hepatic dysfunction (16).
VINCA ALKALOIDS
The vinca alkaloids, derived from the vinca rosea plant, have been a mainstay of the treatment of hematologic malignancies for almost 50 years. They were first discovered in an in vitro antileukemic screen at Eli Lilly and Co. in the early 1950s, and reached prominence in the combination therapy of childhood acute lymphocytic leukemia and Hodgkin disease a decade later. They continue as part of curative regimens for the lymphomas and testicular cancer, and a third derivative, vinorelbine, has proven active in breast and lung cancers. The vinca alkaloids share a common structure and have similar pharmacologic properties, but differ in their profiles of toxicity and specific disease indications (Figure 3-2).
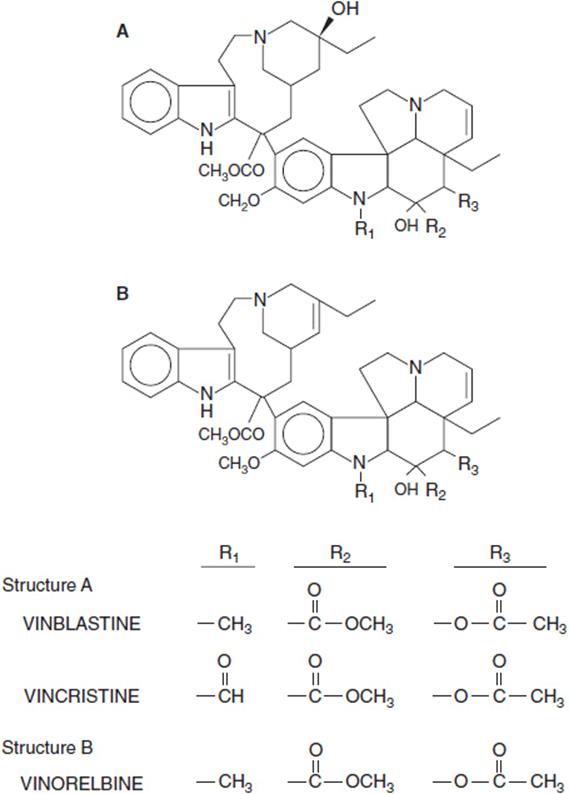
FIGURE 3-2 Metabolic structures of vinca alkaloids.
![]() MECHANISM OF ACTION
MECHANISM OF ACTION
The vincas bind to a common site on β-tubulin and prevent dimerization of tubulin alpha and beta subunits to form microtubules (17). They block cells in mitosis due to the absence of a microtubular apparatus required for chromosomal segregation. Apoptosis follows. An acute cell death dependent on jun kinase activation and potentiated by downregulation of the antiapoptotic protein, Mcl-1, has also been demonstrated experimentally (18).
Resistance to vincas arises through upregulation or amplification of one of several drug exporters, including the MDR gene product (p-glycoprotein) and the breast cancer resistance protein. Resistant cells may manifest a mutation in the vinca binding site on β-tubulin, a mutation that stabilizes microtubules and slows their rate of disassembly. In contrast mutations that destabilize microtubules confer sensitivity to taxanes and resistance to taxanes (19).
![]() CLINICAL PHARMACOLOGY
CLINICAL PHARMACOLOGY
The vincas share a common pharmacokinetic pattern of rapid clearance from plasma, extensive distribution into tissues, slow inactivation through hepatic metabolism by P-450 isoenzymes (primarily CYP3A4) and long half-lives in plasma of up to several days. Vincristine has the longest terminal plasma t½ (up to 85 h), while vinorelbine is intermediate (46 h) and vinblastine is the most rapidly cleared (t½of 24 h). Vinblastine induces CYP3A4 activity. While there is no clear relationship of vinca clearance to any single liver function test, patients with abnormal hepatic function (bili-rubin >1.5 mg/dl or >2-fold AST or ALT levels in serum) should receive no more than 50% of a full dose for their initial infusion, as prolonged ileus, myelosuppression, and neurotoxicity may result (20). No adjustment for renal dysfunction is required. The drugs are given intravenously as bolus infusions every 1–3 weeks, depending on the regimen employed. Usual single doses of the vinca alkaloids are vincristine, 1–2 mg/m2; vinorelbine, 20–30 mg/m2; and vinblastine, 6–8 mg/m2.
![]() TOXICITY
TOXICITY
All vinca alkaloids cause neurotoxicity, primarily a peripheral sensory neuropathy. Vincristine, the most highly neurotoxic, may cause significant motor weakness of the hands and feet in severely toxic patients, and should not be given to patients with significant neurologic dysfunction due to other drugs, diabetes, stroke, or inherited neurologic disease. Neurotoxicity due to vinorelbine occurs with repeated cycles of therapy, but is usually mild and reversible. Vinblastine causes minimal neurotoxicity, but like vinorelbine, is a potent myelosuppressant, with rapid recovery of blood counts in 10–14 days. At usual doses, vincristine has little effect on the bone marrow. In high doses (not used in common practice) vincristine causes abdominal distention and ileus.
Because the vinca alkaloids depend for their clearance on CYP3A4, drug interactions are likely if the vincas are given with inducers or inhibitors of this isoenzyme (21). Thus phenytoin induces vinca clearance, and vincas may accelerate phenantoin metabolism and lead to seizures in patients receiving both drugs. Imidazole antifungal drugs, such as ketoconazole or itraconazole, inhibit CYP3A4 and slow vinca clearance, leading to severe toxicity if the dose of vinca alkaloid is not reduced.
REFERENCES
1. Eisenhauer EA, Vermorken JB. The taxoids: comparative clinical pharmacology and therapeutic potential. Drugs. 1998; 55: 5–30.
2. Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nano-particle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005; 23: 7794–7803.
3. Crown J, O’Leary M, Wei-Seong O. Docetaxel and paclitaxel in the treatment of breast cancer: a review of clinical experience. The Oncologist. 2004; 9 (Suppl 2): 24–32.
4. Wani MC, Taylor HL, Wall ME, et al. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971; 93: 2325–2327.
5. Dumontet C, Sikic BI. Mechanism of action of and resistance to antitubulin agents; microtubule dynamics, drug transport and cell death. J Clin Oncol. 1999; 17: 1061–1070.
6. Wertz IE, Kusam S, Lam C, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011; 471: 122.
7. Gianni L, Kearns CM, Gianni A, et al. Nonlinear pharmacokinetics and metabolism of paclitaxel and it pharmacokinetic/pharmacodynamic relationship in humans. J Clin Oncol. 1995; 13: 2127–2135.
8. Baker AF, Dorr RT. Drug interactions with the taxanes: clinical implications. Cancer Treat Rev. 2001; 27: 221–233.
9. Viganol L, Locatelli A, Granelli G, Gianni L. Drug interactions of paclitaxel and docetaxel and their relevance for the design of combination chemotherapy. Invest New Drugs. 2001; 19: 179–196.
10. Rowinsky EC. Taxanes: paclitaxel (taxol) and docetaxel (taxotere). In Cancer Medicine, 6th edition, BC Decker, Hamilton, Canada, 2003.
11. Ghersi D, Wilcken N, Simers RJ. A systematic review of taxane-containing regimens for metastatic breast cancer. Br J Cancer. 2002; 93: 293–301.
12. Katsumata N, Yasuda M, Takahashi F, et al. Dose dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open-label, randomized controlled trial. Lancet. 2009; 374: 1331–1338.
13. Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III study of nano-particle albumin-bound paclitaxel compared with polyehtylated castor oil–based paclitaxel in women with breast cancer. J Clin Oncol. 2005; 23: 7784–7803.
14. Pean E, Demolis P, Moreau A, et al. The European Medicines Agency review of cabazitaxel (Jevtana) for the treatment of hormone refractory metastatic prostate cancer: summary of the scientific assessment of the Committee for Medicinal Products for Human Use (CHMP). The Oncologist. 2012; 17(4): 543–549.
15. Rivera E, Gomez H. Chemotherapy resistance in metastatic breast cancer: the evolving role of ixabepilone. Breast Canc Res. 2010; 12 (Suppl 2): 52–64.
16. Jain S, Cigler T. Eribulin mesylate in the treatment of metastatic breast cancer. Biologics. 2012; 6: 21–29.
17. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004; 4: 253–265.
18. Salemi BL, Bates DJ, Albershardt TC, et al. Vinblastine induces acute, cell phase-independent apoptosis in some leukemias and lymphomas and can induce acute apoptosis in others when Mcl-1 is suppressed. Mol Cancer Therap. 2011; 9: 791–802.
19. Cheung CHA, Wu S, Lee T, et al. Cancer cells acquire mitotic drug resistance properties through beta 1-tubulin mutations and alterations in expression of beta-tubulin isotypes. PLoS One. 2010; 5: 1–11.4.
20. Robieux I, Sorio R, Borsatti E, et al. Pharmacokinetics of vinorelbine in patients with liver metastases. Clin Pharmacol Ther. 1996; 59: 32–40.
21. Villikka K, Kivisto KT, Maenpaa H, et al. Cytochrome p450-inducing antiepileptics increase the clearance of vincristine in patients with brain tumors. Clin Pharmacol Ther. 1999; 66: 589–593.