Tobias Else, MD, & Gary D. Hammer, MD, PhD
The adrenal medulla secretes catecholamines (epinephrine, norepinephrine, and dopamine). The catecholamines help prepare the individual to deal with emergency situations. The major disorder of the adrenal medulla is pheochromocytoma, a neoplasm characterized by excessive catecholamine secretion.
NORMAL STRUCTURE & FUNCTION OF THE ADRENAL MEDULLA
ANATOMY
The adrenal medulla is the reddish-brown central portion of the adrenal gland. Accessory medullary tissue is sometimes located in the retroperitoneum near the sympathetic ganglia or along the abdominal aorta (paraganglia) (Figure 12-1).
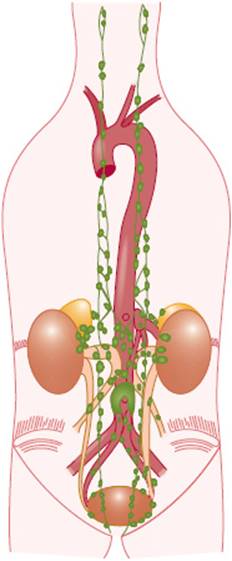
FIGURE 12-1 Anatomic distribution of extra-adrenal chromaffin tissue in the newborn. (Redrawn, with permission, from Coupland R. The Natural History of the Chromaffin Cell. Longman, Green, 1965.)
HISTOLOGY
The adrenal medulla is made up of polyhedral cells arranged in cords or clumps. Embryologically, the adrenal medullary cells derive from neural crest cells. Medullary cells are innervated by cholinergic preganglionic nerve fibers that reach the gland via the splanchnic nerves. The adrenal medulla can be regarded as a specialized sympathetic ganglion, where preganglionic sympathetic nerve fibers (using acetylcholine as a neurotransmitter) directly make contact with postganglionic cells, which secrete catecholamines (mainly epinephrine) directly into the circulation. This relationship is analogous to the other sympathetic paraganglions, which connect preganglionic cholinergic sympathetic nerve fibers with postganglionic fibers using catecholamines (mainly norepinephrine) as neurotransmitters. Medullary parenchymal cells accumulate and store their hormone products in prominent, dense secretory granules, 150–350 nm in diameter. Histologically, these cells and granules have a high affinity for chromium salts (chromaffin reaction) and thus are called chromaffin cells and contain chromaffin granules. The granules contain the catecholamines epinephrine and norepinephrine. Morphologically, two types of medullary cells can be distinguished: epinephrine-secreting cells, which have larger, less dense granules, and norepinephrine-secreting cells, which have smaller, very dense granules. Separate dopamine-secreting cells have not been identified. Ninety percent of medullary cells are the epinephrine-secreting type and 10% are the norepinephrine-secreting type.
PHYSIOLOGY
The catecholamines help to regulate metabolism, contractility of cardiac and smooth muscle, and neurotransmission.
Formation, Secretion, & Metabolism of Catecholamines
The adrenal medulla secretes three catecholamines: epinephrine, norepinephrine, and dopamine. Secretion occurs after release of acetylcholine from the preganglionic neurons that innervate the medullary cells. The major biosynthetic pathways and hormonal intermediates for the catecholamines are shown in Figure 12-2. In humans, most (80%) of the catecholamine output of the adrenal medulla is epinephrine. Norepinephrine is principally found in paraganglionic nerve endings of the sympathetic nervous system and in the CNS, where it functions as a major neurotransmitter.
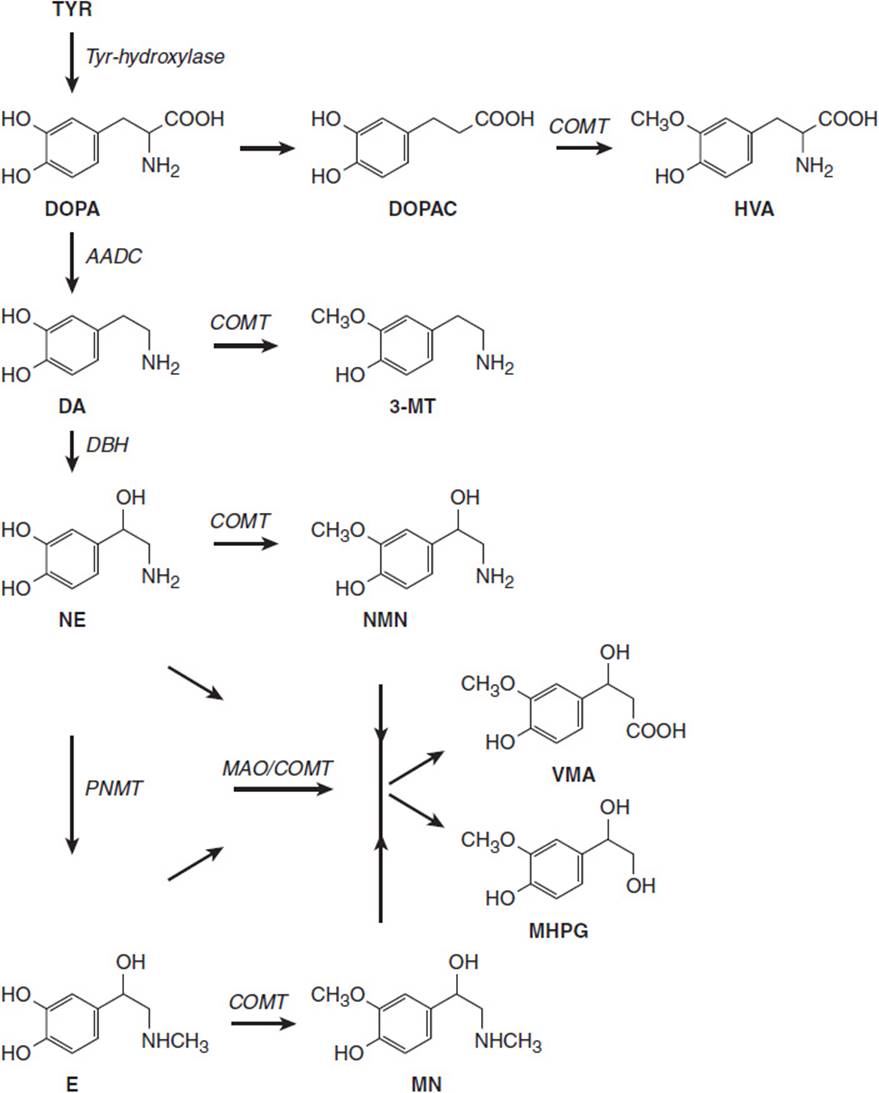
FIGURE 12-2 Biosynthesis and catabolism of catecholamines. The catecholamines are synthesized from tyrosine (TYR). The enzyme catechol-O-methyltransferase (COMT) generates metanephrine (MN) from epinephrine (E) and normetanephrine (NM) from norepinephrine. COMT is constitutively active in pheochromocytomas and paragangliomas and release of these substances is constant rather than episodic. (AADC, aromatic l-amino acid decarboxylase; DA, dopamine; DBH, dopamine beta-hydroxylase; DOPAC, dihydroxyphenylacetic acid; HVA, homovanillic acid; MAO, monoamine oxidase; 3MT, 3-methoxytyramine; NE, norepinephrine; PNMT, phenylethanolamine N-methyltransferase; VMA, vanillylmandelic acid.)
Approximately 70% of the epinephrine and norepinephrine and 95% of the dopamine found in plasma are conjugated to sulfate and inactive. In the supine state, the normal plasma level of free epinephrine is about 30 pg/mL (0.16 nmol/L); there is a 50–100% increase on standing. The normal plasma level of free norepinephrine is about 300 pg/mL (1.8 nmol/L), and the plasma free dopamine level is about 35 pg/mL (0.23 nmol/L).
Most catecholamine metabolism takes place within the same cells where they are synthesized, mainly because of leakage of catecholamines from vesicular stores into the cytoplasm. These vesicular stores exist in a dynamic equilibrium, with outward passive leakage counterbalanced by inward active transport that is controlled by vesicular monoamine transporters. In catecholaminergic neurons, the presence of monoamine oxidase in the cytoplasm leads to formation of reactive catecholaldehydes. Production of these toxic aldehydes is dependent on the dynamics of the vesicular-axoplasmic monoamine exchange and an enzyme-catalyzed conversion to nontoxic acids or alcohols. In sympathetic nerves, the aldehyde produced from norepinephrine is converted to 3,4-dihydroxyphenylglycol. Subsequent extraneuronal O-methylation leads to production of 3-methoxy-4-hydroxyphenylglycol, and its oxidation in the liver catalyzed by alcohol and aldehyde dehydrogenases leads to formation of vanillylmandelic acid (VMA). Compared with intraneuronal deamination, extraneuronal O-methylation of norepinephrine and epinephrine to metanephrines represents minor pathways of metabolism.
The single largest source of metanephrine is the adrenal medulla. In the circulation, the catecholamines have a short half-life of about 2 min. Normally, only very small quantities of free epinephrine (about 6 μg/d) and norepinephrine (about 30 μg/d) are excreted, but about 700 μg of VMA is excreted daily.
Regulation of Catecholamine Secretion
Physiologic stimuli affect medullary secretion through the nervous system. Medullary cells secrete catecholamines after release of acetylcholine from the preganglionic neurons that innervate them. Catecholamine secretion is low in the basal state and is reduced even further during sleep. In emergency situations, there is increased adrenal catecholamine secretion as part of a generalized sympathetic discharge that serves to prepare the individual for stress (“fight-or-flight” response). Physiological stress such as psychological, physical (eg, mechanical, thermal), and metabolic (eg, hypoglycemia, exercise) stress leads to catecholamine secretion.
Mechanism of Action of Catecholamines
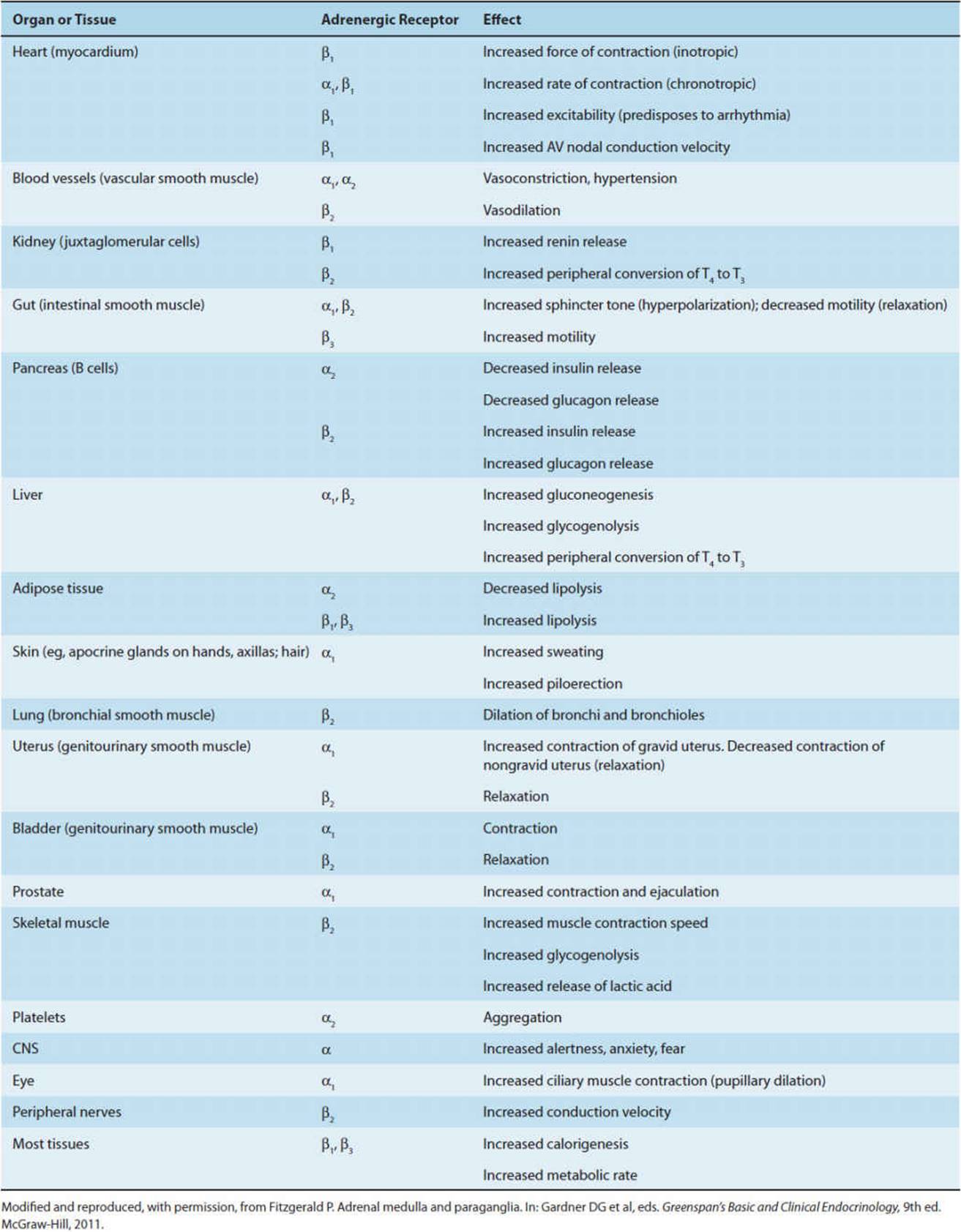
The effects of epinephrine and norepinephrine are mediated by their actions on two classes of receptors: α- and β-adrenergic receptors (Table 12-1). Alpha receptors are subdivided into α1 and α2 receptors and β receptors into β1, β2, and β3 receptors. Alpha1 receptors mediate smooth muscle contraction in blood vessels and the genitourinary (GU) tract and increase glycogenolysis. Alpha2 receptors mediate smooth muscle relaxation in the GI tract and vasoconstriction of some blood vessels. Alpha2 receptors also decrease insulin secretion. Beta1 receptors mediate an increased rate and force of myocardial contraction and stimulate lipolysis and renin release. Beta2 receptors mediate smooth muscle relaxation in the bronchi, blood vessels, GU tract, and GI tract and increase hepatic gluconeogenesis and glycogenolysis, muscle glycogenolysis, and release of insulin and glucagon.
TABLE 12-1 Physiologic effects of catecholamines on adrenergic receptors of selected tissues.
Intracellular post-receptor signaling is different for each subclass of adrenergic receptor. Stimulation of α1-adrenergic receptors results in an increase in intracellular Ca2+ concentrations. First, there is activation of phospholipase C by the guanine nucleotide binding stimulatory protein, Gs. Phospholipase C hydrolyzes the membrane-bound phospholipid, phosphatidylinositol-4,5-bisphosphate, to generate two second messengers: diacylglycerol and inositol-1,4,5-trisphosphate. Diacylglycerol in turn activates protein kinase C, which phosphorylates various cellular substrates. Inositol-1,4,5-trisphosphate stimulates release of intracellular Ca2+, which then initiates various cellular responses.
Activation of α2-adrenergic receptors results in a decrease in intracellular cyclic adenosine 3′,5′-monophosphate (cAMP). The mechanism involves receptor interaction with an inhibitory G protein, Gi, leading to inhibition of adenylyl cyclase. The fall in cAMP level leads to a decrease in activity of the cAMP-dependent protein kinase A. The Gi protein also stimulates K+ channels and inhibits voltage-sensitive calcium channels.
On the other hand, β-adrenergic receptors stimulate adenylyl cyclase through the mediation of Gs. Activation of β-adrenergic receptors thus leads to an increase in cAMP, activation of the cAMP-dependent protein kinase A, and consequent phosphorylation of various cellular proteins. The Gs protein can also directly activate voltage-sensitive Ca2+ channels in the plasma membrane of cardiac and skeletal muscle.
The α1- and β1-adrenergic receptors are generally found in organs and tissues (eg, heart and gut) that are heavily innervated by—and situated so as to be readily activated by stimulation of—the sympathetic nerves. The α1- and β1-adrenergic receptors are preferentially stimulated by norepinephrine, especially that released by nerve endings. In contrast, the α2- and β2-adrenergic receptors are generally situated in postjunctional sites in organs and tissues (eg, uterine and bronchial skeletal muscle) remote from sites of norepinephrine release. The α2- and β2-adrenergic receptors are preferentially stimulated by circulating catecholamines, especially epinephrine.
Differences in tissue distribution, accessibility by nerve fibers, preferences for epinephrine versus norepinephrine, and differences in postreceptor signaling are thus responsible for the diverse effects of catecholamines in an organ- and cell-specific manner.
Effects of Catecholamines
The catecholamines have been termed fight-or-flight hormones because their effects on the heart, blood vessels, smooth muscle, and metabolism assist the organism in responding to stress. The principal physiologic effects of the catecholamines are shown in Table 12-1.
In the peripheral circulation, norepinephrine produces vasoconstriction in most organs (via α1 receptors). Epinephrine produces vasodilation via β2 receptors in skeletal muscle and liver and vasoconstriction elsewhere. The former usually outweighs the latter, and for that reason epinephrine usually lowers total peripheral resistance.
Norepinephrine causes both systolic and diastolic blood pressures to rise. The rise in blood pressure stimulates the carotid and aortic baroreceptors, resulting in reflex bradycardia and a fall in cardiac output. Epinephrine causes a widening of pulse pressure but does not stimulate the baroreceptors to the same degree, so the pulse rises and cardiac output increases.
Hence, pheochromocytomas or other tumors of the adrenal medulla, which usually secrete norepinephrine, lead to vasoconstriction and an increase in blood pressure.
The effects of catecholamines on metabolism include effects on glycogenolysis, lipolysis, and insulin secretion, mediated by both α- and β-adrenergic receptors. These metabolic effects result primarily from the action of epinephrine on four target tissues: liver, muscle, pancreas, and adipose tissue (see Table 12-1). The result is an increase in the levels of circulating glucose and free fatty acids. The increased supply of these two substances helps provide an adequate supply of metabolic fuel to the nervous system and muscle during physiologic stress.
The amount of circulating plasma epinephrine and norepinephrine needed to produce these various effects has been determined by infusing the catecholamines into resting subjects. For norepinephrine, the threshold for the cardiovascular and metabolic effects is a plasma level of about 1500 pg/mL, or about five times the basal level. In normal individuals, the plasma norepinephrine level rarely exceeds this threshold. However, for epinephrine, the threshold for tachycardia occurs at a plasma level of about 50 pg/mL, or about twice the basal level. The threshold for increasing systolic blood pressure and lipolysis is at about 75 pg/mL; for increasing glucose and lactate, about 150 pg/mL; and for increasing insulin secretion, about 40 pg/mL. In healthy individuals, plasma epinephrine levels often exceed these thresholds.
The physiologic effect of circulating dopamine is unknown. Centrally, dopamine acts to inhibit prolactin secretion. Peripherally, in small doses, injected dopamine produces renal vasodilation, probably by binding to a specific dopaminergic receptor. In moderate doses, it also produces vasodilation of the mesenteric and coronary circulation and vasoconstriction peripherally. It has a positive inotropic effect on the heart, mediated by action on the β1-adrenergic receptors. Moderate to large doses of dopamine increase the systolic blood pressure without affecting diastolic pressure.
Overview of Adrenal Medullary Disorders
Pheochromocytoma is an uncommon tumor of adrenal medullary tissue that causes production of excessive amounts of catecholamines. Patients typically present with sustained or episodic hypertension or with a syndrome characterized by episodic palpitations, tachycardia, chest pain, headache, anxiety, pallor, excessive sweating and hyperglycemia. Pheochromocytomas can usually be cured if diagnosed and treated properly. Pheochromocytomas are closely related to paragangliomas, which sometimes are termed extra-adrenal pheochromocytomas. Paragangliomas arise from paraganglia of the autonomous nervous system. Most parasympathetic paragangliomas are found in the head and neck area (eg, carotid body, vagal nerve) and either do not secrete any catecholamines or rarely secrete norepinephrine. Most sympathetic paragangliomas arise in the abdomen and often secrete norepinephrine.
CHECKPOINT
1. What is the embryologic origin of the cells of the adrenal medulla?
2. What nerve fibers innervate the adrenal medulla?
3. Which catecholamines are secreted by the human adrenal medulla? Of these, which is the major product?
4. What are the major physiologic stimuli of catecholamine secretion?
5. What are the subtypes and distribution of catecholamine receptors?
6. What physiologic processes do each subtype of catecholamine receptor control, and how do catecholamines bring about each of these physiologic process?
PATHOPHYSIOLOGY OF SELECTED DISORDERS OF THE ADRENAL MEDULLA
Pheochromocytomas are the main pathological entity of the adrenal medulla. Other tumors of the adrenal medulla or its embryonic precursors include neuroblastomas and ganglioneuromas. Neuroblastomas are one of the most common tumors of early childhood. In response to therapy (or even spontaneously), neuroblastomas can differentiate into ganglioneuromas. Both of these tumors secrete catecholamines, but symptoms due to catecholamine excess are usually absent because they do not reach the levels observed with pheochromocytomas. Absence of the adrenal medulla (eg, after bilateral adrenalectomy) is usually well tolerated, though sometimes symptoms such as orthostatic hypotension may be observed. Closely related, but different from pheochromocytomas, are parasympathetic nervous system paragangliomas, which often arise in the affected patient’s head and neck area.
PHEOCHROMOCYTOMA
Pheochromocytomas are neoplasms of the chromaffin cells of the adrenal medulla or extramedullary sites. These tumors secrete excessive amounts of epinephrine, norepinephrine, or both (rarely dopamine). Most pheochromocytomas secrete norepinephrine and cause sustained or episodic hypertension. Pheochromocytomas that secrete epinephrine cause hypertension less often; more frequently, they produce episodic hyperglycemia, glucosuria, and other metabolic effects.
Table 12–2 summarizes the clinical features of pheochromocytomas. Pheochromocytomas are uncommon, probably found in less than 0.1% of all patients with hypertension and in approximately two individuals per million population. Pheochromocytomas occur in both sexes and in all age groups but are most often diagnosed in the fourth or fifth decade of life. Compared with adults, children with pheochromocytomas are more likely to have multifocal and extra-adrenal tumors, and a causal familial syndrome must always be excluded.
TABLE 12-2 Clinical features of pheochromocytoma.
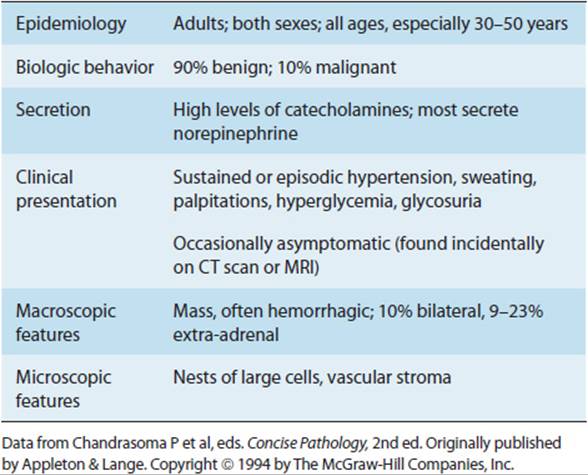
The diagnosis is important because sudden release of catecholamines from these tumors during anesthesia, surgery, or obstetric delivery may prove fatal. Pheochromocytoma was classically referred to as “the 10% tumor” because 10% occur in extra-adrenal paraganglia, 10% are outside the abdomen, 10% are multiple, 10% are bilateral, about 10% are not associated with hypertension, 10% occur in children, and 10% are malignant. Recent research has revised some of these numbers. So, previously, it was thought that about 10% occur as part of a familial syndrome, but now it appears that actually about 20–30% of cases are familial. Also, occurrence at extra-adrenal sites seems to be higher (9–23%) and multifocal pheochromocytomas can be found in roughly one third of childhood cases.
Etiology
Several genetic syndromes, all transmitted in an autosomal dominant fashion, are associated with an increased risk of pheochromocytoma and sympathetic or parasympathetic nervous system paragangliomas. Most familial cases are caused by one of four syndromes: neurofibromatosis type 1, von Hippel–Lindau syndrome, multiple endocrine neoplasia type 2 (MEN-2), and familial paraganglioma syndrome (Table 12-3). The genetic basis of these syndromes is now well defined. Patients with neurofibromatosis type 1 (von Recklinghausen disease) have an increased incidence of pheochromocytoma caused by mutation of the NF1 gene. Pheochromocytoma is a frequent occurrence in families with von Hippel–Lindau disease, which is caused by mutations of the VHL tumor suppressor gene. VHL-associated pheochromocytomas often secrete norepinephrine.
TABLE 12-3 Major genetic syndromes associated with pheochromocytoma.

In MEN-2A syndrome, pheochromocytomas occur in association with calcitonin-producing medullary carcinoma or C-cell hyperplasia of the thyroid and parathyroid hormone (PTH)–producing adenomas of the parathyroid. In MEN-2B, pheochromocytomas occur in association with medullary carcinoma of the thyroid and numerous oral mucosal neuromas. About 50% of patients with MEN-2A and MEN-2B have pheochromocytomas, often bilaterally. The gene responsible for MEN-2A and MEN-2B has now been localized to chromosome 10q11.2. The position of the RET mutation is related to disease phenotype. Any mutation of the RET proto-oncogene at one specific position (codon 634) is associated with MEN-2A and mutations at a different position (codon 918), with MEN-2B. These germline mutations of the RET proto-oncogene were the first examples of a dominantly acting oncogenic point mutation found to cause a heritable neoplasm in humans. These missense mutations can be detected by DNA analysis, allowing identification of MEN carriers.
The most prevalent change involves a cysteine residue in position 634 of the RET proto-oncogene. RET encodes a plasma membrane–associated tyrosine kinase that associates with a number of different related receptors. When these receptors are activated, they dimerize and bring two molecules of the RET tyrosine kinase close together, which initiates the cellular transmission of the signal. The 634 cysteine residue is part of an intramolecular sulfide bridge between associated cysteine residues. When one cysteine is absent, two RET molecules form intermolecular bridges, resulting in the initiation of intracellular signaling even in the absence of receptor association or ligand activation.
Familial paraganglioma syndromes are transmitted in autosomal dominant fashion and are caused by germline mutations in genes coding for subcomponents of the succinate-dehydrogenase complex (SDHD, SDHB, SDHC, and rarely SDHA and SDHAF2). Extra-adrenal paragangliomas of the abdomen (SDHB) and head and neck area (SDHD and SDHC) are more common in patients with familial paraganglioma. SDHD mutations are influenced by genomic imprinting causing a “parent of origin effect.” Affected patients always inherit the defective allele from their father. Indeed, while a carrier of a maternally inherited allele can propagate the defect to offspring, this mutant allele does not increase the risk for a paraganglioma.
SDHx and VHL mutations both cause perturbations in the same intracellular signaling cascade that is usually induced by hypoxia, causing a “pseudo-hypoxia” state. SDHx mutations lead to an accumulation of succinate, which in turn inhibits an enzyme that hydroxylates the transcription factor hypoxia-induced factor α (HIF1α.) Hydroxylated HIF1α is recognized by the VHL gene product and is subjected to destruction. Therefore, SDHx mutations as well as VHL mutations lead to an increase in HIF1α-induced transcription, which is in part responsible for the neoplastic phenotype.
Mutations in several other genes, such as TMEM127 and MAX, have also recently been shown to predispose to the development of pheochromocytoma and paraganglioma.
Germline mutations in RET, VHL, SDHx, and others account for at least 20–30% of cases of isolated pheochromocytomas and paragangliomas. Given the high frequency of germline mutations, genetic counseling and genetic testing are recommended for all patients with pheochromocytomas or paragangliomas, particularly those with a positive family history, multifocal disease, or a diagnosis before age 50 years. Genetic testing may also be useful in screening families of carriers of mutations detected.
Almost all pheochromocytomas (about 90%) occur in the abdomen, and most of these (85%) are in the adrenal medulla. Extra-adrenal paragangliomas (including sympathetic and parasympathetic paragangliomas) are found in the perirenal area, the organ of Zuckerkandl, the urinary bladder, the heart, the neck, and the posterior mediastinum (Figure 12-1). Some of these tumors can lead to very specific symptoms (eg, urinary bladder pheochromocytoma can cause a hypertensive crisis with voiding). Grossly, pheochromocytomas are generally well-circumscribed but vary in size, with weights ranging from less than 1 g to several kilograms (Figure 12-3). They are highly vascular tumors and frequently have cystic, necrotic, or hemorrhagic areas. Microscopically, the tumor consists of large pleomorphic cells arranged in sheets separated by a highly vascular stroma. In the cytoplasm, there are catecholamine-containing storage granules similar to those in normal adrenal medullary cells. Mitoses are rare, but tumor invasion of the adrenal capsule and blood vessels is common even in benign pheochromocytomas. About 10% of pheochromocytomas are malignant. Malignancy is established only when a metastasis is found in a site where chromaffin cells are not usually demonstrated (eg, liver, lung, bone, or brain). Unfavorable prognostic factors suggesting a malignant course include large tumor size, local extension, younger age, DNA aneuploid tumors, dopamine secretion, and SDHB mutation.
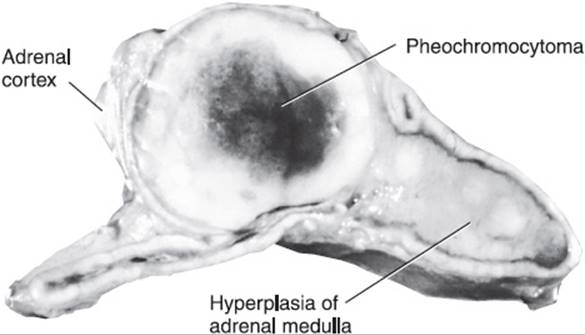
FIGURE 12-3 Cross section of adrenal, showing a pheochromocytoma associated with hyperplasia of the medulla in a patient with multiple endocrine neoplasia type IIa. He also had a medullary carcinoma of the thyroid and a large pheochromocytoma in the opposite adrenal. (Reproduced, with permission, from Chandrasoma P et al, eds. Concise Pathology, 3rd ed. Originally published by Appleton & Lange. Copyright © 1998 by The McGraw-Hill Companies, Inc.)
Pathogenesis
Most pheochromocytomas release predominantly norepinephrine, but most also release epinephrine (Table 12-4). Rarely, a pheochromocytoma releases mostly or only epinephrine and very rarely mostly or only dopamine.
TABLE 12-4 Pathophysiologic and clinical manifestations of catecholamine excess.

In about half of patients with pheochromocytoma, clinical manifestations vary in intensity and occur in an episodic or paroxysmal fashion. The paroxysms are related to sudden catecholamine discharge from the tumor. The sudden catecholamine excess causes hypertension, palpitations, tachycardia, chest pain, headache, anxiety, blanching, and excessive sweating. Such paroxysms usually occur several times a week but may occur only once every few months or up to 25 times daily. Paroxysms typically last for 15 minutes or less but may last for days. As time passes, the paroxysms usually become more frequent but generally do not change in character. A typical paroxysm may be produced by activities that compress the tumor (eg, bending, lifting, exercise, defecation, eating, or deep palpation of the abdomen) and by emotional distress or anxiety.
Other patients have persistently secreting tumors and more chronic symptoms, including sustained hypertension. However, such patients also usually experience paroxysms related to transient increases in catecholamine release. The long-term exposure to high levels of circulating catecholamines seems not to produce the classic hemodynamic responses observed after acute administration of catecholamines. This may be due in part to desensitization of the cardiovascular system to catecholamines and may explain why some patients with pheochromocytomas are entirely asymptomatic.
Clinical Manifestations
The clinical manifestations of pheochromocytoma are due to increased secretion of epinephrine and norepinephrine. Commonly reported manifestations are listed in Table 12-5.
TABLE 12-5 Clinical findings in pheochromocytoma.
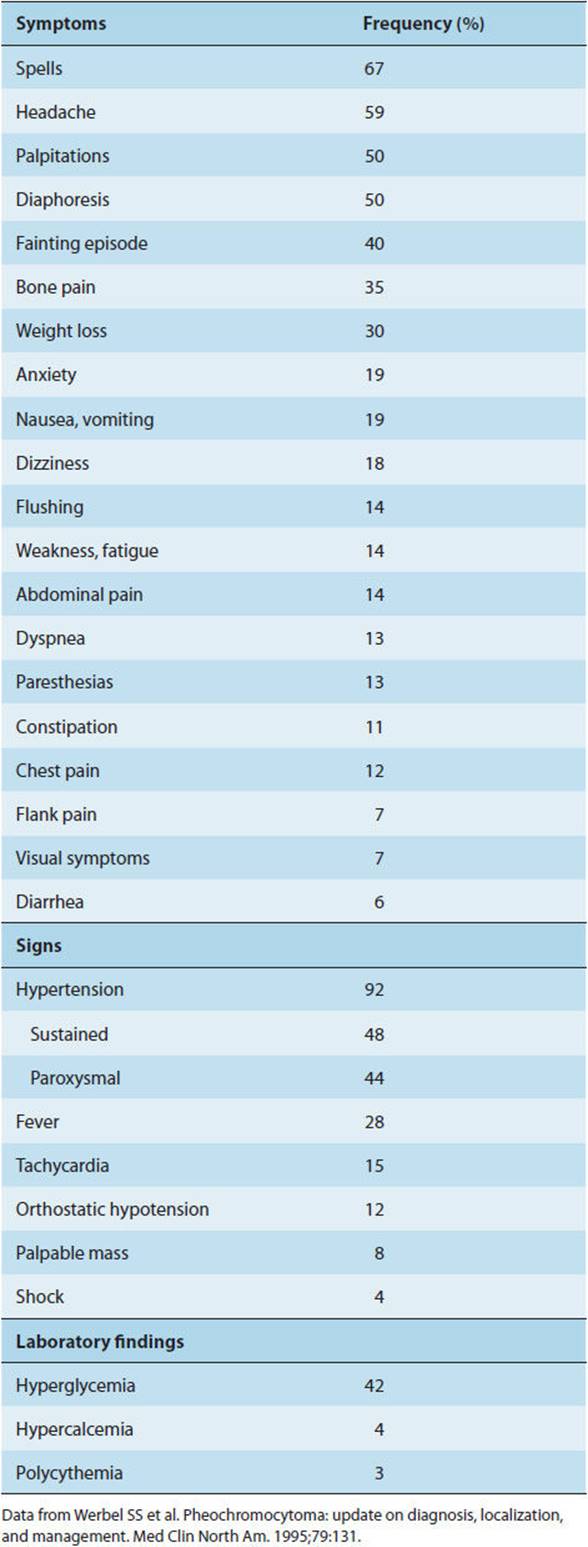
The classical pentad of symptoms in patients with pheochromocytoma consists of: headache, palpitation, perspiration, pallor, and orthostasis. The most common presenting feature of pheochromocytoma is hypertension. In about half of cases, hypertension is sustained but the blood pressure shows marked fluctuations, with peak pressures during symptomatic paroxysms. During a hypertensive episode, the systolic blood pressure can rise to as high as 300 mm Hg. In about one third of cases, hypertension is truly intermittent. In some individuals with pheochromocytoma, hypertension is absent. The blood pressure elevation caused by the catecholamine excess results from two mechanisms: α receptor–mediated vasoconstriction of arterioles, leading to an increase in peripheral resistance; and β1 receptor–mediated increases in cardiac output and in renin release, leading to increased circulating levels of angiotensin II. The increased total peripheral vascular resistance is probably primarily responsible for the maintenance of high arterial pressures.
Hypertensive crisis may be precipitated by a variety of drugs, including tricyclic antidepressants, antidopaminergic agents, metoclopramide, and naloxone. Beta blockers should not be administered until alpha blockade has been established. Otherwise, blockade of β2-adrenergic receptors, which promote vasodilation, will allow unopposed α-adrenergic receptor activation and produce marked vasoconstriction and hypertension.
Peripheral vasoconstriction, mediated by α receptors, causes both facial pallor and cool, moist hands and feet. Chronic vasoconstriction of the arterial and venous beds leads to a reduction in plasma volume and predisposes to postural hypotension. In others, orthostatic hypotension is associated with decreased cardiac stroke volume and an impaired response of total peripheral vascular resistance to changes in posture, perhaps indicative of diminished arteriolar and venous responsiveness. The reduced responsiveness of the vasculature to norepinephrine in patients with pheochromocytoma is probably related to downregulation of α-adrenergic receptors resulting from persistent elevations of norepinephrine levels.
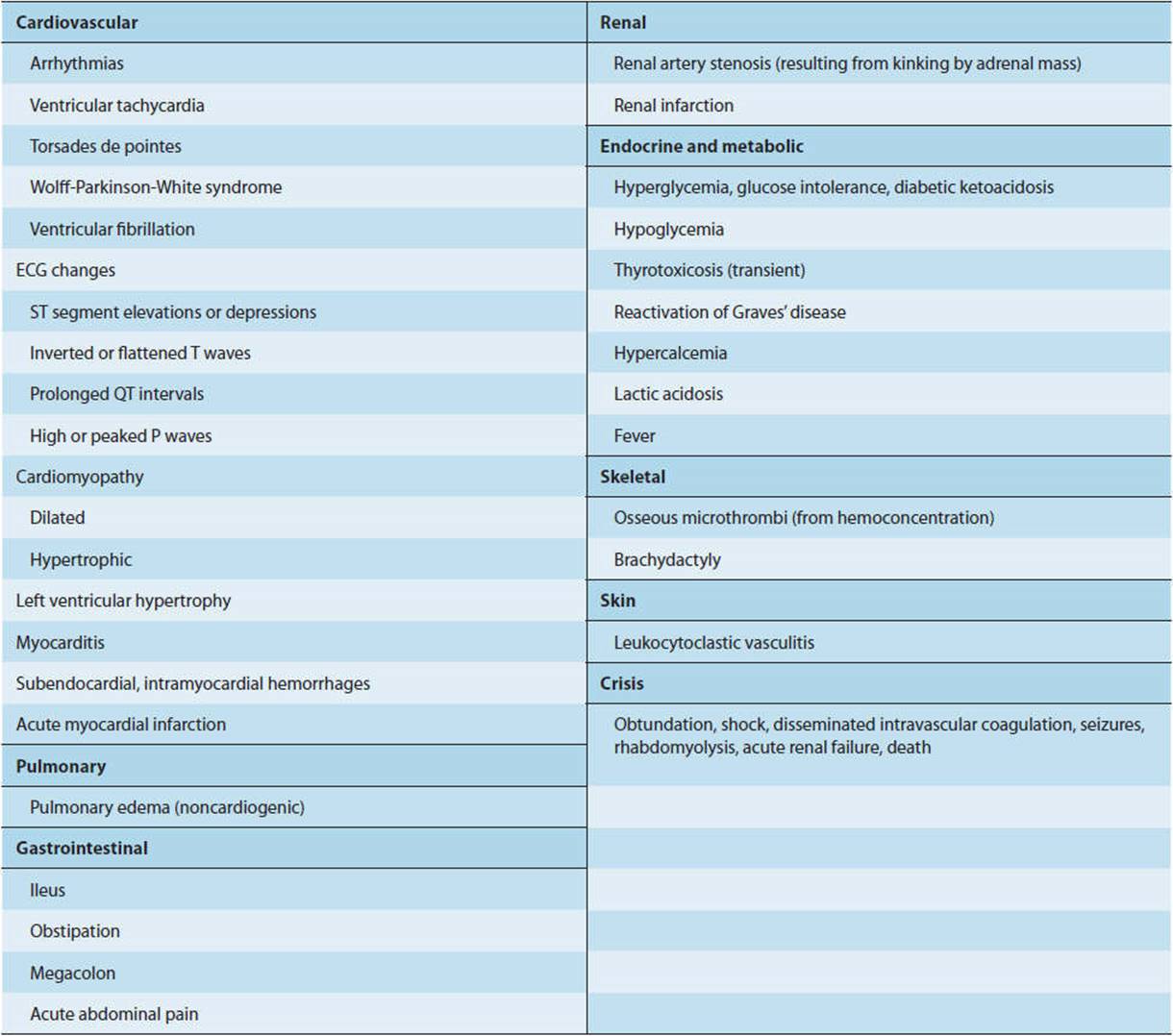
Complications of pheochromocytoma are summarized in Table 12-6. If unrecognized and untreated, pheochromocytoma may be complicated by hypertensive retinopathy (retinal hemorrhages or papilledema); nephropathy; myocardial infarction, resulting from either myocarditis or coronary artery vasospasm; pulmonary edema, secondary either to left-sided heart failure or noncardiogenic causes; and stroke from cerebral infarction, intracranial hemorrhage, or embolism. Cerebral infarction results from hypercoagulability, vasospasm, or both. Hemorrhage occurs secondary to severe arterial hypertension. Emboli can originate in mural thrombi in patients with dilated cardiomyopathy. Cardiomyopathy during a catecholamine surge often resembles so-called takotsubo (stress/catecholamine-induced) cardiomyopathy (the so-called “broken heart” syndrome).
TABLE 12-6 Complications of pheochromocytoma.
In pregnancy, pheochromocytoma may lead to significant maternal morbidity and fetal demise.
The metabolic effects of excessive circulating catecholamines increase both blood glucose and free fatty acid levels. Increased glycolysis and glycogenolysis, combined with an α-adrenergic receptor–mediated inhibition of insulin release, cause the increase in blood sugar levels. In addition, epinephrine stimulates glucose production by gluconeogenesis and decreases insulin-mediated glucose uptake by peripheral tissues such as skeletal muscle. In pheochromocytoma, impaired glucose homeostasis may also result from β-adrenergic receptor desensitization, which produces relative insulin resistance. Glucose intolerance is common, and diabetes mellitus may occur.
Epinephrine raises blood lactate concentrations by stimulation of glycogenolysis and glycolysis. An increase in oxygen consumption from catecholamine stimulation of metabolism occurs in combination with a decrease in oxygen delivery to tissues from vasoconstriction, leading to lactate accumulation.
Occasionally, pheochromocytomas may also produce peptide hormones leading to specific paraneoplastic phenomena. For example, hypercalcemia may occur, related to excessive production of PTH-related peptide (PTHrP) in cases of malignant pheochromocytomas (as in some other malignancies) or to excessive production of PTH itself in cases of pheochromocytoma associated with MEN-2A–related hyperparathyroidism. Very rarely, ectopic production of adrenocorticotropic hormone (ACTH) by pheochromocytoma may lead to “ectopic” Cushing syndrome. Rare cases have been described in which a pheochromocytoma produces vasoactive intestinal peptide (VIP) (causing severe diarrhea), growth hormone–releasing hormone (GHRH) (causing acromegaly), corticotropin-releasing hormone (CRH) (Cushing syndrome), insulin (hypoglycemia), or other peptide hormones.
An increase in metabolic rate may cause weight loss (or, in children, lack of weight gain), and impaired heat loss from peripheral vasoconstriction may cause a mild elevation of basal body temperature, heat intolerance, flushing, or increased sweating.
During paroxysms, patients may experience marked anxiety, and when episodes are prolonged or severe, there may be visual disturbances, paresthesias, or seizures. A feeling of fatigue or exhaustion usually follows these episodes. Some patients present with psychosis or confusion.
There may be abdominal discomfort resulting from a large adrenal mass. Remarkably, some patients with pheochromocytomas are entirely asymptomatic.
Somewhat different clinical manifestations occur with predominantly epinephrine-releasing pheochromocytomas. Symptoms and signs include hypotension, prominent tachycardia, widened pulse pressure, cardiac arrhythmias, and noncardiogenic pulmonary edema. Acute hemorrhagic necrosis of the tumor may present initially as acute abdominal pain with marked hypertension, followed by hypotension, shock, and sudden death as a consequence of sudden cessation of catecholamine production (“fulminant pheochromocytoma crisis”). Death may also result from cardiovascular collapse secondary to prolonged vasoconstriction and loss of blood volume into the interstitium.
Patients with pure epinephrine-producing pheochromocytomas may be hypotensive because of epinephrine-induced peripheral vasodilation. Other patients with severe arterial vasoconstriction may appear to be in shock. In still others, the prolonged vasoconstriction of a hypertensive crisis may lead to shock.
Pheochromocytoma is diagnosed by demonstrating abnormally high concentrations of catecholamines or their breakdown products in the plasma or urine. Increases in plasma metanephrine and normetanephrine concentrations are greater and more consistent than increases in plasma catecholamines or urinary metanephrines. Pheochromocytoma tumor cells produce large amounts of metanephrines from catecholamines leaking from stores and metabolized by catechol-O-methyltransferase (COMT) present in pheochromocytoma cells. Thus, these metabolites are particularly useful for detecting pheochromocytomas. Thus, the elevated plasma levels of free metanephrine and normetanephrine in patients with pheochromocytoma are probably due mostly to metabolism before and not after release of the catecholamines into the circulation.
Plasma levels of chromogranin A (found in chromaffin granules) are significantly higher in patients with pheochromocytomas, especially those with malignant tumors. For malignant pheochromocytomas, serum chromogranin A levels can also be monitored during chemotherapy of malignant pheochromocytomas to gauge tumor response and to detect relapse.
Administration of the antihypertensive agent clonidine can be used to differentiate essential hypertension from hypertension caused by pheochromocytoma. This potent α2 agonist stimulates α2 receptors in the brain, reducing sympathetic outflow and blood pressure. A dose of 0.3 mg is given orally, and blood pressure and plasma catecholamine levels are determined periodically over the next 3 hours. Essential hypertension is partly dependent on centrally mediated catecholamine release. Administering clonidine normally suppresses sympathetic nervous system activity and substantially lowers plasma norepinephrine levels, reducing blood pressure. However, in patients with pheochromocytoma, the drug has little or no effect on plasma catecholamine levels because these tumors, which are not thought to be innervated, behave autonomously. Thus, the blood pressure remains unchanged.
Once a diagnosis of pheochromocytoma is made, the next step is to localize the neoplasm or neoplasms radiographically to permit surgical removal. Computed tomography (CT) or magnetic resonance imaging (MRI) can be used in tumor localization. CT and MRI have good sensitivity but poor specificity for detecting pheochromocytomas. Nuclear imaging studies such as iodine-131–metaiodobenzylguanidine scintigraphy have limited sensitivity but better specificity in diagnosis. For example, the specificity of 131I-metaiodobenzylguanidine scintigraphy is very good for confirming that a tumor is a pheochromocytoma and for ruling out metastatic disease. In addition, 6-[fluorine-18]-fluorodopamine positron emission tomography can aid in both diagnosis and localization of the tumor in patients with positive biochemical test results. Some pheochromocytomas also express somatostatin receptors and can be imaged with an OctreoScan, which uses radiolabeled somatostatin receptor agonists.
Surgery in patients with pheochromocytoma, including resection of the tumor itself, involves the risk of significant complications. Operative and postoperative complications are directly associated with preoperative systolic blood pressure, tumor size, excretion of urinary catecholamines and their metabolites, duration of anesthesia, and number of surgeries. Understanding the pathophysiology of pheochromocytoma is critically important in preparing the patient for surgery. For example, as noted previously, it is important that hypertension not be treated with β blockers, which could cause paradoxic worsening of hypertension by allowing unopposed α stimulation. Instead, an α receptor blocker, such as phenoxybenzamine, can be used effectively.
CHECKPOINT
7. What genetic mutations are found in patients with pheochromocytoma?
8. What are the symptoms and signs of pheochromocytoma?
9. What are some complications of untreated pheochromocytoma?
10. What are the metabolic and neurologic effects of pheochromocytoma?
11. How is the diagnosis of pheochromocytoma made?
CASE STUDIES
Yeong Kwok, MD
(See Chapter 25, p. 722 for Answers)
CASE 61
A 39-year-old woman comes to the office complaining of episodic anxiety, headache, and palpitations. She states that without dieting she has lost 15 pounds over the past 6 months. Physical examination is normal except for a blood pressure of 200/100 mm Hg and a resting pulse rate of 110 bpm. Chart review shows that prior blood pressures have always been normal, including one 6 months ago. A diagnosis of pheochromocytoma is entertained.
Questions
A. What other features of the history should be elicited? Why is family history important?
B. What laboratory tests should be ordered, and what results should be anticipated? If the laboratory tests are nondiagnostic and suspicion is high, what other test can be done?
C. What is the pathogenesis of the symptoms of anxiety, headache, palpitations, and weight loss in pheochromocytomas?
REFERENCES
General
Busaidy NL et al. Endocrine malignancies. In: Kantarjian HM et al, eds. The MD Anderson Manual of Medical Oncology, 2 ed. McGraw-Hill, 2011.
Fitzgerald PA. Adrenal medulla and paraganglia. In: Gardner DG et al, eds. Greenspan’s Basic & Clinical Endocrinology, 9th ed. McGraw-Hill, 2011.
Pheochromocytoma
Agarwal V et al. Takotsubo-like cardiomyopathy in pheochromocytoma. Int J Cardiol. 2011 Dec 15;153(3):241–8. [PMID: 21474192]
Barron J. Phaeochromocytoma: diagnostic challenges for biochemical screening and diagnosis. J Clin Pathol. 2010 Aug;63(8):669–74. [PMID: 20547690]
Eisenhofer G. Screening for pheochromocytomas and paragangliomas. Curr Hypertens Rep. 2012 Apr;14(2):130–7. [PMID: 22258313]
Eisenhofer G et al. Diagnostic tests and biomarkers for pheochromocytoma and extra-adrenal paraganglioma: from routine laboratory methods to disease stratification. Endocr Pathol. 2012 Mar;23(1):4–14. [PMID: 22180288]
Galetta F et al. Cardiovascular complications in patients with pheochromocytoma: a mini-review. Biomed Pharmacother. 2010 Sep;64(7):505–9. [PMID: 20580187]
Gimenez-Roqueplo AP et al. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012 May;44(5):328–33. [PMID: 22328163]
Gimm O et al. Malignant pheochromocytomas and paragangliomas: a diagnostic challenge. Langenbecks Arch Surg. 2012 Feb;397(2):155–77. [PMID: 22124609]
Harari A et al. Malignant pheochromocytoma: a review. Am J Surg. 2011 May;201(5):700–8. [PMID: 20870212]
Prejbisz A et al. Cardiovascular manifestations of phaeochromocytoma. J Hypertens. 2011 Nov;29(11):2049–60. [PMID: 21826022]
Welander J et al. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011 Dec 1;18(6):R253–76. [PMID: 22041710]