INTRODUCTION
Abnormalities in fluid volume and electrolyte composition are common and important clinical disorders. Drugs that block specific transport functions of the renal tubules are valuable clinical tools in the treatment of these disorders. Although various agents that increase urine volume (diuretics) have been described since antiquity, it was not until 1957 that a practical and powerful diuretic agent (chlorothiazide) became available for widespread use.
Technically, a "diuretic" is an agent that increases urine volume, while a "natriuretic" causes an increase in renal sodium excretion. Because natriuretics almost always also increase water excretion, they are usually called diuretics.
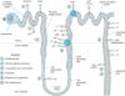
The nephron is divided structurally and functionally into several segments (Figure 15-1, Table 15-1), which are discussed in the first part of this chapter. Many diuretics exert their effects on specific membrane transport proteins in renal tubular epithelial cells. Other diuretics exert osmotic effects that prevent water reabsorption (mannitol), inhibit enzymes (acetazolamide), or interfere with hormone receptors in renal epithelial cells (aldosterone receptor blockers). These effects are discussed in the second part of the chapter. The physiology of each segment is closely linked to the pharmacology of the drugs acting there.
|
|
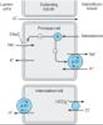
Figure 15-1. Tubule transport systems and sites of action of diuretics. |
I. RENAL TUBULE TRANSPORT MECHANISMS
PROXIMAL TUBULE
Sodium bicarbonate (NaHCO3), sodium chloride (NaCl), glucose, amino acids, and other organic solutes are reabsorbed via specific transport systems in the early proximal tubule (proximal convoluted tubule, PCT). Potassium ions (K+) are reabsorbed via the paracellular pathway. Water is reabsorbed passively, maintaining the osmolality of proximal tubular fluid at a nearly constant level. As tubule fluid is processed along the length of the proximal tubule, the luminal concentrations of these solutes decrease relative to the concentration of inulin, a marker that is filtered but neither secreted nor absorbed by renal tubules. Approximately 66% of total sodium ions (Na+, but 85% of the filtered NaHCO3), 65% of the K+, 60% of the water, and virtually all of the filtered glucose and amino acids are reabsorbed in the proximal tubule.
Of the various solutes reabsorbed in the proximal tubule, the most relevant to diuretic action are NaHCO3 and NaCl. Of the currently available diuretics, only one group (carbonic anhydrase inhibitors, which block NaHCO3 reabsorption) acts predominantly in the PCT. In view of the large quantity of NaCl absorbed in this segment, a drug that specifically blocked proximal tubular absorption of NaCl would be a particularly powerful diuretic. No such drug is currently available.
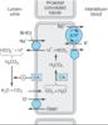
Sodium bicarbonate reabsorption by the PCT is initiated by the action of a Na+/H+ exchanger (NHE3) located in the luminal membrane of the proximal tubule epithelial cell (Figure 15-2). This transport system allows Na+ to enter the cell from the tubular lumen in exchange for a proton (H+) from inside the cell. As in all portions of the nephron, Na+/K+ ATPase in the basolateral membrane pumps the reabsorbed Na+ into the interstitium so as to maintain a low intracellular Na+ concentration. The H+ secreted into the lumen combines with bicarbonate (HCO3-) to form H2CO3 (carbonic acid), which is rapidly dehydrated to CO2 and H2O by carbonic anhydrase. Carbon dioxide produced by dehydration of H2CO3 enters the proximal tubule cell by simple diffusion where it is then rehydrated back to H2CO3, facilitated by intracellular carbonic anhydrase. After dissociation of H2CO3, the H+ is available for transport by the Na+/H+ exchanger, and the HCO3- is transported out of the cell by a basolateral membrane transporter (Figure 15-2). Bicarbonate reabsorption by the proximal tubule is thus dependent on carbonic anhydrase. This enzyme can be inhibited by acetazolamide and related agents.
In the late proximal tubule, as HCO3- and organic solutes have been largely removed from the tubular fluid, the residual luminal fluid contains predominantly NaCl. Under these conditions, Na+ reabsorption continues, but the H+ secreted by the Na+/H+ exchanger can no longer bind to HCO3-. Free H+ causes luminal pH to fall, activating a still poorly defined Cl-/base exchanger (Figure 15-2). The net effect of parallel Na+/H+ exchange and Cl-/base exchange is NaCl reabsorption. As yet, there are no diuretic agents that are known to act on this conjoint process.
Because water is reabsorbed in direct proportion to salt reabsorption in the proximal tubule, luminal fluid osmolality remains nearly constant along its length and an impermeant solute like inulin rises in concentration as water is reabsorbed. If large amounts of an impermeant solute such as mannitol (an osmotic diuretic, see below) are present in the tubular fluid, water reabsorption causes the concentration of the solute and osmolality of tubular fluid to rise, eventually preventing further water reabsorption.
Organic acid secretory systems are located in the middle third of the straight part of the proximal tubule (S2 segment). These systems secrete a variety of organic acids (uric acid, nonsteroidal anti-inflammatory drugs [NSAIDs], diuretics, antibiotics, etc) into the luminal fluid from the blood. These systems thus help deliver diuretics to the luminal side of the tubule, where most of them act. Organic base secretory systems (creatinine, choline, etc) are also present, in the early (S1) and middle (S2) segments of the proximal tubule.
|
|
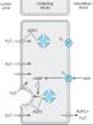
Figure 15-2. Apical membrane Na+/H+ exchange (via NHE3) and bicarbonate reabsorption in the proximal convoluted tubule cell. Na+/K+ ATPase is present in the basolateral membrane to maintain intracellular sodium and potassium levels within the normal range. Because of rapid equilibration, concentrations of the solutes are approximately equal in the interstitial fluid and the blood. Carbonic anhydrase (CA) is found in other locations in addition to the brush border of the luminal membrane. |
LOOP OF HENLE
At the boundary between the inner and outer stripes of the outer medulla, the proximal tubule empties into the thin descending limb of Henle's loop. Water is extracted from the descending limb of this loop by osmotic forces found in the hypertonic medullary interstitium. As in the proximal tubule, impermeant luminal solutes such as mannitol oppose this water extraction. The thin ascending limb is relatively water-impermeable.
The thick ascending limb (TAL) of the loop of Henle actively reabsorbs NaCl from the lumen (about 25% of the filtered sodium), but unlike the proximal tubule and the thin limb of Henle's loop, it is nearly impermeable to water. Salt reabsorption in the TAL therefore dilutes the tubular fluid, and it is called a "diluting segment." Medullary portions of the thick ascending limb contribute to medullary hypertonicity and thereby also play an important role in concentration of urine by the collecting duct.
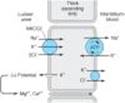
The NaCl transport system in the luminal membrane of the TAL is a Na+/K+/2Cl- cotransporter (called NKCC2 or NK2CL) (Figure 15-3). This transporter is selectively blocked by diuretic agents known as "loop" diuretics (see below). Although the Na+/K+/2Cl- transporter is itself electrically neutral (two cations and two anions are cotransported), the action of the transporter contributes to excess K+ accumulation within the cell. Back diffusion of this K+ into the tubular lumen causes a lumen-positive electrical potential that provides the driving force for reabsorption of cations¾including magnesium and calcium¾via the paracellular pathway. Thus, inhibition of salt transport in the thick ascending limb by loop diuretics, which reduces the lumen-positive potential, causes an increase in urinary excretion of divalent cations in addition to NaCl.
|
|
Figure 15-3. Ion transport pathways across the luminal and basolateral membranes of the thick ascending limb cell. The lumen positive electrical potential created by K+back diffusion drives divalent (and monovalent) cation reabsorption via the paracellular pathway. NKCC2 is the primary transporter in the luminal membrane. |
DISTAL CONVOLUTED TUBULE
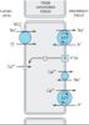
Only about 10% of the filtered NaCl is reabsorbed in the distal convoluted tubule (DCT). Like the thick ascending limb of Henle's loop, this segment is relatively impermeable to water and NaCl reabsorption further dilutes the tubular fluid. The mechanism of NaCl transport in the DCT is an electrically neutral thiazide-sensitive Na+ and Cl- cotransporter (NCC, Figure 15-4).
Because K+ does not recycle across the apical membrane of the DCT as it does in the TAL, there is no lumen-positive potential in this segment, and Ca2+ and Mg2+ are not driven out of the tubular lumen by electrical forces. Instead, Ca2+ is actively reabsorbed by the DCT epithelial cell via an apical Ca2+ channel and basolateral Na+/Ca2+ exchanger (Figure 15-4). This process is regulated by parathyroid hormone.
|
|
Figure 15-4. Ion transport pathways across the luminal and basolateral membranes of the distal convoluted tubule cell. As in all tubular cells, Na+/K+ ATPase is present in the basolateral membrane. NCC is the primary sodium and chloride transporter in the luminal membrane. (R, parathyroid hormone [PTH] receptor.) |
COLLECTING TUBULE
The collecting tubule (CCT) is responsible for only 2-5% of NaCl reabsorption by the kidney. Despite this small contribution, the CCT plays an important role in renal physiology and in diuretic action. As the final site of NaCl reabsorption, the collecting tubule is responsible for tight regulation of body fluid volume and for determining the final Na+ concentration of the urine. Furthermore, the collecting tubule is a site at which mineralocorticoids exert a significant influence. Lastly, the collecting tubule is the most important site of K+ secretion by the kidney and the site at which virtually all diuretic-induced changes in K+ balance occur.
The mechanism of NaCl reabsorption in the CCT is distinct from the mechanisms found in other tubule segments. The principal cells are the major sites of Na+, K+, and water transport (Figure 15-5), and the intercalated cells are the primary sites of H+ secretion. Unlike cells in other nephron segments, the principal cells do not contain cotransport systems for Na+ and other ions in their apical membranes. Principal cell membranes exhibit separate ion channels for Na+ and K+. Since these channels exclude anions, transport of Na+ or K+leads to a net movement of charge across the membrane. Because Na+ entry into the principal cell predominates over K+ secretion, a 10-50 mV lumen-negative electrical potential develops. Na+ that enters the principal cell from the tubular fluid is then transported back to the blood via the basolateral Na+/K+ ATPase (Figure 15-5). The 10-50 mV lumen-negative electrical potential drives the transport of Cl- back to the blood via the paracellular pathway and draws K+ out of cells through the apical membrane K+ channel. Thus, there is an important relationship between Na+ delivery to the CCT and the resulting secretion of K+. Diuretics that act upstream of the CCT will increase Na+ delivery to this site and will enhance K+ secretion. If the Na+ is delivered with an anion that cannot be reabsorbed as readily as Cl- (eg, HCO3-), the lumen-negative potential is increased, and K+ secretion will be enhanced. This mechanism, combined with enhanced aldosterone secretion due to volume depletion, is the basis for most diuretic-induced K+ wasting.
Reabsorption of Na+ via the epithelial Na channel (ENaC) and its coupled secretion of K+ is regulated by aldosterone. This steroid hormone, through its actions on gene transcription, increases the activity of both apical membrane channels and the basolateral Na+/K+ ATPase. This leads to an increase in the transepithelial electrical potential and a dramatic increase in both Na+ reabsorption and K+ secretion.
The collecting tubule is also the site at which the final urine concentration is determined. Antidiuretic hormone (ADH, also called arginine vasopressin, AVP) controls the permeability of this segment to water by regulating the insertion of preformed water channels (aquaporin-2, AQP2) into the apical membrane via a G protein-coupled cAMP-mediated process (Figure 15-6). In the absence of ADH, the collecting tubule (and duct) is impermeable to water and dilute urine is produced. ADH markedly increases water permeability and this leads to the formation of a more concentrated final urine. ADH also stimulates the insertion of urea transporter UT1 molecules into the apical membranes of medullary collecting tubule cells. Urea concentration in the medulla plays an important role maintaining the high osmolarity of the medulla and in the concentration of urine. ADH secretion is regulated by serum osmolality and by volume status.
|
|
Figure 15-5. Ion transport pathways across the luminal and basolateral membranes of collecting tubule and collecting duct cells. Inward diffusion of Na+ via the epithelial sodium channel (ENaC) leaves a lumen-negative potential, which drives reabsorption of Cl- and efflux of K+. (R, aldosterone receptor; ADH, antidiuretic hormone.) |
|
|
Figure 15-6. Water transport across the luminal and basolateral membranes of collecting duct cells. Above, low water permeability exists in the absence of antidiuretic hormone (ADH). Below, in the presence of ADH, aquaporins are inserted into the apical membrane, greatly increasing water permeability. (V2, vasopressin V2 receptor; AQP2, apical aquaporin water channels; AQP3, 4, basolateral aquaporin water channels.) |
II. BASIC PHARMACOLOGY OF DIURETIC AGENTS
CARBONIC ANHYDRASE INHIBITORS
Introduction
Carbonic anhydrase is present in many nephron sites, but the predominant location of this enzyme is the luminal membrane of the PCT (Figure 15-2), where it catalyzes the dehydration of H2CO3 as described above. By blocking carbonic anhydrase, inhibitors block NaHCO3 reabsorption and cause diuresis.
Carbonic anhydrase inhibitors were the forerunners of modern diuretics. They were discovered when it was found that bacteriostatic sulfonamides caused an alkaline diuresis and hyperchloremic metabolic acidosis. With the development of newer agents, carbonic anhydrase inhibitors are now rarely used as diuretics, but they still have several specific applications that are discussed below. The prototypical carbonic anhydrase inhibitor is acetazolamide.
Pharmacokinetics
The carbonic anhydrase inhibitors are well absorbed after oral administration. An increase in urine pH from the HCO3- diuresis is apparent within 30 minutes, maximal at 2 hours, and persists for 12 hours after a single dose. Excretion of the drug is by secretion in the proximal tubule S2 segment. Therefore, dosing must be reduced in renal insufficiency.
Pharmacodynamics
Inhibition of carbonic anhydrase activity profoundly depresses HCO3- reabsorption in the PCT. At its maximal safely administered dosage, 85% of the HCO3- reabsorptive capacity of the superficial PCT is inhibited. Some HCO3- can still be absorbed at other nephron sites by carbonic anhydrase-independent mechanisms, so the overall effect of maximal acetazolamide dosage is only about 45% inhibition of whole kidney HCO3- reabsorption. Nevertheless, carbonic anhydrase inhibition causes significant HCO3- losses and hyperchloremic metabolic acidosis (Table 15-2). Because of reduced HCO3- in the glomerular filtrate and the fact that HCO3- depletion leads to enhanced NaCl reabsorption by the remainder of the nephron, the diuretic efficacy of acetazolamide decreases significantly with use over several days.
At present, the major clinical applications of acetazolamide involve carbonic anhydrase-dependent HCO3- and fluid transport at sites other than the kidney. The ciliary body of the eye secretes HCO3- from the blood into the aqueous humor. Likewise, formation of cerebrospinal fluid by the choroid plexus involves HCO3- secretion. Although these processes remove HCO3- from the blood (the direction opposite to that in the proximal tubule), they are similarly inhibited by carbonic anhydrase inhibitors.
Clinical Indications & Dosage (Table 15-3)
A. GLAUCOMA
The reduction of aqueous humor formation by carbonic anhydrase inhibitors decreases the intraocular pressure. This effect is valuable in the management of glaucoma, making it the most common indication for use of carbonic anhydrase inhibitors. Topically active carbonic anhydrase inhibitors (dorzolamide, brinzolamide) are also available. These topical compounds reduce intraocular pressure, but plasma levels are undetectable. Thus, diuretic and systemic metabolic effects are eliminated for the topical agents.
B. URINARY ALKALINIZATION
Uric acid, cystine, and other weak acids are most easily reabsorbed from acidic urine. Therefore, renal excretion of cystine (in cystinuria) and other weak acids can be enhanced by increasing urinary pH with carbonic anhydrase inhibitors. In the absence of continuous HCO3- administration, these effects of acetazolamide last only 2-3 days. Prolonged therapy requires HCO3- administration.
C. METABOLIC ALKALOSIS
Metabolic alkalosis is generally treated by correction of abnormalities in total body K+, intravascular volume, or mineralocorticoid levels. However, when the alkalosis is due to excessive use of diuretics in patients with severe heart failure, replacement of intravascular volume may be contraindicated. In these cases, acetazolamide can be useful in correcting the alkalosis as well as producing a small additional diuresis for correction of volume overload. Acetazolamide can also be used to rapidly correct the metabolic alkalosis that may develop in the setting of respiratory acidosis.
D. ACUTE MOUNTAIN SICKNESS
Weakness, dizziness, insomnia, headache, and nausea can occur in mountain travelers who rapidly ascend above 3000 m. The symptoms are usually mild and last for a few days. In more serious cases, rapidly progressing pulmonary or cerebral edema can be life-threatening. By decreasing cerebrospinal fluid formation and by decreasing the pH of the cerebrospinal fluid and brain, acetazolamide can increase ventilation and diminish symptoms of mountain sickness.
E. OTHER USES
Carbonic anhydrase inhibitors have been used as adjuvants in the treatment of epilepsy, in some forms of hypokalemic periodic paralysis, and to increase urinary phosphate excretion during severe hyperphosphatemia.
Toxicity
A. HYPERCHLOREMIC METABOLIC ACIDOSIS
Acidosis predictably results from chronic reduction of body HCO3- stores by carbonic anhydrase inhibitors (Table 15-2) and limits the diuretic efficacy of these drugs to 2 or 3 days. Unlike the diuretic effect, acidosis persists as long as the drug is continued.
B. RENAL STONES
Phosphaturia and hypercalciuria occur during the bicarbonaturic response to inhibitors of carbonic anhydrase. Renal excretion of solubilizing factors (eg, citrate) may also decline with chronic use. Calcium salts are relatively insoluble at alkaline pH, which means that the potential for renal stone formation from these salts is enhanced.
C. RENAL POTASSIUM WASTING
Potassium wasting can occur because Na+ presented to the collecting tubule is partially reabsorbed, increasing the lumen-negative electrical potential in that segment and enhancing K+secretion. This effect can be counteracted by simultaneous administration of potassium chloride.
D. OTHER TOXICITIES
Drowsiness and paresthesias are common following large doses of acetazolamide. Carbonic anhydrase inhibitors may accumulate in patients with renal failure, leading to nervous system toxicity. Hypersensitivity reactions (fever, rashes, bone marrow suppression, and interstitial nephritis) may also occur.
Contraindications
Carbonic anhydrase inhibitor-induced alkalinization of the urine will decrease urinary excretion of NH4+ and may contribute to the development of hyperammonemia and hepatic encephalopathy in patients with cirrhosis.
LOOP DIURETICS
Introduction
Loop diuretics selectively inhibit NaCl reabsorption in the TAL. Due to the large NaCl absorptive capacity of this segment and the fact that the diuretic action of these drugs is not limited by development of acidosis, as is the case with the carbonic anhydrase inhibitors, loop diuretics are among the most efficacious diuretic agents available.
Chemistry
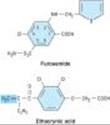
The two prototypical drugs of this group are furosemide and ethacrynic acid. The structures of these diuretics are shown in Figure 15-7. In addition to furosemide, bumetanide and torsemide are sulfonamide loop diuretics.
Ethacrynic acid¾not a sulfonamide derivative¾is a phenoxyacetic acid derivative containing an adjacent ketone and methylene group (Figure 15-7). The methylene group (shaded) forms an adduct with the free sulfhydryl group of cysteine. The cysteine adduct appears to be an active form of the drug.
Organic mercurial diuretics also inhibit salt transport in the TAL but are no longer used because of their toxicity.
|
|
Figure 15-7. Two loop diuretics. The shaded methylene group on ethacrynic acid is reactive and may combine with free sulfhydryl groups. |
Pharmacokinetics
The loop diuretics are rapidly absorbed. They are eliminated by the kidney by glomerular filtration and tubular secretion. Absorption of oral torsemide is more rapid (1 hour) than that of furosemide (2-3 hours) and is nearly as complete as with intravenous administration. The duration of effect for furosemide is usually 2-3 hours and that of torsemide is 4-6 hours. Half-life depends on renal function. Since loop agents act on the luminal side of the tubule, their diuretic activity correlates with their secretion by the proximal tubule. Reduction in the secretion of loop diuretics may result from simultaneous administration of agents such as NSAIDs or probenecid, which compete for weak acid secretion in the proximal tubule. Metabolites of ethacrynic acid and furosemide have been identified, but it is not known if they have any diuretic activity. Torsemide has at least one active metabolite with a half-life considerably longer than that of the parent compound.
Pharmacodynamics
These drugs inhibit NKCC2, the luminal Na+/K+/2Cl- transporter in the thick ascending limb of Henle's loop. By inhibiting this transporter, the loop diuretics reduce the reabsorption of NaCl and also diminish the lumen-positive potential that comes from K+ recycling (Figure 15-3). This positive potential normally drives divalent cation reabsorption in the loop (Figure 15-3), and by reducing this potential, loop diuretics cause an increase in Mg2+ and Ca2+ excretion. Prolonged use can cause significant hypomagnesemia in some patients. Since vitamin D-induced intestinal absorption of Ca2+ can be increased and Ca2+ is actively reabsorbed in the DCT, loop diuretics do not generally cause hypocalcemia. However, in disorders that cause hypercalcemia, Ca2+ excretion can be usefully enhanced by treatment with loop diuretics combined with saline infusions.
Loop diuretics induce synthesis of renal prostaglandins, which participate in the renal actions of these diuretics. NSAIDs (eg, indomethacin) can interfere with the actions of the loop diuretics by reducing prostaglandin synthesis in the kidney. This interference is minimal in otherwise normal subjects but may be significant in patients with nephrotic syndrome or hepatic cirrhosis.
In addition to their diuretic activity, loop agents have direct effects on blood flow through several vascular beds. Furosemide increases renal blood flow. Both furosemide and ethacrynic acid have also been shown to reduce pulmonary congestion and left ventricular filling pressures in heart failure before a measurable increase in urinary output occurs, and in anephric patients.
Clinical Indications & Dosage (Table 15-4)
The most important indications for the use of the loop diuretics include acute pulmonary edema, other edematous conditions, and acute hypercalcemia. The use of loop diuretics in these conditions is discussed in Section III, Clinical Pharmacology. Other indications for loop diuretics include hyperkalemia, acute renal failure, and anion overdose.
A. HYPERKALEMIA
In mild hyperkalemia¾or after acute management of severe hyperkalemia by other measures¾loop diuretics can significantly enhance urinary excretion of K+. This response is enhanced by simultaneous NaCl and water administration.
B. ACUTE RENAL FAILURE
Loop agents can increase the rate of urine flow and enhance K+ excretion in acute renal failure. However, they do not shorten the duration of renal failure. If a large pigment load has precipitated acute renal failure (or threatens to), loop agents may help flush out intratubular casts and ameliorate intratubular obstruction. On the other hand, loop agents can theoretically worsen cast formation in myeloma and light chain nephropathy.
C. ANION OVERDOSE
Loop diuretics are useful in treating toxic ingestions of bromide, fluoride, and iodide, which are reabsorbed in the thick ascending limb. Saline solution must be administered to replace urinary losses of Na+ and to provide Cl-, so as to avoid extracellular fluid volume depletion.
Toxicity
A. HYPOKALEMIC METABOLIC ALKALOSIS
By inhibiting salt reabsorption in the TAL, loop diuretics increase delivery to the collecting duct. Increased delivery leads to increased secretion of K+ and H+ by the duct, causing hypokalemic metabolic alkalosis (Table 15-2). This toxicity is a function of the magnitude of the diuresis and can be reversed by K+ replacement and correction of hypovolemia.
B. OTOTOXICITY
Loop diuretics occasionally cause dose-related hearing loss that is usually reversible. It is most common in patients who have diminished renal function or who are also receiving other ototoxic agents such as aminoglycoside antibiotics.
C. HYPERURICEMIA
Loop diuretics can cause hyperuricemia and precipitate attacks of gout. This is caused by hypovolemia-associated enhancement of uric acid reabsorption in the proximal tubule. It may be prevented by using lower doses to avoid development of hypovolemia.
D. HYPOMAGNESEMIA
Magnesium depletion is a predictable consequence of the chronic use of loop agents and occurs most often in patients with dietary magnesium deficiency. It can be reversed by administration of oral magnesium preparations.
E. ALLERGIC & OTHER REACTIONS
Except for ethacrynic acid, the loop diuretics are sulfonamides. Therefore skin rash, eosinophilia and, less often, interstitial nephritis are occasional side effects of these drugs. This toxicity usually resolves rapidly after drug withdrawal. Allergic reactions are much less common with ethacrynic acid.
Because Henle's loop is normally responsible for so much salt and water reabsorption, loop diuretics can cause severe dehydration. Hyponatremia is less common than with the thiazides (see below), but patients who increase water intake in response to hypovolemia-induced thirst can become severely hyponatremic with loop agents. Loop agents are sometimes used for their calciuric effect, but hypercalcemia can occur in volume-depleted patients who have another¾previously occult¾cause for hypercalcemia, such as metastatic breast or squamous cell lung carcinoma.
Contraindications
Furosemide, bumetanide, and torsemide may exhibit allergic cross-reactivity in patients who are sensitive to other sulfonamides but this appears to be very rare. Overzealous use of any diuretic is dangerous in hepatic cirrhosis, borderline renal failure, or heart failure (see below).
THIAZIDES
Introduction
The thiazide diuretics emerged from efforts to synthesize more potent carbonic anhydrase inhibitors. It subsequently became clear that the thiazides inhibit NaCl transport predominantly in the DCT. However, some members of this group retain significant carbonic anhydrase inhibitory activity. The prototypical thiazide is hydrochlorothiazide.
Chemistry & Pharmacokinetics
Like carbonic anhydrase inhibitors and many loop diuretics, all of the thiazides have an unsubstituted sulfonamide group (Figure 15-8).
All of the thiazides can be administered orally, but there are differences in their metabolism. Chlorothiazide, the parent of the group, is not very lipid-soluble and must be given in relatively large doses. It is the only thiazide available for parenteral administration. Chlorthalidone is slowly absorbed and has a longer duration of action. Although indapamide is excreted primarily by the biliary system, enough of the active form is cleared by the kidney to exert its diuretic effect in the DCT.
All of the thiazides are secreted by the organic acid secretory system in the proximal tubule and compete with the secretion of uric acid by that system. As a result, thiazide use may blunt uric acid secretion and elevate serum uric acid level.
|
|
Figure 15-8. Hydrochlorothiazide and related agents. |
Pharmacodynamics
Thiazides inhibit NaCl reabsorption from the luminal side of epithelial cells in the DCT by blocking the Na+/Cl- transporter (NCC). In contrast to the situation in the TAL, where loop diuretics inhibit Ca2+ reabsorption, thiazides actually enhance Ca2+ reabsorption. This enhancement has been postulated to result from effects in both the proximal and distal convoluted tubules. In the proximal tubule, thiazide-induced volume depletion leads to enhanced Na+ and passive Ca2+ reabsorption. In the DCT, lowering of intracellular Na+ by thiazide-induced blockade of Na+ entry enhances Na+/Ca2+ exchange in the basolateral membrane (Figure 15-4), and increases overall reabsorption of Ca2+. While thiazides rarely cause hypercalcemia as the result of this enhanced reabsorption, they can unmask hypercalcemia due to other causes (eg, hyperparathyroidism, carcinoma, sarcoidosis). Thiazides are useful in the treatment of kidney stones caused by hypercalciuria.
The action of thiazides depends in part on renal prostaglandin production. As described above for the loop diuretics, the actions of thiazides can also be inhibited by NSAIDs under certain conditions.
Clinical Indications & Dosage (Table 15-5)
The major indications for thiazide diuretics are (1) hypertension, (2) heart failure, (3) nephrolithiasis due to idiopathic hypercalciuria, and (4) nephrogenic diabetes insipidus. Use of the thiazides in each of these conditions is described in Section III, Clinical Pharmacology.
Toxicity
A. HYPOKALEMIC METABOLIC ALKALOSIS AND HYPERURICEMIA
These toxicities are similar to those observed with loop diuretics (see above and Table 15-2).
B. IMPAIRED CARBOHYDRATE TOLERANCE
Hyperglycemia may occur in patients who are overtly diabetic or who have even mildly abnormal glucose tolerance tests. The effect is due to both impaired pancreatic release of insulin and diminished tissue utilization of glucose. Hyperglycemia may be partially reversible with correction of hypokalemia.
C. HYPERLIPIDEMIA
Thiazides cause a 5-15% increase in total serum cholesterol and low-density lipoproteins (LDL). These levels may return toward baseline after prolonged use.
D. HYPONATREMIA
Hyponatremia is an important adverse effect of thiazide diuretics. It is due to a combination of hypovolemia-induced elevation of ADH, reduction in the diluting capacity of the kidney, and increased thirst. It can be prevented by reducing the dose of the drug or limiting water intake.
E. ALLERGIC REACTIONS
The thiazides are sulfonamides and share cross-reactivity with other members of this chemical group. Photosensitivity or generalized dermatitis occurs rarely. Serious allergic reactions are extremely rare but do include hemolytic anemia, thrombocytopenia, and acute necrotizing pancreatitis.
F. OTHER TOXICITIES
Weakness, fatigability, and paresthesias similar to those of carbonic anhydrase inhibitors may occur. Impotence has been reported but is probably related to volume depletion.
Contraindications
Excessive use of any diuretic is dangerous in hepatic cirrhosis, borderline renal failure, or heart failure (see below).
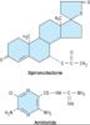
POTASSIUM-SPARING DIURETICS
Introduction
These diuretics prevent K+ secretion by antagonizing the effects of aldosterone at the late distal and cortical collecting tubules. Inhibition may occur by direct pharmacologic antagonism of mineralocorticoid receptors (spironolactone, eplerenone) or by inhibition of Na+ influx through ion channels in the luminal membrane (amiloride, triamterene).
Chemistry & Pharmacokinetics
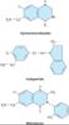
The structures of spironolactone and amiloride are shown in Figure 15-9.
Spironolactone is a synthetic steroid that acts as a competitive antagonist to aldosterone. Onset and duration of its action are determined by the kinetics of the aldosterone response in the target tissue. Substantial inactivation of spironolactone occurs in the liver. Overall, spironolactone has a rather slow onset of action, requiring several days before full therapeutic effect is achieved. Eplerenone is a spironolactone analog with greater selectivity for the aldosterone receptor.
Amiloride and triamterene are direct inhibitors of Na+ influx in the CCT. Triamterene is metabolized in the liver, but renal excretion is a major route of elimination for the active form and the metabolites. Because triamterene is extensively metabolized, it has a shorter half-life and must be given more frequently than amiloride (which is not metabolized).
|
|
Figure 15-9. Potassium-sparing diuretics. |
Pharmacodynamics
Potassium-sparing diuretics reduce Na+ absorption in the collecting tubules and ducts. Na+ absorption (and K+ secretion) at this site is regulated by aldosterone, as described above. Aldosterone antagonists interfere with this process. Similar effects are observed with respect to H+ handling by the intercalated cells of the collecting tubule, in part explaining the metabolic acidosis seen with aldosterone antagonists (Table 15-2).
Spironolactone and eplerenone bind to aldosterone receptors and may also reduce the intracellular formation of active metabolites of aldosterone. Amiloride and triamterene do not block the aldosterone receptor but instead directly interfere with Na+ entry through the epithelial sodium ion channels (ENaC) in the apical membrane of the collecting tubule. Since K+secretion is coupled with Na+ entry in this segment, these agents are also effective potassium-sparing diuretics.
The actions of the aldosterone antagonists depend on renal prostaglandin production. As described above for loop diuretics and thiazides, the actions of K+-sparing diuretics can be inhibited by NSAIDs under certain conditions.
Clinical Indications & Dosage (Table 15-6)
Potassium-sparing diuretics are most useful in states of mineralocorticoid excess or hyperaldosteronism (also called aldosteronism), due either to primary hypersecretion (Conn's syndrome, ectopic adrenocorticotropic hormone production) or to secondary hyperaldosteronism (evoked by heart failure, hepatic cirrhosis, nephrotic syndrome, or other conditions associated with diminished effective intravascular volume). Use of diuretics such as thiazides or loop agents can cause or exacerbate volume contraction and may cause secondary hyperaldosteronism. In the setting of enhanced mineralocorticoid secretion and excessive delivery of Na+ to distal nephron sites, renal K+ wasting occurs. Potassium-sparing diuretics of either type may be used in this setting to blunt the K+ secretory response.
Toxicity
A. HYPERKALEMIA
Unlike other diuretics, K+-sparing diuretics can cause mild, moderate, or even life-threatening hyperkalemia (Table 15-2). The risk of this complication is greatly increased by renal disease (in which maximal K+ excretion may be reduced) or by the use of other drugs that reduce renin (b blockers, NSAIDs) or angiotensin II activity (angiotensin-converting enzyme inhibitors, angiotensin receptor inhibitors). Since most other diuretic agents lead to K+ losses, hyperkalemia is more common when K+-sparing diuretics are used as the sole diuretic agent, especially in patients with renal insufficiency. With fixed-dosage combinations of K+-sparing and thiazide diuretics, the thiazide-induced hypokalemia and metabolic alkalosis are ameliorated. However, owing to variations in the bioavailability of the components of fixed-dosage forms, the thiazide-associated adverse effects often predominate. Therefore, it is generally preferable to adjust the doses of the two drugs separately.
B. HYPERCHLOREMIC METABOLIC ACIDOSIS
By inhibiting H+ secretion in parallel with K+ secretion, the K+-sparing diuretics can cause acidosis similar to that seen with type IV renal tubular acidosis.
C. GYNECOMASTIA
Synthetic steroids may cause endocrine abnormalities by actions on other steroid receptors. Gynecomastia, impotence, and benign prostatic hyperplasia have all been reported with spironolactone. Such effects have not been reported with eplerenone.
D. ACUTE RENAL FAILURE
The combination of triamterene with indomethacin has been reported to cause acute renal failure. This has not been reported with other K+-sparing diuretics.
E. KIDNEY STONES
Triamterene is only slightly soluble and may precipitate in the urine, causing kidney stones.
Contraindications
These agents can cause severe, even fatal hyperkalemia in susceptible patients. Oral K+ administration should be discontinued if K+-sparing diuretics are administered. Patients with chronic renal insufficiency are especially vulnerable and should rarely be treated with these diuretics. Concomitant use of other agents that blunt the renin-angiotensin system (b blockers or ACE inhibitors) increases the likelihood of hyperkalemia. Patients with liver disease may have impaired metabolism of triamterene and spironolactone, so dosing must be carefully adjusted. Strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole) can markedly increase blood levels of eplerenone.
AGENTS THAT ALTER WATER EXCRETION
1. Osmotic Diuretics
Introduction
The proximal tubule and descending limb of Henle's loop are freely permeable to water (Table 15-1). Any osmotically active agent that is filtered by the glomerulus but not reabsorbed causes water to be retained in these segments and promotes a water diuresis. Such agents can be used to reduce intracranial pressure and to promote prompt removal of renal toxins. The prototypic osmotic diuretic is mannitol.
Pharmacokinetics
Osmotic diuretics are poorly absorbed, which means that they must be given parenterally. If administered orally, mannitol causes osmotic diarrhea. Mannitol is not metabolized and is excreted by glomerular filtration within 30-60 minutes, without any important tubular reabsorption or secretion.
Pharmacodynamics
Osmotic diuretics have their major effect in the proximal tubule and the descending limb of Henle's loop. Through osmotic effects, they also oppose the action of ADH in the collecting tubule. The presence of a nonreabsorbable solute such as mannitol prevents the normal absorption of water by interposing a countervailing osmotic force. As a result, urine volume increases. The increase in urine flow rate decreases the contact time between fluid and the tubular epithelium, thus reducing Na+ as well as water reabsorption. The resulting natriuresis is of lesser magnitude than the water diuresis, leading eventually to excessive water loss and hypernatremia.
Clinical Indications & Dosage
A. TO INCREASE URINE VOLUME
Osmotic diuretics are used to increase water excretion in preference to sodium excretion. This effect can be useful when avid Na+ retention limits the response to conventional agents. It can be used to maintain urine volume and to prevent anuria that might otherwise result from presentation of large pigment loads to the kidney (eg, from hemolysis or rhabdomyolysis). Some oliguric patients do not respond to osmotic diuretics. Therefore, a test dose of mannitol (12.5 g intravenously) should be given prior to starting a continuous infusion. Mannitol should not be continued unless there is an increase in urine flow rate to more than 50 mL/h during the 3 hours following the test dose. Mannitol (12.5-25 g) can be repeated every 1-2 hours to maintain urine flow rate greater than 100 mL/h. Prolonged use of mannitol is not advised.
B. REDUCTION OF INTRACRANIAL AND INTRAOCULAR PRESSURE
Osmotic diuretics alter Starling forces so that water leaves cells and reduces intracellular volume. This effect is used to reduce intracranial pressure in neurologic conditions and to reduce intraocular pressure before ophthalmologic procedures. A dose of 1-2 g/kg mannitol is administered intravenously. Intracranial pressure, which must be monitored, should fall in 60-90 minutes.
Toxicity
A. EXTRACELLULAR VOLUME EXPANSION
Mannitol is rapidly distributed in the extracellular compartment and extracts water from cells. Prior to the diuresis, this leads to expansion of the extracellular volume and hyponatremia. This effect can complicate heart failure and may produce florid pulmonary edema. Headache, nausea, and vomiting are commonly observed in patients treated with osmotic diuretics.
B. DEHYDRATION, HYPERKALEMIA, AND HYPERNATREMIA
Excessive use of mannitol without adequate water replacement can ultimately lead to severe dehydration, free water losses, and hypernatremia. As water is extracted from cells, intracellular K+ concentration rises, leading to cellular losses and hyperkalemia. These complications can be avoided by careful attention to serum ion composition and fluid balance.
2. Antidiuretic Hormone (ADH) Agonists
Vasopressin and desmopressin are used in the treatment of central diabetes insipidus. They are discussed in Chapter 37. Their renal action appears to be mediated primarily via V2receptors although V1a receptors may also be involved.
3. Antidiuretic Hormone (ADH) Antagonists
Introduction
A variety of medical conditions, including congestive heart failure and syndrome of inappropriate ADH (SIADH), cause water retention as the result of ADH excess. Dangerous hyponatremia can result. Several nonpeptide ADH receptor antagonists (vaptans) have been studied, with encouraging clinical results, but only conivaptan has been approved for use. Two nonselective agents, lithium and demeclocycline (a tetracycline antimicrobial drug), also have anti-ADH effects.
Pharmacokinetics
Conivaptan, lithium, and demeclocycline are orally active. Conivaptan and demeclocycline have half-lives of 5-10 hours. Lithium (discussed in detail in Chapter 29) is never used as an ADH antagonist.
Pharmacodynamics
Antidiuretic hormone antagonists inhibit the effects of ADH in the collecting tubule. Conivaptan is a pharmacologic antagonist at V1a and V2 receptors. Both lithium and demeclocycline appear to reduce the formation of cyclic adenosine monophospate (cAMP) in response to ADH and also to interfere with the actions of cAMP in the collecting tubule cells, but the mechanisms of these effects is not known.
Clinical Indications & Dosage
A. SYNDROME OF INAPPROPRIATE ADH SECRETION (SIADH)
Antidiuretic hormone antagonists are used to manage SIADH when water restriction has failed to correct the abnormality. This generally occurs in the outpatient setting, where water restriction cannot be enforced, or in the hospital when large quantities of intravenous fluid are needed for other purposes. Lithium carbonate has been used to treat this syndrome, but the response is unpredictable. Demeclocycline, in dosages of 600-1200 mg/d, yields a more predictable result and is less toxic. Appropriate plasma levels (2 mcg/mL) should be maintained by monitoring. Unlike demeclocycline, conivaptan is administered by IV injection, so it is not suitable for chronic use in outpatients.
B. OTHER CAUSES OF ELEVATED ANTIDIURETIC HORMONE (ADH)
Antidiuretic hormone is also elevated in response to diminished effective circulating blood volume, as often occurs in congestive heart failure. When treatment by volume replacement is not desirable, hyponatremia may result. As for SIADH, water restriction is the treatment of choice, but if it is not successful, demeclocycline or conivaptan may be used.
Toxicity
A. NEPHROGENIC DIABETES INSIPIDUS
If serum Na+ is not monitored closely, ADH antagonists can cause severe hypernatremia and nephrogenic diabetes insipidus. If lithium is being used for a psychiatric disorder, nephrogenic diabetes insipidus can be treated with a thiazide diuretic or amiloride (see below).
B. RENAL FAILURE
Both lithium and demeclocycline have been reported to cause acute renal failure. Long-term lithium therapy may also cause chronic interstitial nephritis.
C. OTHER
Adverse effects associated with lithium therapy are discussed in Chapter 29. Demeclocycline should be avoided in patients with liver disease (see Chapter 44) and in children younger than 12 years.
DIURETIC COMBINATIONS
LOOP AGENTS & THIAZIDES
Some patients are refractory to the usual dose of loop diuretics or become refractory after an initial response. Since these agents have a short half-life (2-6 hrs), refractoriness may be due to an excessive interval between doses. Renal Na+ retention may be greatly increased during the time period when the drug is no longer active. After the dosing interval for loop agents is minimized or the dose is maximized, the use of two drugs acting at different nephron sites may exhibit dramatic synergy. Loop agents and thiazides in combination will often produce diuresis when neither agent acting alone is even minimally effective. There are several reasons for this phenomenon. First, salt and water reabsorption in either the TAL or the DCT can increase when the other is blocked. Inhibition of both can therefore produce more than an additive diuretic response. Second, thiazide diuretics often produce a mild natriuresis in the proximal tubule that is usually masked by increased reabsorption in the TAL. The combination of loop diuretics and thiazides can therefore block Na+ reabsorption, to some extent, from all three segments.
Metolazone is the usual thiazide-like drug used in patients refractory to loop agents alone, but it is likely that other thiazides would be as effective as metolazone. Moreover, metolazone is available only in an oral preparation, while chlorothiazide can be given parenterally.
The combination of loop diuretics and thiazides can mobilize large amounts of fluid, even in patients who have not responded to single agents. Therefore, close hemodynamic monitoring is essential. Routine outpatient use is not recommended. Furthermore, K+-wasting is extremely common and may require parenteral K+ administration with careful monitoring of fluid and electrolyte status.
POTASSIUM-SPARING DIURETICS & LOOP AGENTS OR THIAZIDES
Hypokalemia eventually develops in many patients who are placed on loop diuretics or thiazides. This can usually be managed with dietary NaCl restriction or with dietary KCl supplements. When hypokalemia cannot be managed in this way, the addition of a K+-sparing diuretic can significantly lower K+ excretion. While this approach is generally safe, it should be avoided in patients with renal insufficiency and in those receiving angiotensin antagonists such as ACE inhibitors, in whom life-threatening hyperkalemia can develop in response to K+-sparing diuretics.
III. CLINICAL PHARMACOLOGY OF DIURETIC AGENTS
INTRODUCTION
A summary of the effects of diuretics on urinary electrolyte excretion is shown in Table 15-2.
EDEMATOUS STATES
INTRODUCTION
A common reason for diuretic use is for reduction of peripheral or pulmonary edema that has accumulated as a result of cardiac, renal, or vascular diseases that reduce blood delivery to the kidney. This reduction is sensed as insufficient "effective" arterial blood volume and leads to salt and water retention and edema formation. Judicious use of diuretics can mobilize this interstitial edema without significant reductions in plasma volume. However, excessive diuretic therapy may lead to further compromise of the effective arterial blood volume with reduction in perfusion of vital organs. Therefore, the use of diuretics to mobilize edema requires careful monitoring of the patient's hemodynamic status and an understanding of the pathophysiology of the underlying illness.
HEART FAILURE
When cardiac output is reduced by heart failure, the resultant changes in blood pressure and blood flow to the kidney are sensed as hypovolemia and lead to renal retention of salt and water. This physiologic response initially increases intravascular volume and venous return to the heart and may partially restore the cardiac output toward normal (see Chapter 13).
If the underlying disease causes cardiac output to deteriorate despite expansion of plasma volume, the kidney continues to retain salt and water, which then leaks from the vasculature and becomes interstitial or pulmonary edema. At this point, diuretic use becomes necessary to reduce the accumulation of edema, particularly in the lungs. Reduction of pulmonary vascular congestion with diuretics may actually improve oxygenation and thereby improve myocardial function. Reduction of preload can reduce the size of the heart, allowing it to work at a more efficient fiber length. Edema associated with heart failure is generally managed with loop diuretics. In some instances, salt and water retention may become so severe that a combination of thiazides and loop diuretics is necessary.
In treating the heart failure patient with diuretics, it must always be remembered that cardiac output in these patients is being maintained in part by high filling pressures and that excessive use of diuretics may diminish venous return and further impair cardiac output. This is especially critical in right ventricular heart failure. Systemic, rather than pulmonary vascular, congestion is the hallmark of this disorder. Diuretic-induced volume contraction will predictably reduce venous return and can severely compromise cardiac output if left ventricular filling pressure is reduced below 15 mm Hg (see Chapter 13).
Diuretic-induced metabolic alkalosis is another adverse effect that may further compromise cardiac function. While this complication can be treated with replacement of K+ and restoration of intravascular volume with saline, severe heart failure may preclude the use of saline even in patients who have received excessive diuretic therapy. In these cases, adjunctive use of acetazolamide will help correct the alkalosis.
Another serious toxicity of diuretic use, particularly in the cardiac patient, is hypokalemia. Hypokalemia can exacerbate underlying cardiac arrhythmias and contribute to digitalis toxicity. This can usually be avoided by having the patient reduce Na+ intake, thus decreasing Na+ delivery to the K+-secreting collecting tubule. Patients who are noncompliant with a low Na+diet must take oral KCl supplements or a K+-sparing diuretic.
KIDNEY DISEASE
A variety of renal diseases interfere with the kidney's critical role in volume homeostasis. Although some renal disorders cause salt wasting, most kidney diseases cause retention of salt and water. When loss of renal function is severe, diuretic agents are of little benefit, because there is insufficient glomerular filtration to sustain a natriuretic response. However, a large number of patients with milder degrees of renal insufficiency can be treated with diuretics when they retain sodium.
Many glomerular diseases, such as those associated with diabetes mellitus or systemic lupus erythematosus, exhibit renal retention of salt and water. The cause of this sodium retention is not precisely known, but it probably involves disordered regulation of the renal microcirculation and tubular function through release of vasoconstrictors, prostaglandins, cytokines, and other mediators. When edema or hypertension develops in these patients, diuretic therapy can be very effective. If heart failure is also present, see the warnings mentioned above.
Certain forms of renal disease, particularly diabetic nephropathy, are frequently associated with development of hyperkalemia at a relatively early stage of renal failure. In these cases, a thiazide or loop diuretic will enhance K+ excretion by increasing delivery of salt to the K+-secreting collecting tubule.
Patients with renal diseases leading to the nephrotic syndrome often present complex problems in volume management. These patients may exhibit fluid retention in the form of ascites or edema but have reduced plasma volume due to reduced plasma oncotic pressures. This is very often the case in patients with "minimal change" nephropathy. In these patients, diuretic use may cause further reductions in plasma volume that can impair glomerular filtration rate and may lead to orthostatic hypotension. Most other causes of nephrotic syndrome are associated with primary retention of salt and water by the kidney, leading to expanded plasma volume and hypertension despite the low plasma oncotic pressure. In these cases, diuretic therapy may be beneficial in controlling the volume-dependent component of hypertension.
In choosing a diuretic for the patient with kidney disease, there are a number of important limitations. Acetazolamide must usually be avoided because it can exacerbate acidosis. Potassium-sparing diuretics may cause hyperkalemia. Thiazide diuretics are generally ineffective when glomerular filtration rate falls below 30 mL/min. Thus, loop diuretics are often the best choice in treating edema associated with kidney failure. Lastly, although excessive use of diuretics can impair renal function in all patients, the consequences are more serious in those with underlying renal disease.
HEPATIC CIRRHOSIS
Liver disease is often associated with edema and ascites in conjunction with elevated portal hydrostatic pressures and reduced plasma oncotic pressures. Mechanisms for retention of Na+ by the kidney in this setting include diminished renal perfusion (from systemic vascular alterations), diminished plasma volume (due to ascites formation), and diminished oncotic pressure (hypoalbuminemia). In addition, there may be primary Na+ retention due to elevated plasma aldosterone levels.
When ascites and edema become severe, diuretic therapy can be very useful. However, cirrhotic patients are often resistant to loop diuretics because of decreased secretion of the drug into the tubular fluid and because of high aldosterone levels. In contrast, cirrhotic edema is unusually responsive to spironolactone and eplerenone. The combination of loop diuretics and an aldosterone receptor antagonist may be useful in some patients.
It is important to note that, even more than in heart failure, overly aggressive use of diuretics in this setting can be disastrous. Vigorous diuretic therapy can cause marked depletion of intravascular volume, hypokalemia, and metabolic alkalosis. Hepatorenal syndrome and hepatic encephalopathy are the unfortunate consequences of excessive diuretic use in the cirrhotic patient.
IDIOPATHIC EDEMA
Despite intensive study, the pathophysiology of this disorder (fluctuating salt retention and edema) still remains obscure. Some studies, but not all, suggest that intermittent diuretic use may actually contribute to the syndrome. Idiopathic edema should probably be managed with moderate salt restriction alone if possible.
NONEDEMATOUS STATES
HYPERTENSION
The diuretic and mild vasodilator actions of the thiazides are useful in treating virtually all patients with essential hypertension, and may be sufficient in many. Loop diuretics are usually reserved for patients with renal insufficiency or heart failure. Moderate restriction of dietary Na+ intake (60-100 mEq/d) has been shown to potentiate the effects of diuretics in essential hypertension and to lessen renal K+ wasting.
A recent very large study (over 30,000 participants) has shown that inexpensive diuretics like thiazides result in similar or superior outcomes to those found with ACE inhibitor or calcium channel blocker therapy. This important result reinforces the importance of thiazide therapy in hypertension.
Although diuretics are often successful as monotherapy, they also play an important role in patients who require multiple drugs to control blood pressure. Diuretics enhance the efficacy of many agents, particularly the ACE inhibitors. Patients being treated with powerful vasodilators such as hydralazine or minoxidil usually require simultaneous diuretics because the vasodilators cause significant salt and water retention.
NEPHROLITHIASIS
Approximately two thirds of kidney stones contain Ca2+ phosphate or Ca2+ oxalate. Many patients with such stones exhibit a defect in proximal tubular Ca2+ reabsorption that causes hypercalciuria. This can be treated with thiazide diuretics, which enhance Ca2+ reabsorption in the distal convoluted tubule and thus reduce the urinary Ca2+ concentration. Salt intake must be reduced in this setting, as excess dietary NaCl will overwhelm the hypocalciuric effect of thiazides. Calcium stones may also be caused by increased intestinal absorption of Ca2+, or they may be idiopathic. In these situations, thiazides are also effective, but should be used as adjunctive therapy with decreased Ca2+ intake and other measures.
HYPERCALCEMIA
Hypercalcemia can be a medical emergency. Because loop diuretics reduce Ca2+ reabsorption significantly, they can be quite effective in promoting Ca2+ diuresis. However, loop diuretics alone can cause marked volume contraction. If this occurs, loop diuretics are ineffective (and potentially counterproductive) because Ca2+ reabsorption in the proximal tubule would be enhanced. Thus, saline must be administered simultaneously with loop diuretics if an effective Ca2+ diuresis is to be maintained. The usual approach is to infuse normal saline and furosemide (80-120 mg) intravenously. Once the diuresis begins, the rate of saline infusion can be matched with the urine flow rate to avoid volume depletion. Potassium chloride may be added to the saline infusion as needed.
DIABETES INSIPIDUS
Diabetes insipidus is due either to deficient production of ADH (neurogenic or central diabetes insipidus) or inadequate responsiveness to ADH (nephrogenic diabetes insipidus). Administration of supplementary ADH or one of its analogs is only effective in central diabetes insipidus. Thiazide diuretics can reduce polyuria and polydipsia in both types of diabetes insipidus. This seemingly paradoxic beneficial effect is mediated through plasma volume reduction, with an associated fall in glomerular filtration rate, enhanced proximal reabsorption of NaCl and water, and decreased delivery of fluid to the downstream diluting segments. Thus, the maximum volume of dilute urine that can be produced is lowered and thiazides can significantly reduce urine flow in the polyuric patient. Dietary sodium restriction can potentiate the beneficial effects of thiazides on urine volume in this setting. Lithium (Li+), used in the treatment of manic-depressive disorder, is a common cause of nephrogenic diabetes insipidus and thiazide diuretics have been found to be helpful in treating it. Serum Li+ levels must be carefully monitored in these patients, because diuretics may reduce renal clearance of Li+ and raise plasma Li+ levels into the toxic range (see Chapter 29). Lithium-induced polyuria can also be partially reversed by amiloride, which blocks Li+ entry into collecting duct cells, much as it blocks Na+ entry.
PREPARATIONS AVAILABLE
Acetazolamide (generic, Diamox)
Oral: 125, 250 mg tablets
Oral sustained-release: 500 mg capsules
Parenteral: 500 mg powder for injection
Amiloride (generic, Midamor, combination drugs)
Oral: 5 mg tablets
Bendroflumethiazide (Naturetin, combination drugs)
Oral: 5, 10 mg tablets
Brinzolamide (Azopt) (For ocular conditions)
Ophthalmic: 1% suspension
Bumetanide (generic, Bumex)
Oral: 0.5, 1, 2 mg tablets
Parenteral: 0.5 mg/2 mL ampule for IV or IM injection
Chlorothiazide (generic, Diuril)
Oral: 250, 500 mg tablets; 250 mg/5 mL oral suspension
Parenteral: 500 mg for injection
Chlorthalidone (generic, Hygroton, Thalitone, combination drugs)
Oral: 25, 50, 100 mg tablets
Conivaptan (Vaprisol)
Parenteral: 5 mg/mL for IV injection
Demeclocycline (Declomycin)
Oral: 150 mg tablets and capsules; 300 mg tablets
Dichlorphenamide (Daranide)
Oral: 50 mg tablets
Dorzolamide (Trusopt) (For ocular conditions)
Ophthalmic: 2% solution
Eplerenone (Inspra)
Oral: 25, 50 mg tablets
Ethacrynic acid (Edecrin)
Oral: 25, 50 mg tablets
Parenteral: 50 mg IV injection
Furosemide (generic, Lasix, others)
Oral: 20, 40, 80 mg tablets; 8, 10 mg/mL oral solutions
Parenteral: 10 mg/mL for IM or IV injection
Hydrochlorothiazide (generic, Esidrix, Hydro-DIURIL, combination drugs)
Oral: 12.5 mg capsules; 25, 50, 100 mg tablets; 10, 100 mg/mL solution
Hydroflumethiazide (generic, Saluron)
Oral: 50 mg tablets
Indapamide (generic, Lozol)
Oral: 1.25, 2.5 mg tablets
Mannitol (generic, Osmitrol)
Parenteral: 5, 10, 15, 20, for injection
Methazolamide (generic, Neptazane) (For ocular conditions)
Oral: 25, 50 mg tablets
Methyclothiazide (generic, Aquatensen, Enduron)
Oral: 2.5, 5 mg tablets
Metolazone (Mykrox, Zaroxolyn) (Note: Bioavailability of Mykrox is greater than that of Zaroxolyn.)
Oral: 0.5 (Mykrox); 2.5, 5, 10 mg (Zaroxolyn) tablets
Polythiazide (Renese, combination drugs)
Oral: 1, 2, 4 mg tablets
Quinethazone (Hydromox)
Oral: 50 mg tablets
Spironolactone (generic, Aldactone)
Oral: 25, 50, 100 mg tablets
Torsemide (Demadex)
Oral: 5, 10, 20, 100 mg tablets
Parenteral: 10 mg/mL for injection
Triamterene (Dyrenium)
Oral: 50, 100 mg capsules
Trichlormethiazide (generic, Diurese, Naqua, others)
Oral: 2, 4 mg tablets
REFERENCES
Abdallah JG et al: Loop diuretic infusion increases thiazide-sensitive Na(+)/Cl(-)-cotransporter abundance: Role of aldosterone. J Am Soc Nephrol 2001;12:1335.
ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group: Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002;288:2981.
Alvarez-Guerra M, Garay RC: Renal Na-K-Cl transporter NKCC2 in Dahl salt-sensitive rats. J Hypertens 2002;20:721.
Brenner BM (editor): Brenner & Rector's The Kidney, 7th ed. Saunders, 2003.
Costello-Boerrigter LC, Boerrigter G, Burnett JC Jr: Revisiting salt and water retention: New diuretics, aquaretics, and natriuretics. Med Clin North Am 2003;87:475.
Fall PJ: Hyponatremia and hypernatremia. A systematic approach to causes and their correction. Postgrad Med 2000;107:75.
Gottlieb SS et al: BG9719 (CVT-124), an A1 adenosine receptor antagonist, protects against the decline in renal function observed with diuretic therapy. Circulation 2002;105:1348.
Greenberg A: Diuretic complications. Am J Med Sci 2000;319:10.
Hackett PH, Roach RC: High-altitude illness. N Engl J Med 2001;345:107.
Kalra PR et al: The regulation and measurement of plasma volume in heart failure. J Am Coll Cardiol 2002;39:1901.
Kaplan NM: The place of diuretics in preventing cardiovascular events. J Hum Hypertens 2004;18:S29.
Knepper MA, Brooks HL: Regulation of the sodium transporters NHE3, NKCC2, and NCC in the kidney. Curr Opin Nephrol Hypertens 2001;10:655.
Na KY et al: Upregulation of Na+ transporter abundance in response to chronic thiazide or loop diuretic treatment in rats. Am J Physiol 2003;284:F133.
Nijenhuis T et al: Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 2005;115:1651.
Rejnmark L et al: Effects of long-term treatment with loop diuretics on bone mineral density, calciotropic hormones and bone turnover. J Intern Med 2005;257:176.
Schrot RJ, Muizelaar JP: Mannitol in acute traumatic brain injury. Lancet 2002;359:1633.
Shlipak MG, Massie BM: The clinical challenge of cardiorenal syndrome. Circulation 2004;110:1514.
Sica DA, Gehr TWB: Diuretic use in stage 5 chronic kidney disease and end-stage renal disease. Curr Opin Nephrol Hypertens 2003;12:483.
Tovar-Palacio C et al: Ion and diuretic specificity of chimeric proteins between apical Na+-K+-2CL- and Na+-Cl- cotransporters. Am J Physiol 2004;287:F570.
Wilcox C: New insights into diuretic use in patients with chronic renal disease. J Am Soc Nephrol 2002;13:798.