Adrien Bougle
Djillali Annane
During critical illness, the stressors are multiple and include emotional and physical stress such as trauma or infection, and various therapeutic or diagnostic interventions such as surgery, arterial or venous catheterization, laryngeal intubation and mechanical ventilation, and drugs. It is also paramount to recognize that stress is sustained at a certain level of intensity for several days with additive and unpredictable surges. Thus, the host has to adapt to counteract prolonged stress while maintaining ability to adjust to unpredictable surges of stress. The integrity and flexibility of host response to these stressors is essential to survive critical illness.
The development of molecular biology has provided scientists with tools to demonstrate the close interaction between the neuroendocrine and immune system. It is now recognized that the “stress system” has two main components: the corticotropin-releasing hormone/vasopressin neurons of the hypothalamus and the locus ceruleus noradrenaline/autonomic neurons of the brainstem (1). In this chapter, we will summarize recent knowledge on how immune molecules such as interleukin or nitric oxide signal the brain to generate both neurologic and hormonal responses aimed at downgrading the immune system when the inflammatory response is no longer needed. Abbreviations used in this chapter are presented in Table 162.1.
Physiology of the endocrine response
Two pathways are used by the organism for interorgan communication. First the central nervous system and its peripheral arms allow rapid communication between different tissues. The endocrine system is made of several organs or tissues distributed throughout the body. All endocrine tissues are directly linked to the pituitary and the hypothalamus. The anatomic organization of the endocrine system highlights its pivotal role in the interorgan communication. After Claude Bernard discovered that glucose is delivered from the liver to the blood, Bayliss and Starling in 1904 identified for the first time that chemical substances can be secreted by one organ (e.g., secretin from the intestinal mucosa) and carried by the bloodstream to act on specific distant tissues. They introduced the term hormone to characterize such chemical substances. In the vertebrates, the endocrine organs include anterior and posterior pituitary, ovary and testis, adrenal cortex and medulla, thyroids and parathyroids, islets of Langerhans in the pancreas, and various parts of the intestinal mucosa (Fig. 162.1). The pineal and thymus can also be considered endocrine organs. In addition, many other organs have some endocrine properties. For example, the kidney secretes rennin and angiotensin, and the heart secretes natriuretic factors. The main known hormones are listed in Table 162.2 including the organ from which they are secreted, their carrier in the bloodstream (if any), their target tissues, and main actions. Hormones are divided into steroids (cholesterol-derived proteins), peptides, and amines.
General Organization of the Endocrine System
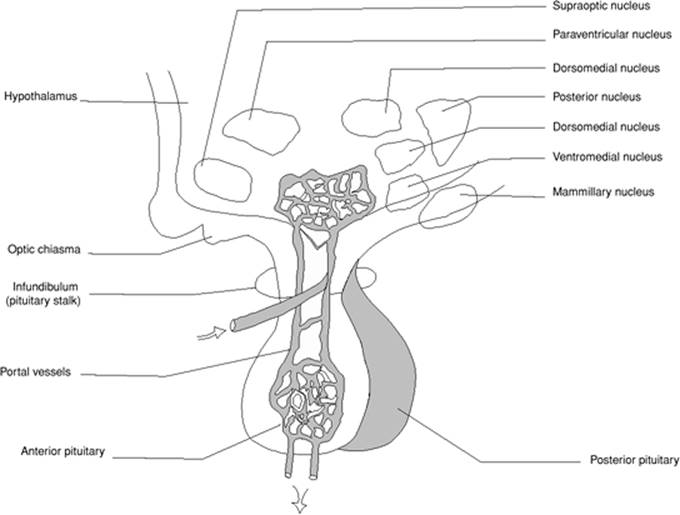
The pituitary gland (hypophysis) can be considered a pivotal endocrine organ. It is connected to the central nervous system by the pituitary stalk and is divided into two parts: the anterior pituitary (adenohypophysis) as it originates from the Rathke pouch, and the posterior pituitary (neurohypophysis), which is developed from the brain (Fig. 162.2). The anterior pituitary is made up of the pars distalis which is richly vascular and has three cell types (chromophobe and two types of chromophil cells), the pars intermedia which is the least vascular part and contains only a few cells, and the pars tuberalis. The anterior pituitary produces seven hormones: gonadotropic hormones (follicle-stimulating hormones (FSH), luteinizing hormone (LH) and luteotropic hormone (LTH)), thyrotropic (thyroid-stimulating) hormone (TSH), adrenocorticotropic hormone (ACTH), growth hormone (GH), and prolactin (PRL). The posterior pituitary has three parts: the median eminence, infundibular stem, and infundibular process or neural lobe. Its tissues consist of unmyelinated fibers, fusiform cells, neurons, and mast cells. The posterior pituitary produces mainly vasopressin, also called the antidiuretic hormone (ADH). It is obvious from this brief description that the anterior pituitary exerts control of most endocrine organs, and insufficiency of the anterior pituitary results in atrophy of ovary or testis, adrenal cortex, and thyroid.
The thyroid and adrenal cortex have no specific target organ, and the hormones they secrete act broadly to contribute to homeostasis. The gonads also produce hormones that act on tissues in general and on specific target organs (urogenital apparatus). The adrenal medulla secretes adrenalin and noradrenalin, which are released during stressed states enhancing the sympathetic system in a “fight or flight” response. It is possible that the release of these hormones depends on direct sympathetic activation rather than on ACTH stimulation.
|
Table 162.1 Abbreviations |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Physiologic Control of the Endocrine System
There is probably no uniform mechanism for the regulation of hormone activity. For example, there is no evidence that parathormone is released from the parathyroid glands on innervating nerve stimulation or action of a specific trophic hormone. This is in contrast to the thyroid, adrenals, and gonads. We will focus on the main hormones involved in response to stress, i.e., steroids, catecholamines, and vasopressin (Fig. 162.3). One has to consider two main mechanisms of regulation of the endocrine activity: feedback loops, and interaction with the central nervous system. Experiments in which peripheral glands are disconnected from the pituitary showed full cessation of gonad function whereas the thyroid and adrenal cortex continue to secrete hormones at a lower level depicting their intrinsic activity. Similarly, the anterior pituitary has an intrinsic activity specific to thyroid and adrenal cortex function.
The feedback mechanisms allow circulating hormones from the target organs as well as from the anterior pituitary to down- or up-regulate the release of hypothalamic molecules. The feedback loop also involves the central nervous structures such as the hippocampus, which contains the highest concentration of glucocorticoid receptors in the central nervous system. This self-balancing system stabilizes the endocrine activity under resting conditions but is insufficient in case of enhanced endocrine activity. In the latter case, the neurologic control is the key regulator of the endocrine activity, and the hypothalamus plays a key role in the regulation of these hormones. First, it is directly connected to the posterior pituitary and the adrenal medulla. Second, it influences the anterior pituitary by releasing in synchronous pulses (approximately hourly) stimulatory or inhibitory hormones in the hypophyseal portal vessels of the pituitary stalk. This hypothalamic influence is demonstrated by dramatic falls in hormone activity when the pituitary gland is disconnected from the hypothalamus.
The hypothalamus is organized into three regions including the lateral, medial, and periventricular hypothalamus with each having distinct morphologic and functional features. The paraventricular nuclei is organized into three cellular divisions: a medial group that produces corticotropin-releasing hormone (CRH) that is released into the hypophyseal portal system; an intermediate group that secretes vasopressin in association with the supraoptic nuclei that is stored in the posterior pituitary gland; and a lateral group that produces CRH and innervates noradrenergic neurons in the brainstem. Hypothalamic-derived peptides that stimulate the pituitary gland include corticotropin-releasing hormone (CRH), LH-releasing hormone (LHRH), FSH-releasing factor (FSHRF), GH-releasing factor (GHRF), prolactin (PRL)-stimulating factor, and thyrotropin-releasing hormone (TRH). Other peptides are inhibiting factors such as GH-inhibiting hormone (somatostatin) and PRL-inhibiting hormone (2). Vasopressin, natriuretic peptides, and catecholamines also influence the pituitary function by direct action on the gland. The effect of CRH on ACTH release by the pituitary is permissive, and vasopressin acts in synergy with CRH. There are tight interconnections between projections of CRH-synthesizing neurons from the parvocellular nuclei to the brainstem, and reciprocally noradrenergic projections originating from the locus coeruleus and ending in the parvocellular reticular nuclei. Thereby, noradrenaline, CRH, and vasopressin can stimulate each other. Through collateral fibers, ultrashort negative feedback loops allow permanent adaptation of the synergy between the two systems. Finally, CRH, vasopressin, and noradrenaline are on the stimulatory control of the serotoninergic, cholinergic, and histaminergic systems, and are inhibited by the gamma-aminobutyric acid, benzodiazepine, and opioids.
Potential mechanisms of regulation of the endocrine activity during critical illness
The Stressors
Critical illness is a condition involving multiple stressors of both emotional and physical types. Critically ill patients are unexpectedly faced with a life-threatening situation and a hostile environment (the intensive care unit). Physical stressors can be classified into disease related (e.g., infection, burns, trauma) and interventions related (e.g., surgery, invasive diagnostic or therapeutic procedures, drugs). The unpredictable nature, duration, and intensity of the stressors renders the host response more problematic.
|
|
|
Figure 162.1. The endocrine organs are distributed throughout the body. There is an electrochemical connection from the hypothalamus to all organs, controlling body metabolism, growth and development, and reproduction. |
Mechanisms of Neuroendocrine Activation in Acute Inflammation
Signals Sent to the Hypothalamic-Pituitary Axis
Acute inflammatory response to insults such as lipopolysaccharide (LPS) include the release of a number of mediators such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6, IL-8, nitric oxide (NO), and the late mediators, macrophage migration-inhibiting factors (MIF), and high mobility group box (HMGB)-1 (3) (Fig. 162.4). These mediators reach the hypophyseal portal capillaries in the median eminence via the anterior hypophyseal arteries. Cytokines can diffuse into the pituitary as these areas are free of blood–brain barrier (4) and be carried to the hypothalamus and the brain areas lacking a blood–brain barrier (organum vasculosum lamina terminalis, subfornical organ, subcommissural organ, area postrema, pineal gland, and probably the plexus choroids), as well as through specific transport systems (5). In addition to the bloodborne cytokines, glial cells can produce a number of cytokines, such as IL-1, IL-2, and IL-6 (6). Similarly, LPS challenge induces IL-6 expression in the anterior pituitary in animals (7). In humans, sepsis induces hypothalamic expression of TNF and IL-1β within the parvocellular and supraoptic nuclei (8). Intraperitoneal injection of LPS induces IL-1β followed by inducible NO synthase (iNOS) mRNA within 2 hours, peaking in 4 to 6 hours and then returning to basal values by 24 hours (9). The induction of IL-1β and iNOS occur in the meninges, areas lacking a blood–brain barrier, and also in the parvocellular nuclei and the arcuate nucleus, which contain the hypothalamic-releasing and -inhibiting hormones. Because the latter nuclei are inside the blood–brain barrier, active transport of cytokines is required (5). Alternately, the neurons from the parvocellular and arcuate nuclei may have projections to the median eminence and may express LPS receptors on their surface (9). LPS can also be carried by the cerebrospinal fluid to the third ventricle where it crosses the ependyma or acts on projections from parvocellular neurons. Thus, it is likely that delayed overexpression of NO through iNOS activation prolongs the synthesis of hypothalamic hormones induced by LPS (9). Similar overexpression of iNOS has been detected in walls of vessels neighboring the parvocellular nuclei in patients who died from septic shock (10). Evidence for brain expression of anti-inflammatory mediators, particularly IL-10, IL-13, and IL-1 receptor antagonists in the pituitary and pineal gland, suggest that they antagonize the stimulatory effects of the proinflammatory mediators on the neurohormones (11). In addition, cytokines, via activation of GABAergic neurons, block NO-induced LHRH but not FSH release, inhibit GHRH release, and stimulate somatostatin and prolactin release (9). The regulatory action of NO is mediated by the combined activation of guanylate cyclase, cyclo-oxygenase, and lipoxygenase (12). Cytokines can also directly act on the anterior pituitary, particularly to stimulate ACTH synthesis and release (13).
A second pathway of activation of the hypothalamic-pituitary axis is the neural route. Various afferent neurons of the peripheral system sense the threat at the inflammatory sites and stimulate the noradrenergic system and the hypothalamus (1). Stimulation of vagal afferent fibers by LPS results in activation of the locus coeruleus where neurons have projections that synapse on cholinergic interneurons in the parvocellular nucleus (14). It has been shown that CRH is released on acetylcholine stimulation of muscarinic receptors (15), and that this effect is prevented by nonspecific NO antagonists (14). Nitric oxide diffuses in the parvocellular cells and induces the generation of both cyclo-oxygenase and lipoxygenase, which in turn stimulate CRH synthesis (12).
|
Table 162.2 List of hormones |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figure 162.2. The hypothalamus lies directly above the pituitary gland. The hypothalamic neurohypophysial tract defines the neuronal system terminating in the posterior pituitary and is best known for its secretion of vasopressin and oxytocin into the peripheral circulation. The pars distalis of the anterior pituitary is supplied by venous blood delivered through the long portal veins that descend along the ventral surface of the pituitary stalk and interconnect capillary beds in the pars distalis with specialized capillary beds of the portal capillary system in the base of the hypothalamus called the median eminence. Venous drainage from the anterior pituitary to the systemic circulation is through adenohypophyseal veins. |
More recently, the role of endogenous gas in the modulation of the hypothalamic pituitary response to stress has been suggested. The effects of carbon monoxide (CO) are inconsistent with data showing that incubation of hypothalamic cells with a CO-enriched milieu resulted in huge release of CRH (16), but another study reported inhibitory effects (17). Carbon monoxide generation was shown to prevent the release of vasopressin from hypothalamic explants (18). Hydrogen sulfide is another endogenous gaseous neuromodulator that was shown both in vitro and in vivo to inhibit CRH and vasopressin release (19).
Factors Influencing Cytokine-Induced Activation of the Hypothalamic-Pituitary Axis
All cytokines do not exert the same effect on CRH release. IL-1 was the first cytokine that was shown to activate the hypothalamus to release CRH (20). IL-1 injection is associated with a strong and sustained activation of the hypothalamic-pituitary axis, whereas IL-6 and TNF induce weak and transient hypothalamic responses, and IL-2 and interferon alpha have no effect (21). The route of cytokine administration also influences their stimulatory effects on the hypothalamus. IL-1 given intravenously increased ACTH and corticosterone in plasma within 15 minutes and the effect lasted 1 hour (22), whereas when given intraperitoneally, ACTH and corticosterone increased within 30 minutes and peaked after 2 hours (20). Cyclo-oxygenase inhibitors prevented hypothalamic response to intravenous IL-1 challenge but prevented only the early response to intraperitoneal IL-1 challenge, suggesting a biphasic response to IL-1 (20,23). Cyclo-oxygenase inhibitors also attenuated hypothalamic response to TNF but not to IL-6 (21). The noradrenergic system, mainly substance P, neuropeptides Y, or galanin, modulate the hypothalamic response to IL-1 whereas the serotoninergic system has no effect (24).
|
|
|
Figure 162.3. Integrative approach of the endocrine system. AVP, arginine vasopressin; CRH, corticotropin-releasing hormone; GABA, gamma-aminobutyric acid. |
Patterns of endocrine activity during critical illness
Infection, LPS challenge, major surgery, trauma, or burns elicit very similar patterns of pituitary hormone secretion. Plasma ACTH and prolactin increase within a few minutes following the insult and are associated with a rapid inhibition of LH and TSH but not FSH. Growth hormone secretion is also stimulated in humans but inhibited in rats (13).
Hypothalamic-Pituitary-Adrenal Axis
Acute stress is associated with an immediate increase in the amplitude of hypothalamic hormones, mainly CRH and vasopressin, resulting in increases in amplitude and frequency of ACTH and cortisol pulses and the loss of the circadian rhythm (1). To achieve this enhanced and accelerated release of hormones, the host recruits additional secretagogues of CRH, vasopressin, and ACTH, mainly magnocellular vasopressin and angiotensin II. In addition, catecholamines, neuropeptides Y, and to a lesser extent CRH released from the adrenal medulla, as well as direct autonomic neural input to the adrenal cortex, stimulate glucocorticoid secretion. The common feature is characterized by high circulating levels of ACTH and cortisol, which remain in plateau as long as the stressful condition is maintained. However, in critical illness, circulating levels of cortisol reflect not only cortisol release, but also cortisol clearance from plasma (25). In critically ill patients, cortisol levels may vary from less than 5 µg/dL to more than 100 µg/dL and do not reflect the hypothalamic-pituitary-adrenal (HPA) axis function (26).
Vasopressin (ADH)
Circulating vasopressin levels are regulated through various stimuli including changes in blood volume or blood pressure, plasma osmolality (27), cytokines, and other neuromediators (as discussed above). Acute illness may be associated with inappropriately high circulating levels of vasopressin resulting in water retention and hyponatremia (28). In sepsis, vasopressin levels in plasma seem to follow a biphasic response with initially high concentrations, probably related to the release from posterior pituitary stores on strong baroreflex activation, followed by a decline in 72 hours with relative vasopressin insufficiency occurring in about one third of cases (29). The delayed decrease in vasopressin levels in plasma is not related to altered hormone clearance from plasma (29) and may result from NO overexpression in the parvocellular nuclei (10).
The Hypothalamic-Pituitary Thyroid Axis
Low T3-T4 syndrome has been described for more than 20 years in fasting conditions and in a wide variety of diseases (e.g., sepsis, surgery, myocardial infarction, transplantation, heart, renal and hepatic failure, cancers, malnutrition, inflammatory diseases) and is also called euthyroid sick syndrome or nonthyroidal illness syndrome (NTIS) (30). In the early phase following an acute stress, there is a decrease in serum tri-iodothyronine (T3) level, an increase in rT3 level. Then serum thyroxine (T4) levels decrease within 24 to 48 hours, and TSH levels remain within normal range without a circadian rhythm (31). Underlying mechanisms include the following: (i) a decrease in conversion of T4 and T3 in extrathyroid tissues due to inhibition of the hepatic 5′-monodeiodination; (ii) the lack of substrates due to the presence of transport protein inhibitors preventing T4 fixation on the protein; (iii) a dysfunction of the thyrotrophic negative feedback on the hypothalamic-pituitary axis; (iv) cytokines (IL-1, IL-6, TNF-α, IFN-γ) inhibiting the thyrotrophic centers and/or affecting the expression of thyroid hormone nuclear receptors (32); (v) the presence of other inhibitory substances such as dopamine. Prolonged critical illness is associated with centrally induced hypothyroidism as suggested by restoration of T3 and T4 pulses by exogenous TRH infusion (33). In addition, as compared with patients who died from acute illness, postmortem examination of patients who died from chronic critical illness showed diminished thyroid gland weight and follicular size (34), low expression of TRH mRNA in the hypothalamic paraventricular nuclei, and low concentrations of tissue T3 (35,36).
|
|
|
Figure 162.4. Signals to the hypothalamus–pituitary axis. There are two different pathways of activation of the hypothalamus–pituitary axis. The humoral pathway acts by (a) cytokines produced in the blood and carried to the hypothalamus and the areas of the brain lacking a blood-brain barrier, (b) cytokines produced by glial cells, and (c) cytokines produced in the pituitary gland and the hypothalamus. The second pathway is the neural route. It acts by activation of various afferent neurons of the peripheral system such as vagal fibers. Finally, endogenous gas such as hydrogen sulfide (H2S) or carbon monoxide (CO) may play a role. Refer to Table 162.1 for definitions of abbreviations. |
Growth Hormone
The acute phase of critical illness is characterized by high growth hormone levels with attenuated oscillatory activity associated with low levels of insulinlike growth factor (IGF)-1 (37). Serum concentrations of GH effectors IGF-1 are low during this phase (38). This pattern is interpreted as a state of resistance to GH that is mainly related to decreased expression of GH receptors (39). This GH resistance seems to be beneficial: the direct lipolysis and anti-insulin effects may be enhanced, liberating metabolic substrates such as free fatty acids and glucose to vital organs, while costly metabolism mediated by IGF-1 is postponed. When critical illness–related stress is sustained, the pattern of GH secretion shows less pulsatile fraction and elevated nonpulsatile fraction (40). It correlates with the low circulating levels of peripheral effectors such as IGF-1. However, contrary to the acute phase of critical illness, this reduced secretion of IGF-1 does not reflect resistance to GH, but rather, suggests a hypothalamic origin as confirmed by the restoration of the pulsatile GH secretion pattern by infusion of GH secretagogues (41).
Adrenal Medulla Hormones
It is well known that under resting conditions very small amounts of adrenaline and noradrenaline are released from the adrenal medulla (i.e., less than 50 ng/kg per minute in the dog). Therefore, removing the adrenal medulla allows an animal to survive indefinitely (42). However, exposure to stressors like fright, pain, anesthesia, exercise, cold, or endotoxin cause within seconds an increase in circulating adrenaline and noradrenaline concentrations by 2 to 3 logs, an effect that was prevented by removal of the adrenal medulla. The release of the catecholamines in the circulation account for the tachycardia, pupillary dilation, pilar erection, and spitting observed in the stressed animals (43). Following his experiments in the early twentieth century, Cannon introduced the concept of “fight or flight” and established its relationship with catecholamines release. Catecholamine synthesis results from an enzymatic cascade starting from the hydroxylation of the amino acid tyrosine leading to the production of dihydroxyphenylalanine (dopa). Dopa is decarboxylated to form dopamine. Dopamine is then hydroxylated in the side chain to form noradrenaline. Finally, noradrenaline is N-methylated by phenylethanolamine-N-methyltransferase to form adrenaline. Adrenaline is stored in the adrenal medulla in vesicles. Noradrenaline is present in the subcellular granules of the sympathetic nervous endings. Catecholamines have a very short half-life (10–20 seconds for adrenaline) and are metabolized through captation, enzymatic inactivation (methylation in liver or kidney; oxidative deamination by monoamine oxydase), or renal excretion. The regulation of catecholamine secretion involves hormonal and nervous factors, and negative feedback through calcium channels. The hormonal regulation depends on cortisol, which is necessary for the enzymatic degradation of catecholamine synthesis. This interaction relies on nerve transmission, paracrine effect, and the local vascular system. The neurogenic regulation involves the cholinergic preganglionic parasympathetic nervous system through the splanchnic nerves. Like cortisol, catecholamine levels in plasma can remain elevated as long as the stress is maintained, even up to a few months after recovery. Nonetheless, obtaining circulating plasma levels of adrenaline and noradrenaline is useless to assess the appropriateness of the catecholamine response to critical illness.
|
|
|
Figure 162.5. Decision tree for the diagnosis and treatment of adrenal insufficiency. |
Insulin
Insulin is involved in glucose metabolism through (i)) mobilization of the store of glucose transport molecules in target cells such as muscle and fat tissue, (ii) activation of hepatic glucokinase gene transcription, and (iii) activation of glycogen synthetase and inhibition of glycogen phosphorylase. Other actions of insulin include growth stimulation, cellular differentiation, intracellular traffic, increased lipogenesis, glycogenesis, and protein synthesis. These effects result from insulin fixation to a ubiquitous membrane receptor belonging to the tyrosine kinase family, the insulinlike growth factor receptor (IGF-1) and insulin receptor–related receptor (IRR). Insulin levels in plasma are rapidly increased following an acute stress as a result of both increased secretion and tissue resistance. Insulin suppresses and antagonizes the effects of TNF (44), macrophage migration inhibitory factor (MIF), and superoxide anions (45), and decreases the synthesis of the acute phase reactants (46). Moreover, insulin modulates leptin and other adipokine release from fat cells (47).
Clinical consequences of endocrine activity during critical illness
The main objective of the neuroendocrine response to critical illness is fight or flight. The immediate activation for the endocrine system, mainly the sympathoadrenal hormones, results in alertness, insomnia, hyperactivity, pupillary dilation, pilar erection, sweating, salivary secretion, tachycardia, rise in blood pressure with vasodilation of skeletal muscle and coronary arteries, bronchiolar dilation, skin vasoconstriction, mobilization of glucose from liver with hyperglycemia, increased oxygen capacity of the blood via spleen constriction and mobilization of red blood cells, and shortening of coagulation time. However, in the intensive care unit where flight is not possible, fighting is the only option, and the appropriateness of the neuroendocrine activity to the intensity and duration of the stress determines host survival and recovery (Table 162.3). The clinical consequences of the stress system activation include behavioral changes and cardiovascular, metabolic, and immune adaptations.
|
Table 162.3 Clinical consequences of endocrine activity during critical illness |
|||||||||||||||
|
Behavioral Changes
In animals, infections are associated with anorexia and body weight loss, hypersomnia, psychomotor retardation, fatigue, and impaired cognitive abilities (48). Similar behavioral changes are consistently reported in humans after cytokine or LPS challenge (49). The so-called depression due to a general medical condition is likely mediated through release of peripheral and brain cytokines. When glucocorticoids and catecholamine responses are insufficient, critically ill patients will develop brain dysfunction that can result to coma.
Cardiovascular Changes
The cardiovascular adaptation is mainly driven by the sympathoadrenal hormones even though thyroid hormones and vasopressin contribute to cardiac adaptation, blood volume, and vasomotor tone regulation. Corticosteroids exert important actions of the various elements of the cardiovascular system including the capillaries, the arterioles, and the myocardium. Numerous studies in various animal models, in healthy volunteers challenged with LPS, and in patients consistently showed that corticosteroids enhanced vessel responsiveness to various vasoactive agents, particularly catecholamines (50). The underlying mechanisms are not fully understood and may involve direct mobilization of intracellular calcium, enzymatic metabolism of adrenaline, increased binding affinity of adrenaline for its receptor, or facilitation of the intracellular signalization that follows the coupling of adrenaline to its receptor. Corticosteroids also have catecholamine-independent effects on the heart and the vessels, and by retaining salt and water contribute to maintain an appropriate blood volume. This results in the fight or flight response: tachycardia, high blood pressure, oliguria, skin vasoconstriction, and cardiac output being redistributed toward the brain, the heart, the liver, and skeletal muscles.
However, whenever the hypothalamic-pituitary adrenal axis or the noradrenergic responses are inappropriate, critically ill patients will develop cardiovascular dysfunction. It has been shown that septic shock patients with adrenal insufficiency, as defined by an increase in cortisol of 9 µg/dL or less to ACTH challenge, have more pronounced hypotension than those with presumed normal function and are more likely to develop refractory shock (51). Adrenal failure also resulted in a more prolonged cardiovascular dysfunction and death in the septic patients (52). Although more common in septic shock, adrenal insufficiency can develop in other critical illnesses (26), particularly in chronically ventilated patients where an association with weaning failure has been observed (53). Adrenal insufficiency is at best diagnosed in critically ill patients by either low baseline cortisol levels (of 10 µg/dL or less) or cortisol increment after 250 µg of corticotropin (ACTH) of 9 µg/dL or less (26). Failure of the noradrenergic system will also result in cardiovascular dysfunction during critical illness. Both animal and human studies have shown that LPS challenge or sepsis is associated with decrease in the noradrenergic activity that precedes cardiovascular dysfunction (54,55,56). The decrease of the pulsatile activity of the HPA axis and the noradrenergic system result in regularity within the circulatory and respiratory function enabling the subject to adjust to stressful conditions, losing the interorgan communications resulting in multiple organ dysfunction and death (57). Finally, inappropriately low vasopressin levels contribute to the vasodilatory shock associated with many critical illnesses (58).
Metabolic Changes
The net result from the activation of the endocrine system is hyperglycemia. The rise in blood glucose follows the activation of the so-called counterregulatory hormones (glucocorticoids, adrenaline, and glucagon) and results in mobilization of glucose mainly from the liver. Subsequently, tissues that are insulin dependent cannot uptake glucose which is then available for insulin-independent tissues like the brain or inflammatory cells (47). The main reason for critical illness–associated insulin resistance is impairment in glucose transporter isoform (GLUT)-4 metabolism (59). Proinflammatory cytokines such as TNF result in the generation of intracellular ceramides that block the transcription of the gene coding for GLUT-4, preventing the translocation of GLUT-4 to the cell's membrane and entry of glucose into the cells (60). Hyperglycemia has been shown to increase mortality in critical illness (61,62). The mechanisms underlying glucose toxicity for the cells is still unknown and may include an overloading of the insulin-independent cells such as neurons. Subsequent to low ATP levels in the cells, the excess of intracellular glucose cannot enter the Krebs cycle and result in the generation of free radicals and peroxynitrites, which in turn block complex IV of the mitochondria. By destroying the mitochondria of insulin-independent cells, hyperglycemia may facilitate acquisition of superinfection, damage the central and peripheral nervous system and the liver, and eventually cause multiple organ dysfunction (62). Excess in the catabolic hormones (cortisol, adrenaline, and glucagon) will also elicit an imbalance between muscle protein breakdown rate and the rate of muscle protein synthesis, resulting in a net catabolism of muscle protein (63), which may contribute to critical illness–induced muscle weakness and affect long-term prognosis (64).
Immune Changes
The changes in the immune function are mainly related to the sympathoadrenal hormones even though insulin and vasopressin can also influence immunity. Glucocorticoids suppress most, if not all, T-cell–derived cytokines and change the T-helper (Th)1/Th2 balance toward excess Th2 cells (50). Glucocorticoids up-regulate lymphocyte-derived IL-10 but do not effect IL-10 synthesis. They also inhibit the synthesis of many other inflammatory mediators such as cyclo-oxygenase and inducible NOS and down-regulate cell surfaces markers such as endotoxin receptor and adhesion molecules. Finally, they enhance the occurrence of apoptosis of thymocytes, mature T lymphocytes, eosinophils, epithelial cells, and precursors of dermal/interstitial dendritic cells, but delay apoptosis of neutrophils (50). Catecholamines also drive a Th2 shift in both antigen-presenting cells and Th1 cells. In LPS-stimulated human blood cultures, noradrenaline and adrenaline inhibit IL-12 synthesis and enhance IL-10 release, an effect that is mediated via beta-adrenergic receptors (65). These effects of beta-adrenergic stimulation on the Th1/Th2 balance are also observed in vivo. However, through alpha-adrenergic stimulation, catecholamines increase TNF and IL-1 synthesis from LPS-stimulated peritoneal macrophages or lung mononuclear cells (1). Thus, while the stress hormones glucocorticoids and catecholamines induce systemically a shift of the Th1/Th2 balance in favor of Th2 cells, catecholamines also promote locally at the level of inflamed tissues the synthesis of proinflammatory mediators. In addition, at the inflammatory sites, tight cross talk between cytokines and the cortisone/cortisol shuttle, with TNF and IL-1 converting cortisone to cortisol and IL-4 and IL-13 inactivating cortisol into cortisone, helps balance the proinflammatory and anti-inflammatory responses (66).
When critical illness is associated with an impaired HPA axis, the Th1/Th2 shift favors the release of proinflammatory mediators in the circulation and in body tissues, allowing cytokine-induced cell deaths either through ischemic or apoptotic mechanisms. In acute lung injury, sepsis, or trauma, or in patients with an adrenal insufficiency, there are large amounts of circulating proinflammatory cytokines that contribute to the development of multiple organ dysfunction and death (67,68). By contrast, too high circulating cortisol levels, such as after a bolus of a high dose of methylprednisolone, may eventually induce systemic immune suppression and favor superinfection and death (69).
Manipulation of endocrine activity during critical illness
The use of exogenous hormones in critical illness has become a standard of care (Table 162.4). It was shown a long time ago that hypotension can be corrected by administration of catecholamines. Even though there are no randomized controlled trials of adrenaline, noradrenaline, or dopamine versus a placebo or no treatment, these drugs are routinely administered in critically ill patients with cardiovascular dysfunction (70). A recent multicenter randomized trial has shown that adrenaline and noradrenaline are equally effective in restoring cardiovascular homeostasis during septic shock (71). Several trials have also demonstrated that administration of vasopressin can improve cardiovascular function in vasodilatory shock (58,72,73). Vasopressin administration increases systemic vascular resistance and mean arterial pressure and improves cardiac performance and renal function. Although it is clear that exogenous administration of catecholamines or vasopressin can restore hemodynamic stability in critical illness, whether manipulating these hormones helps survival remains uncertain.
The effects of corticosteroid administration have been studied, particularly in patients with severe infections. There is enough evidence in the literature supporting the benefit of corticosteroids on hemodynamic and systemic inflammation (74). In a meta-analysis of all randomized controlled trials of hydrocortisone for septic shock, it was demonstrated that hydrocortisone at a dose of 200 to 300 mg per day for 5 to 11 days improved systemic hemodynamic instability and hastened shock recovery. The benefit of treatment on systemic inflammation during sepsis was characterized by prompt and important reduction in circulating proinflammatory mediators, in expression of adhesion molecules, in endothelial activation, and in cell capacity to produce late inflammatory mediators such as MIF (75,76). It is well recognized that a short course of high-dose glucocorticoids should be avoided (74). In contrast, combined administration of low doses of glucocorticoid and mineralocorticoid improved survival in septic shock patients with demonstrable failure of the HPA axis (77) although there are conflicting results.
|
Table 162.4 Manipulation of endocrine activity during critical illness |
||||||||||||||||
|
||||||||||||||||
In another prospective randomized trial (Corticus trial), there was no difference in the 28-day mortality between steroid recipients and the placebo group (78). The use of corticosteroids in patients with septic shock has been controversial for several decades and continues to be controversial despite these two large well-performed studies (77,78). The two studies evaluated different patient populations and came to opposite conclusions. Similarities between the two studies included steroids' beneficial effects on time to shock reversal, and no evidence for increased risk of neuromuscular weakness and hyperglycemia. Differences between the two studies include the following for Annane et al. (77) and Sprung et al. (78), respectively: entry window (8 vs. 72 hours; systolic blood pressure [SBP] <90 mm Hg [>1 hour vs. <1 hour]); additional treatment (fludrocortisone vs. no fludrocortisone); treatment duration (7 vs. 11 days); weaning (none vs. present); SAPS II scores (Simplified Acute Physiology Score) (59 vs. 49); nonresponders to corticotropin (77% vs. 47%); differences in steroid effects according to the response to corticotropin (yes vs. no); increased risk of superinfection (no vs. yes), and the study occurred after practice guidelines recommended steroids (no vs. yes). The updated Surviving Sepsis campaign has given the following recommendation: “We suggest intravenous hydrocortisone be given only to adult septic shock patients after blood pressure is identified to be poorly responsive to fluid resuscitation and vasopressor therapy” (79). Additional recommendations are as follows: fludrocortisone is optional when hydrocortisone is used, and steroid therapy should not be guided by the corticotropin test results (79). In fact, another international task force came up with similar recommendations: “Hydrocortisone should be considered in the management strategy of patients with septic shock, particularly those patients who have responded poorly to fluid resuscitation and vasopressor agents” (80). The benefit of hydrocortisone in refractory shock can also be seen in neonates (81). Finally, in critically ill patients who failed to wean from the ventilator because of adrenal insufficiency, hydrocortisone replacement significantly improved outcome (53).
Recently, intensive treatment with insulin targeting a blood glucose of 4.4 to 6 mmol/L was shown to significantly improve morbidity and mortality in both surgical (82) and medical (83) patients. The benefit is mainly observed in the chronic phase of critical illness (after 72 hours) and may be related to protection of cells from glucose toxicity rather than from direct anti-inflammatory effects of insulin. However, this manipulation of glucose metabolism is extremely difficult and limited by the risk of hypoglycemia. A recent multicenter study did not find any benefit for a tight glucose control with intensive insulin therapy in patients with severe sepsis (84), but the problem may have been in the method of glucose measurement since capillary samples may not read similar levels as whole blood samples, particularly in critically ill patients. One may also suggest that the very early increase in blood glucose mainly relates to stress hormones and should not be counteracted whereas later hyperglycemia relates more to cytokine-induced insulin resistance and should be treated. In other words, the lack of benefit observed in the VISEP study may result from a too-early intervention (84).
Other attempts to manipulate the endocrine system during critical illness have been less successful. Many studies have tried to replace thyroid hormones in various critical illnesses including in patients with cardiac disease, sepsis, acute respiratory distress syndrome, or with burn and trauma patients. Although hormone replacement was associated with some hemodynamic improvement, there was evidence for side effects–related increased risk of death (85). Similarly, the attempt to treat critically ill patients with growth hormone was associated with increased mortality (86), although there is speculation that elevation of blood sugars may have contributed to increased mortality. Nonetheless, it was suggested that combined activation of the GH and thyroid axis with treatment with GH-releasing peptide, TRH, and GNRH elicited beneficial metabolic effect in chronic critically ill patients (87).
Summary
The neuroendocrine response to critical illness is the determinant of host survival and recovery. The sympathoadrenal hormones are the key actors in maintaining homeostasis, and they are very tightly controlled by the brain. When the neuroendocrine response to an acute event such as trauma, infection, burns, and surgery is appropriate both in time and in intensity, then critical illness does not develop and recovery is easy. Otherwise, if the neuroendocrine response is insufficient for the intensity or duration of the stressful episode-related inflammation, then multiple organ failure syndrome develops. By contrast, if the host response is too excessive when compared to the intensity or duration of the inflammatory process, then persistent changes in behavior and mood, in metabolic state, and in immune function cause increased susceptibility to superinfection, risk for chronic muscle fatigue, and posttraumatic stress disorders. Whether the neuroendocrine system can be manipulated to be adjusted to the inflammatory process remains a controversial issue.
Pearls
Physiology of the endocrine system:
Interorgan communication depends on:
· The noradrenergic and vagal systems
· Hormones
The pituitary is made of:
· Anterior pituitary, which produces:
1. Gonadotrophic hormones (follicle-stimulating hormones, luteinizing hormone, and luteotrophic hormone)
2. Thyrotrophic hormone
3. Adrenocorticotropic hormone
4. Growth hormone
5. Prolactin
· Posterior pituitary, which produces mainly vasopressin
Physiologic control of the endocrine activity:
· Feedback loops modulate hormone synthesis and release under resting conditions
· Hypothalamus regulates the endocrine activity in stress conditions
Signalization to the hypothalamic-pituitary axis involves five mechanisms:
· Bloodborne cytokines diffuse into the hypothalamic pituitary axis through areas lacking blood–brain barriers, mainly circumventricular organs
· Bloodborne cytokines are actively transported across the blood–brain barrier via specific carriers
· Cells inside the blood–brain barrier can produce cytokines
· Activated afferent vagal fibers activate the locus ceruleus with subsequent cholinergic-mediated release of CRH
· Gaseous neuromediators such as carbon monoxide and hydroxide sulphide modulate vasopressin and CRH release
Patterns of endocrine activity during critical illness:
· Loss of pulsatile release of hypothalamic hormones
· Increased and sustained ACTH and cortisol levels in plasma
· Increased and sustained circulating catecholamine levels
· Biphasic response in vasopressin with initial high circulating levels and subsequent progressive decline
· Inhibition of TRH release, normal TSH levels with low T3 and T4 concentrations in plasma
· High GH levels and low IGF-1 levels in plasma
· High insulin levels and high blood glucose levels
Endocrine system failure contributes to cardiovascular dysfunction and exaggerated immune response in critical illness:
· Abnormal cortisol response is associated with loss in pressure sensitivity to catecholamines
· Loss in noradrenergic-mediated cardiovascular variability precedes shock in sepsis and contributes to multiple organ dysfunction
· Low vasopressin levels aggravate the impaired noradrenergic system–related vasodilatory shock
· Impaired cortisol and autonomic nervous system induce a shift of the Th1/Th2 balance in favor of Th1 response
Excessive endocrine response to critical illness results in organ dysfunction and immune suppression:
· Excess in counterregulatory hormones induces glucose overload in insulin-independent cells
· Subsequently, via generation of peroxynitrites, mitochondrial function is stopped, predisposing to cell death and organ dysfunction, mainly long-term sequels such as neurologic impairment
· Excess activation of cortisol and autonomic nervous system induces a systemic shift of the Th1/Th2 balance toward excessive Th2 response resulting in immune suppression and predisposing to superinfection
References
1. Chrousos GP. The stress response and immune function: clinical implications. The 1999 Novera H. Spector Lecture. Ann N Y Acad Sci. 2000;917:38.
2. McCann SM, Ojeda SR. The anterior pituitary and hypothalamus. In: Griffin JE, Ojeda SR, eds. Textbook of Endocrine Physiology. 3rd ed. Oxford, England: Oxford University Press; 1996:101–133.
3. Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365:63.
4. Porter JC, Sisom JF, Arita J, et al. The hypothalamic-hypophysial vasculature and its relationship to secretory cells of the hypothalamus and pituitary gland. Vitam Horm. 1983;40:145.
5. Banks WA, Kastin AJ, Huang W, et al. Leptin enters the brain by a saturable system independent of insulin. Peptides. 1996;17:305.
6. Koenig JI. Presence of cytokines in the hypothalamic-pituitary axis. Prog Neuroendocrinoimmunol. 1991;4:143.
7. Spangelo BL, MacLeod RM, Isakson PC. Production of interleukin-6 by anterior pituitary cells in vitro. Endocrinology. 1990;126:582.
8. Sharshar T, Annane D, Lorin de la Grandmaison G, et al. The neuropathology of septic shock. Brain Pathol. 2004;14:21.
9. McCann SM, Kimura M, Karanth S, et al. The mechanism of action of cytokines to control the release of hypothalamic and pituitary hormones in infection. Ann N Y Acad Sci. 2000;917:4.
10. Sharshar T, Gray F, Lorin de la Grandmaison G, et al. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362:1799.
11. Wong ML, Bongiorno PB, Rettori, et al. Interleukin (IL) 1b, IL-1 receptor antagonist, IL-10, IL-13 gene expression in the central nervous system and anterior pituitary during systemic inflammation: pathophysiological implications. Proc Natl Acad Sci U S A. 1997;93:227.
12. Lyson K, McCann SM. Involvement of arachidonic acid cascade pathways in interleukin-6 stimulated corticotropin-releasing factor release in vitro. Neuroendocrinology. 1992;55:708.
13. Rettori V, Dees WL, Hiney JK, et al. An interleukin-1a-like neuronal system in the preoptic-hypothalamic region and its induction by bacterial lipopolysaccharide in concentrations which alter pituitary hormone release. Neuroimmunomodulation. 1994;1:251.
14. Karanth S, Lyson K, McCann SM. Role of nitric oxide in interleukin 2 induced corticotropin-releasing factor release from incubated hypothalami. Proc Natl Acad Sci U S A. 1993;90:3383.
15. Karanth S, Lyson K, McCann SM. Effects of cholinergic agonists and antagonists on interleukin-2-induced corticotropin-releasing hormone release from the mediobasal hypothalamus. Neuroimmunomodulation. 1999;6:168.
16. Parkes D, Kasckow J, Vale W. Carbon monoxide modulates secretion of corticotropin-releasing factor (CRF) from rat hypothalamic cell cultures. Brain Res. 1993;646:315.
17. Pozzoli G, Mancuso C, Mirtella A, et al. Carbon monoxide as a novel neuroendocrine modulator: inhibition of stimulated corticotropin-releasing hormone release from acute rat hypothalamic explants. Endocrinology. 1994;135:2314.
18. Kostoglou-Athanassiou I, Forsling ML, Navarra P, et al. Oxytocin release is inhibited by the generation of carbon monoxide from the rat hypothalamus—further evidence for carbon monoxide as a neuromodulator. Mol Braisn Res. 1996;42:301.
19. Dello Russo C, Tringali G, Ragazzoni N, et al. Evidence that hydrogen sulfide can modulate hypothalamo-pituitary-adrenal axis function: in vitro and in vivo studies in the rat. J Neuroendocrinol. 2000;12:225.
20. Besedovsky HO, Del Rey A, Sorkin E, et al. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science. 1986;233:652.
21. Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. 2000;917:608.
22. Dunn AJ, Chuluyan H. The role of cyclo-oxygenase and lipoxygenase in the interleukin-1 induced activation of the HPA axis: dependence on the route of the injection. Life Sci. 1992;51:219.
23. Rivier C, Vale W. Stimulatory effect of interleukin-1 on adrenocorticotropin secretion in the rat: is it modulated by prostaglandins? Endocrinology. 1991;129:384.
24. Dunn AJ. Endotoxin-induced activation of cerebral catecholamine and serotonin metabolism: comparison with interleukin-1. J Pharmacol Exp Ther. 1992;261:964.
25. Melby JC, Spink WW. Comparative studies on adrenal cortical function and cortisol metabolism in healthy adults and in patients with shock due to infection. J Clin Invest. 1958;37:1791.
26. Annane D, Maxime V, Ibrahim F, et al. Diagnosis of adrenal insufficiency in severe sepsis and septic shock. Am J Respir Crit Care Med. 2006;174(12):1319–1326. Epub 2006 Sep 14.
27. Holmes CL, Patel BM, Russell JA, et al. Physiology of vasopressin relevant to management of septic shock. Chest. 2001;120:989.
28. Dreyfuss D, Leviel F, Paillard M, et al. Acute infectious pneumonia is accompanied by a latent vasopressin-dependent impairment of renal water excretion. Am Rev Respir Dis. 1988;138:583.
29. Sharshar T, Blanchard A, Paillard M, et al. Circulating vasopressin levels in septic shock. Crit Care Med. 2003;31:1752.
30. De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999;84:151.
31. Romijn JA, Wiersinga WM. Decreased nocturnal surge of thyrotropin in nonthyroidal illness. J Clin Endocrinol Metab. 1990;70:35.
32. Michalaki M, Vagenakis AG, Makri M, et al. Dissociation of the early decline in serum T(3) concentration and serum IL-6 rise and TNFalpha in nonthyroidal illness syndrome induced by abdominal surgery. J Clin Endocrinol Metab. 2001;86:4198.
33. De Jongh FE, Jobsis AC, Elte JW. Thyroid morphology in lethal non-thyroidal illness: a post-mortem study. Eur J Endocrinol. 2001;144:221.
34. Van den Berghe G, de Zegher F, Baxter RC, et al. Neuroendocrinology of prolonged critical illness: effects of exogenous thyrotropin-releasing hormone and its combination with growth hormone secretagogues. J Clin Endocrinol Metab. 1998;83:309.
35. Fliers E, Guldenaar SE, Wiersinga WM, et al. Decreased hypothalamic thyrotropin-releasing hormone gene expression in patients with nonthyroidal illness. J Clin Endocrinol Metab. 1997;82:4032.
36. Arem R, Wiener GJ, Kaplan SG, et al. Reduced tissue thyroid hormone levels in fatal illness. Metabolism. 1993;42:1102.
37. Ross R, Miell J, Freeman E, et al. Critically ill patients have high basal growth hormone levels with attenuated oscillatory activity associated with low levels of insulin-like growth factor-I. Clin Endocrinol (Oxf). 1991;35:47.
38. Baxter RC, Hawker FH, To C, et al. Thirty-day monitoring of insulin-like growth factors and their binding proteins in intensive care unit patients. Growth Horm IGF Res. 1998;8:455.
39. Hermansson M, Wickelgren RB, Hammarqvist F, et al. Measurement of human growth hormone receptor messenger ribonucleic acid by a quantitative polymerase chain reaction-based assay: demonstration of reduced expression after elective surgery. J Clin Endocrinol Metab. 1997;82:421.
40. Van den Berghe G, de Zegher F, Lauwers P, et al. Growth hormone secretion in critical illness: effect of dopamine. J Clin Endocrinol Metab. 1994;79:1141.
41. Van den Berghe G, de Zegher F, Veldhuis JD, et al. The somatotropic axis in critical illness: effect of continuous growth hormone (GH)-releasing hormone and GH-releasing peptide-2 infusion. J Clin Endocrinol Metab. 1997;82:590.
42. Witek-Janusek L, Yelich MR. Role of the adrenal cortex and medulla in the young rats' glucoregulatory response to endotoxin. Shock. 1995;3:434.
43. Cannon WB. Am J Physiol. 1914;33:356.
44. Satomi N, Sakurai A, Haranaka K. Relationship of hypoglycemia to tumor necrosis factor production and antitumor activity: role of glucose, insulin, and macrophages. J Natl Cancer Inst. 1985;74:1255.
45. Das UN. Is insulin an antiinflammatory molecule? Nutrition. 2001;17:409.
46. Seshadri V, Fox PL, Mukhopadhyayi CK. Dual role of insulin in transcriptional regulation of the acute phase reactant ceruloplasmin. J Biol Chem. 2002;277:27903.
47. Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799.
48. Maier SF, Watkins LR. Cytokines for psychologists: implications of bi-directional immune-to-brain communication for understanding behaviour, mood, and cognition. Psychol Rev. 1998;105:83.
49. Meyers CA. Mood and cognitive disorders in cancer patients receiving cytokine therapy. In: Dantzer R, Wollman EE, Yirmiya R, eds, Cytokines, Stress and Depression. New York, NY: Kluwer Academic/Plenum Publishers; 1999;75–82.
50. Annane D, Cavaillon JM. Corticosteroids in sepsis: from bench to bedside? Shock. 2003;20:197.
51. Annane D, Bellissant E, Lesieur O, et al. Impaired pressor sensitivity to norepinephrine in septic shock patients with and without impaired adrenal function reserve. Br J Clin Pharmacol. 1998;46:589.
52. Annane D, Sébille V, Troché G, et al. A three-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283:1038.
53. Huang CJ, Lin HC. Association between adrenal insufficiency and ventilator weaning. Am J Respir Crit Care Med. 2006;173:276.
54. Goldstein B, Kempski MH, Stair D, et al. Autonomic modulation of heart rate variability during endotoxin shock in rabbits. Crit Care Med. 1995;23:1694.
55. Godin PJ, Fleisher LA, Eidsath A, et al. Experimental human endotoxemia increases cardiac regularity: results from a prospective, randomized, crossover trial. Crit Care Med. 1996;24:1117.
56. Annane D, Trabold F, Sharshar T, et al. Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med. 1999;160:458.
57. Godin PJ, Buchman TG. Uncoupling of biological oscillators: a complementary hypothesis concerning the pathogenesis of multiple organ dysfunction syndrome. Crit Care Med. 1996;24:1107.
58. Landry DW, Levin HR, Gallant EM, et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997;95:1122.
59. Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Bio Chem. 2003;278:33609.
60. QI C, Pekala PH. Tumor necrosis factor-alpha-induced insulin resistance in adipocytes. Proc Soc Exp Biol Med. 2000;223:128.
61. Umpierrez JE, Isaacs SD, Bazargan N, et al. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab. 2002;87:978.
62. Van den Berghe G. How does blood glucose control with insulin save lives in intensive care? J Clin Invest. 2004;114:1187.
63. Gore DC, Jahoor F, Wolfe RR, et al. Acute response of human muscle protein to catabolic hormones. Ann Surg. 1993;218:679.
64. Herridge MS, Cheung AM, Tansey CM, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348:683.
65. Elenkov IJ, Papanicolaou RL, Wilder GP, et al. Modulatory effects of glucocorticoids and catecholamines on human interleukin-12 and interleukin-10 production: clinical implications. Proc Assoc Am Physiol. 1996;108:374.
66. Rook G, Baker R, Walker B, et al. Local regulation of glucocorticoid activity in sites of inflammation: insights from the study of tuberculosis. Ann N Y Acad Sci. 2000;917:913.
67. Hoen S, Asehnoune K, Brailly-Tabard S, et al. Cortisol response to corticotropin stimulation in trauma patients: influence of hemorrhagic shock. Anesthesiology. 2002;97:807.
68. Annane D, Sebille V, Bellissant E, et al. Effect of low doses of corticosteroids in septic shock patients with or without early acute respiratory distress syndrome. Crit Care Med. 2006;34:22.
69. Bone RC, Fischer CJ Jr, Clemmer TP, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653.
70. Hollenberg SM, Ahrens TS, Annane D, et al. Practice parameters for hemodynamic support of sepsis in adult patients: 2004 update. Crit Care Med. 2004;32:1928.
71. Annane D, Vignon P, Bollaert PE, et al. Norepinephrine plus dobutamine versus epinephrine alone for the management of septic shock [abstract]. Intensive Care Med. 2005;31:S18.
72. Argenziano M, Choudhry AF, Oz MC, et al. A prospective randomized trial of arginine vasopressin in the treatment of vasodilatory shock after left ventricular assist device placement. Circulation. 1997;96(Suppl 9):286.
73. Dunser MW, Mayr AJ, Ulmer H, et al. Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation. 2003;107:2313.
74. Annane D, Bellissant E, Bollaert PE, et al. Corticosteroids for severe sepsis and septic shock: a systematic review and meta-analysis. BMJ. 2004;329:480.
75. Keh D, Boehnke T, Weber-Cartens S, et al. Immunologic and hemodynamic effects of “low-dose” hydrocortisone in septic shock: a double-blind, randomized, placebo-controlled, crossover study. Am J Respir Crit Care Med. 2003;167:512.
76. Maxime V, Fitting C, Annane D, et al. Corticoids normalize leukocyte production of macrophage migration inhibitory factor in septic shock. J Infect Dis. 2005;191:138.
77. Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862.
78. Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111.
79. Dellinger EP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296.
80. Marik PE, Pastores SM, Annane D, et al. Clinical practice guidelines for the diagnosis and management of corticosteroid insufficiency in critical illness: recommendations of an international task force. Submitted for publication.
81. Ng PC, Lee CH, Bnur FL, et al. A double-blind, randomized, controlled study of a “stress dose” of hydrocortisone for rescue treatment of refractory hypotension in preterm infants. Pediatrics. 2006;117:367.
82. Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359.
83. Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449.
84. Brunkhorst FM, Engel C, Bloos F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358:125.
85. Stathatos N, Levetan C, Burman KD, et al. The controversy of the treatment of critically ill patients with thyroid hormone. Best Pract Res Clin Endocrinol Metab. 2001;15:465.
86. Takala J, Ruokonen E, Webster NR, et al. Increased mortality associated with growth hormone treatment in critically ill adults. N Engl J Med. 1999;341:785.
87. Van den Berghe G, Baxter RC, Weekers F, et al. The combined administration of GH-releasing peptide-2 (GHRP-2), TRH and GnRH to men with prolonged critical illness evokes superior endocrine and metabolic effects compared to treatment with GHRP-2 alone. Clin Endocrinol (Oxf). 2002;56:655.