Jack D. Shannon
Kevin W. Hatton
Elamin M. Elamin
Pearls
· Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic nonketotic syndrome (HHS) result from dysregulation of normal glucose homeostasis caused by one of many precipitating conditions in patients with diabetes mellitus.
· DKA (minimal intrinsic insulin secretion) is associated with hyperglycemia and ketone body formation, whereas HHS (intact intrinsic insulin secretion) is associated with hyperglycemia without ketone body formation.
· Both DKA and HHS may present with various and vague complaints and progress to severe shock with cardiovascular collapse, severe metabolic acidosis, and death.
· The treatment of DKA and HHS involves intravascular fluid resuscitation and insulin replacement with additional electrolyte supplementation.
· Patients, during treatment of DKA and HHS, should be closely monitored for evidence of hypoglycemia, hyperglycemia, electrolyte disturbances, worsening lactic acidosis, intravascular volume overload, cerebral edema, acute respiratory distress syndrome (ARDS), severe coagulopathy, rhabdomyolysis, and thromboembolism.
· Recent evidence has suggested that intensified glycemic control of intensive care unit (ICU) patients by using intravenous insulin infusions results in improved outcomes, including decreased mortality rate. While these observations led to dramatic changes in the management of diabetic patients in the ICU, the optimal range of glycemic control for patients without systemic disorders of glucose metabolism and the clinical implication of the increased risk of iatrogenic hypoglycemia are currently under investigation.
Disordered glucose metabolism is a significant medical problem in patients in the outpatient, emergency room, ward, and intensive care settings. Diabetes mellitus (DM), the most important group of medical conditions resulting from disordered glucose metabolism, results in significant short-term and chronic morbidity, and is increasing in prevalence around the globe. In the United States alone, nearly 6% of the population (18.2 million people) is estimated to have this disease (1). Additionally, a significant number of these patients may not be diagnosed, which can further negatively impact their health. Despite increasing public awareness over the last few years, the prevalence of diabetes continues to increase (2).
Due to a combination of the sheer volume of patients with DM and the associated chronic health conditions, the disease is important for physicians, nurses, health care administrators, insurers, and public health advocates. More than 3.8 million hospitalizations per year are associated with diabetes, which is an increase of more than 70% over the last 20 years (3). Of note, the average length of stay is 6.5 days for these hospitalizations (3). Undeniably, these facts alone place an enormous economic and workforce burden on the entire health care system.
In addition to the many associated chronic health conditions developed by patients with DM, there are several acute and life-threatening conditions that also develop in these patients. Hyperglycemic emergencies, such as DKA and HHS, are important causes of morbidity and mortality in patients with DM who are admitted to the intensive care unit. Additionally, new research indicates that the prevention of hyperglycemia—with or without the diagnosis of diabetes—may be a key component of appropriate intensive care support of patients with numerous medical and surgical conditions.
Diabetic Ketoacidosis
Since the discovery of insulin by Frederick Banting in 1921, outcomes for patients with diabetic ketoacidosis have steadily improved. Nevertheless, DKA remains a serious and potentially fatal complication of DM. Overall mortality from DKA is less than 5%; however, mortality increases substantially with extremes of age, the presence of coma, or the development of hypotension (4).
Hospital and ICU admissions for DKA and related conditions are increasing, and cause a significant burden on current health care delivery systems (5). With an annual incidence of 4.6 to 8 episodes per 1,000 patients with diabetes, DKA is the initial presentation of DM in up to 30% of patients overall, with approximately 40% of children and 17% of adults presenting in DKA without prior diagnosis of DM (4,5). While most patients presenting with DKA have type 1 DM, those with type 2 DM can also develop DKA during times of significant physiologic stress.
Pathophysiology
DKA results from a serious dysregulation of normal glucose homeostasis, leading to hyperglycemia and ketone body formation. Excess glucose and ketones launch a host of subsequent systemic sequelae. A deficiency, either relative or absolute, in insulin production, combined with an excess production of certain insulin counterregulatory hormones—glucagon, catecholamines, cortisol, and growth hormone, is responsible for these changes in serum glucose control. Most patients with type 1 DM who develop DKA have an absolute or near-absolute insulin deficiency, whereas most patients with type 2 DM have either normal or elevated insulin levels (6,7). Because of the aberrant hormonal milieu, protein, lipid, and carbohydrate metabolism are all disrupted, and culminate in the production of proinflammatory cytokines, such as interleukin-6, interleukin-1β, interleukin-8, and tumor necrosis factor-α; free fatty acids; and plasminogen activator inhibitor-1, resulting in significant morbidity and mortality (8).
Normal glucose metabolism is typically tightly regulated to maintain a serum glucose concentration between 70 and 115 mg/dL (about 3.9–6.4 mm/L) by carefully balancing glucose production in the liver and glucose utilization in peripheral tissues (9). Insulin, a 51-amino-acid peptide, is mainly responsible for this tight glucose control by stimulating hepatic glucose uptake and storage (glycogen synthesis), and suppressing hepatic gluconeogenesis and glycogenolysis. Insulin also affects peripheral muscle tissue by promoting peripheral glucose uptake, promoting glycogen synthesis, and inhibiting peripheral glycogenolysis.
In DKA, either relative or absolute insulin deficiency combined with increased counterregulatory hormones (CRHs) promotes metabolic pathways opposite to insulin in both hepatic and peripheral tissues (10,11,12,13). These changes are typically the result of a precipitating event in patients with severely imbalanced DM (Table 163.1). Infection accounts for 30% to 50% of precipitating causes of DKA, with urinary tract and pulmonary infections making up the vast majority (14). Myocardial infarction, cerebrovascular accident, pulmonary embolism, pancreatitis, trauma, alcohol abuse, and drugs that affect carbohydrate metabolism can also precipitate DKA (15).
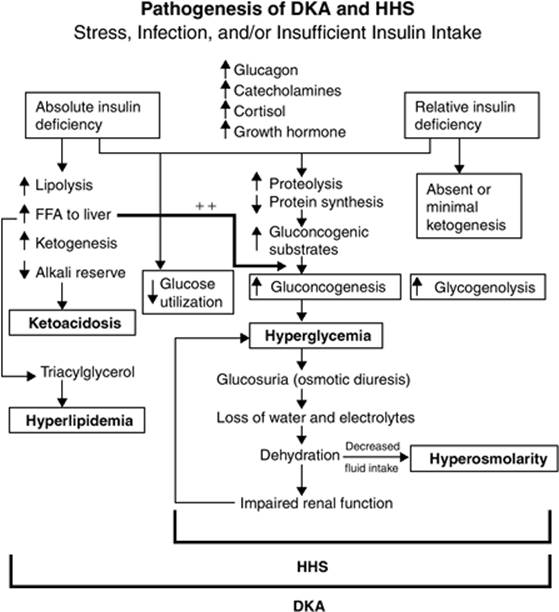
The end result of these changes is a substantial increase in serum glucose—through increased hepatic gluconeogenesis, glycogenolysis, and lipolysis—with inappropriately decreased peripheral insulin uptake (Fig. 163.1). DKA is also associated with ketosis, an additional product of worsening glucose homeostatic decompensation, which occurs as a result of increased lipolysis from increased action of hormone-sensitive lipase, an enzyme that causes increased triglyceride breakdown and free fatty acid release into the systemic circulation. Hormone-sensitive lipase is highly up-regulated during periods of insulin deficiency and elevations in CRH. Hepatic oxidation of free fatty acids induced by hormone-sensitive lipase produces ketone bodies, mainly β-hydroxybutyrate (β-OHB) and acetoacetic acid, strong acids that present a large hydrogen ion load to the body. The normal buffering systems are rapidly overwhelmed by the ongoing hydrogen load, and an anion gap acidosis develops.
Hyperglycemia and ketonemia produce a hypertonic intravascular environment, resulting in an intracellular water shift into the intravascular and interstitial compartments. The ensuing cellular dehydration is accompanied by electrolyte shifts as well. When the renal glucose reabsorption rate is exceeded, an osmotic diuresis of water and electrolytes occurs. Sodium, potassium, magnesium, calcium, chloride, and phosphate are all lost during this osmotic diuresis. Commonly, water and electrolyte deficits are compounded by poor oral intake and protracted vomiting. The effects of hypovolemia are responsible for the clinical picture as the depletion of the intravascular space produces the life-threatening signs and symptoms. The body's response is a further increase in CRH, and the cycle is perpetuated.
|
Table 163.1 Precipitating Factors in Diabetic Ketoacidosis and Hyperosmolar Hyperglycemic Nonketotic Syndrome |
|||||
|
Presentation and Diagnosis
The presentation and diagnosis of DKA is typically straightforward and relies on a thorough patient history, focused physical examination, and appropriate laboratory analysis. Patients typically report a history of poor glucose control and symptoms associated with hyperglycemia, such as polyuria, polydipsia, weight loss, and lethargy that may progress over the course of days to weeks. Nausea, vomiting, and abdominal pain are also common presenting complaints and frequently signify the progression from symptomatic hyperglycemia to overt DKA. Physical examination may reveal evidence of dehydration—for example, tachycardia, hypotension, prolonged capillary refill time, poor skin turgor, dry mucous membranes, and weight loss. Additionally, Kussmaul respirations (very deep, gasping breaths taken in response to severe metabolic acidosis), an acetone or fruity breath odor, depressed mental status, and even focal neurologic deficits or coma may also be seen.
Laboratory analysis is usually confirmatory of DKA in these patients (Table 163.2). A complete blood count, blood glucose, serum electrolytes, serum osmolality, blood urea nitrogen, serum creatinine, arterial or venous blood gas, serum ketones, and urinalysis should be ordered in patients with suspected DKA. Caution should be exercised when using the serum sodium levels in patients with DKA, as the reported laboratory value can be artificially low, normal, or elevated, depending on the spurious effects of glucose and triglycerides in these patients and the relative loss of water compared to sodium. In the presence of hyperglycemia, serum sodium measurement can be corrected by adding 1.6 mg/dL to the measured serum sodium for each 100 mg/dL increase of glucose above normal (16). Appropriate cultures should be requested if infectious triggers of DKA are suspected. Other tests, such as serum lactate, β-human chorionic gonadotropin (β-HCG), electrocardiography, chest radiography, and computed tomography may be indicated, depending on the clinical scenario.
|
|
||||||||||||||||||||||||||||||||||||
|
Figure 163.1. Pathogenesis of diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic nonketotic syndrome (HHS). FFA, free fatty acid. (Copyright © 2001, American Diabetes Association, from Diabetes Care 2001;24:131–153. Reprinted with permission from the American Diabetes Association.) |
||||||||||||||||||||||||||||||||||||
|
Table 163.2 Diagnostic Criteria for Diabetic Ketoacidosis (DKA) and Hyperosmolar Hyperglycemic Nonketotic Syndrome (HHS) |
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
A plasma anion gap–associated metabolic acidosis is typically seen in laboratory analysis of patients with DKA, as ketosis and ketone body accumulation are responsible for an increase in the anion gap. However, in up to 11% of patients, a nongap hyperchloremic metabolic acidosis may occur instead (17). Typically, the normal anion gap is between 7 and 9 mEq/L and reflects unmeasured ions in the serum. In patients with DKA, an elevated anion gap occurs because of the high ketone concentration. Other causes of anion gap–associated metabolic acidosis, which must be excluded during DKA evaluation, include alcoholic ketoacidosis, starvation ketoacidosis, and lactic acidosis, as well as methanol, ethylene glycol, paraldehyde, and salicylate ingestion.
In patients with possible DKA, these other causes of anion gap–associated metabolic acidosis should be excluded through further history and laboratory analysis. For example, alcoholic ketoacidosis may present with profound metabolic acidosis, but typically has a characteristic history, an elevated blood alcohol content, and only mildly elevated serum glucose concentrations. Likewise, starvation ketosis is accompanied by a significant history and only mild acidosis.
Ketonemia and ketonuria can both be assessed semiquantitatively with the nitroprusside reaction test. This test estimates the relative levels of acetoacetate and acetone in the blood, but does not detect the presence of β-OHB, potentially underestimating the degree of ketosis. Because the ratio of β-OHB to acetoacetate may increase from 1:1 to as much as 5:1 during the development of DKA, β-OHB may represent the predominant ketone during illness (18). Of note, β-OHB monitoring may significantly improve the diagnostic specificity in DKA patients with euglycemia or only mild hyperglycemia—as with prolonged vomiting, starvation, pregnancy, hepatic insufficiency, or following insulin administration—where blood glucose levels can be misleading (19).
|
|
|
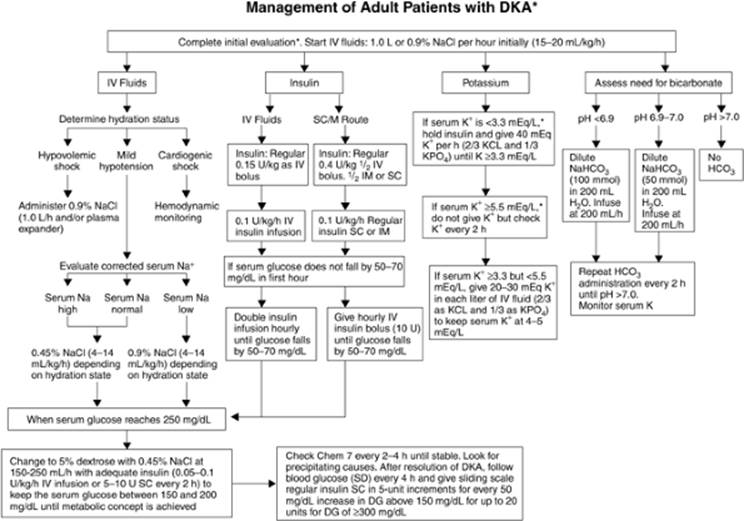
Figure 163.2. Management of adult patients with diabetic ketoacidosis (DKA). DG,; SD,. (Copyright © 2001, American Diabetes Association, from Diabetes Care 2001;24:131–153. Reprinted with permission from the American Diabetes Association.) |
Management
Management of DKA includes the phases of initial resuscitation, correction of hyperglycemia and resolution of ketosis, and treatment of any precipitating causes (Figs. 163.2 and 163.3). Following these phases, it is essential to also provide chronic therapy to prevent repeated episodes and secondary sequelae of diabetes mellitus.
As with all resuscitations, evaluation and treatment of airway and breathing dysfunction should be done first. DKA can cause loss of protective airway reflexes, hypoxia, and hyperventilation. In patients with a severely depressed mental status, appropriate care should be taken to protect the airway so that pulmonary aspiration of gastric contents does not occur. If the patient's Glasgow coma scale is 8 or less, or in situations that require sedation and transport away from the acute care environment for further evaluation, tracheal intubation may be necessary to ensure adequate airway protection and ventilation. Because these patients have a high incidence of gastroparesis, the placement of a decompressive gastric tube may also be warranted in the presence of an altered level of consciousness, and elevation of the head of bed to between 30 and 40 degrees may serve to prevent passive regurgitation. Mechanical ventilation, if utilized, should be set to maintain respiratory compensation of the accompanying severe metabolic acidosis initially and adjusted appropriately as the acidosis corrects.
|
|
|
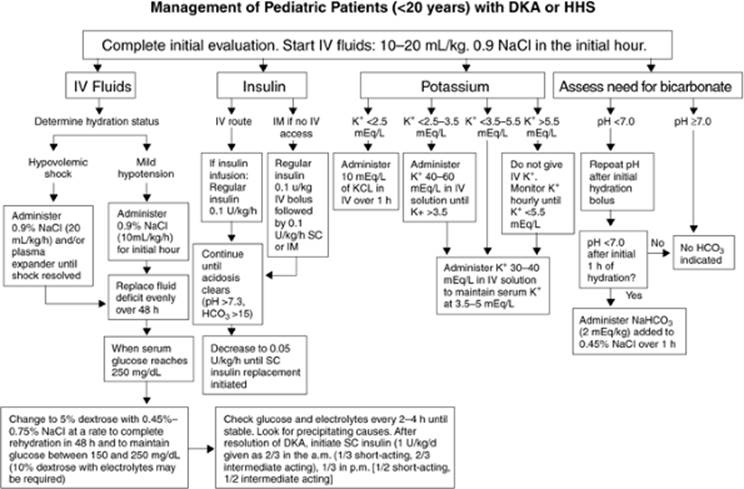
Figure 163.3. Management of pediatric patients with diabetic ketoacidosis (DKA) or hyperosmolar hyperglycemic nonketotic syndrome (HHS). (Copyright © 2001, American Diabetes Association, from Diabetes Care 2001;24:131–153. Reprinted with permission from the American Diabetes Association.) |
Following airway and respiratory care, initial therapy should be directed at restoring adequate blood volume and organ perfusion with intravascular volume resuscitation. In addition to correcting the hemodynamic insults associated with severe hypovolemia, appropriate volume administration can also decrease CRH levels and plasma glucose concentration (20,21). The goal during this phase is to replace the fluid deficit over the first 24 hours, half of which should be replaced in the first 6 to 8 hours. Estimation of fluid deficit can be based on body weight or general guidelines and response characteristics. Typically 1 to 2 liters of isotonic saline in the first 1 to 2 hours is sufficient for initial resuscitation; however, in more severe cases the resuscitation may require larger volumes, and some prefer to add colloids. The following clinical estimations of volume deficit using orthostatic blood pressure and heart rate may also be used to guide initial fluid replacement, although these criteria may be less reliable in patients with neuropathy and/or impaired cardiovascular reflexes (14):
· An increase in pulse without change in blood pressure with orthostatic position change indicates approximately a 10% decrease in extracellular volume (i.e., 2 liters).
· A decrease in blood pressure (>15/10 mm Hg) with position change indicates approximately a 15% to 20% decrease in extracellular volume (i.e., 3–4 liters).
· Supine hypotension indicates a decrease of >20% in extracellular fluid volume (i.e., >4 liters).
After the initial resuscitation phase, both the rate of infusion and type of intravenous fluid must be adjusted. Current recommendations are to decrease the infusion rate to 250 mL/hour or to 4 to 14 mL/kg/hour, depending on the patient's hydration status and goal replacement volume. Depending on the patient's corrected serum sodium, isotonic saline is continued or changed to hypotonic saline. If the patient's corrected serum sodium is low, 0.9% saline solution should be continued as the replacement fluid; however, if the patient's corrected serum sodium is normal or elevated, the fluid should be changed to 0.45% saline solution in order to continue free water deficit replacement. Additionally, once plasma glucose levels reach 250 mg/dL, either 5% or 10% dextrose solution should be added to the replacement fluids to maintain serum glucose levels between 150 and 200 mg/dL, allow the insulin infusion to continue until ketosis is reversed, and prevent the too rapid correction of serum glucose levels. The addition of potassium to the fluids, once adequate renal function is assured, can help prevent hypokalemia during DKA treatment.
Concomitant with aggressive fluid resuscitation, insulin therapy to decrease glucose production and increase glucose utilization with subsequent improvements in ketosis, acidosis, and hyperglycemia should be instituted. Most experts recommend low-dose insulin infusion over intermittent intravenous or subcutaneous administration, as the former is more physiologic, is more therapeutically reliable, and decreases the risk of hypoglycemia and hypokalemia (22,23,24,25,26). Continuous insulin infusion is the preferred route of insulin in all but the mildest cases of DKA. Additionally, in patients with significant hemodynamic compromise or severe hypokalemia (i.e., with a serum potassium level less than 3.3 mEq/L), insulin should be temporarily withheld as resuscitation occurs, because the reduction in plasma glucose levels and acidosis can cause significant intracellular fluid and potassium shifts that may worsen cardiovascular function to the point of collapse (14,27).
For continuous insulin infusion, many experts recommend an initial bolus of regular insulin of 0.1 to 0.15 U/kg followed by an infusion at 0.05 to 0.1 U/kg/hour, with a goal of decreasing plasma glucose levels by no more than 50 to 75 mg/dL/hour. If the plasma glucose level does not respond appropriately in the first few hours, and if the intravascular volume status is adequate, the infusion rate may be doubled every hour until a constant decline in plasma glucose level is achieved. As previously mentioned, once the plasma glucose concentration reaches 250 mg/dL, the insulin infusion rate should be decreased by 50%, and dextrose should be added to the replacement fluid. Continuous adjustment of the insulin infusion rate and dextrose concentration is required to maintain appropriate plasma glucose concentration.
In addition to serum glucose and bicarbonate levels, the American Diabetic Association recommends evaluation of β-OHB levels as the preferred method of monitoring DKA, which may become the preferred method for rapid diagnosis. Typically, β-OHB concentrations are less than 1 mmol/L; however, in patients with DKA, plasma β-OHB concentration can be elevated to concentrations in excess of 4 to 12 mmol/L (mean 7 mmol/L). Adequate DKA treatment should prompt a decrease in β-OHB concentration by approximately 1 mmol/L/hour, and should return to baseline (<1 mmol/L) (18,29,30,31,32,33). As has been documented, serum bicarbonate levels are slower to correct, and β-OHB is a more appropriate measure of therapy. It is also noted that as appropriate treatment progresses, β-OHB is oxidized to acetoacetate, which may worsen the results of the nitroprusside reaction test and lead to incorrect conclusions. For these reasons, it is now recommended to directly measure β-OHB for diagnosis and treatment monitoring in DKA (14,28).
Despite massive potassium losses (3–5 mEq/kg) in patients presenting with DKA, the serum potassium concentration may be normal due to intravascular volume contraction and intracellular electrolyte shifts, and should not be used as an indicator of potassium homeostasis in the early phases of treatment. As insulin is provided and acidosis is corrected, potassium may quickly shift back into the intracellular compartment. Additionally, initial resuscitation with normal saline may lower the serum potassium concentration. Severe hypokalemia may potentially cause life-threatening cardiac dysrhythmias and respiratory muscle weakness (13,34,35). Potassium replacement should begin once the serum potassium concentration falls below 5.5 mEq/L, assuming adequate urine output, using the following guidelines:
· If the serum potassium is 4 to 5 mEq/L, 20 to 30 mEq of potassium should be added to each liter of IV fluid.
· If the serum potassium is 3 to 4 mEq/L, 30 to 40 mEq of potassium should be added to each liter of IV fluid.
· If the serum potassium is less than 3 mEq/L, 40 to 60 mEq of potassium should be added to each liter of IV fluid.
In addition to potassium, other electrolytes such as magnesium, phosphate, and calcium are also depleted in patients with DKA. Inadequate serum concentrations of these electrolytes may cause respiratory depression, cardiac dysfunction, and alteration of tissue oxygenation that can be avoided by aggressive monitoring and appropriate replacement. Measurement of these electrolytes should be done at presentation to identify significant abnormalities in order to facilitate appropriate correction early as clinically indicated. They should be repeated as necessary, depending on the clinical scenario. While several studies have failed to show an outcome benefit to phosphate replacement, some experts continue to recommend replacement of phosphate by using potassium phosphate as a portion of the total potassium replacement (14,36). Of note, hypocalcemia may become problematic in the face of overaggressive phosphate replacement.
Adequate treatment of DKA—intravascular volume repletion with reversal of hyperglycemia and ketosis—is generally associated with improvements in both physiologic and laboratory parameters. Criteria for resolution of DKA include plasma glucose <200 mg/dL, serum bicarbonate concentration ≥18 mEq/L, venous pH >7.3, anion gap <12 mEq/L, and, recently, β-OHB <1 mmol/L (19). After resolution of the DKA episode, when the patient is able to tolerate enteral nutrition, a multidose subcutaneous insulin regimen that includes a combination of short- and intermediate- or long-acting insulin should be instituted. To allow for sufficient insulin plasma levels, intravenous insulin should be continued for 1 to 2 hours following the first dose of subcutaneous insulin. Patients with previously diagnosed diabetes may restart their previous insulin schedule with additional adjustment and coverage as needed.
These treatments should ideally be provided in an environment that has adequate nursing care and monitoring capabilities, as well as rapid turnaround of laboratory tests. Invasive monitoring should be provided as necessary; patients with mild to moderate DKA may require only noninvasive blood pressure monitoring, continuous electrocardiography, pulse oximetry, and a urinary catheter, whereas patients with the most severe disease states and comorbidities may, in addition, require arterial and central venous catheterization. Additionally, patients with oliguria or hypotension refractory to initial rehydration, mental obtundation, sepsis, respiratory insufficiency, pregnancy, or significant comorbidities or precipitating events, such as myocardial infarction or decompensated congestive heart failure, should be managed in a critical care environment (14,34).
Complications
Common complications encountered during DKA treatment include hypoglycemia and hyperglycemia, various electrolyte disturbances, lactic acidosis, intravascular volume overload, cerebral edema, acute respiratory distress syndrome, coagulopathy, rhabdomyolysis, and thromboembolism (Table 163.3).
|
Table 163.3 Complications of Diabetic Ketoacidosis and Hyperosmolar Hyperglycemic Nonketotic Syndrome Treatment |
|
|
Hypoglycemia and hyperglycemia are common complications of DKA treatment. Because intensive intravenous insulin therapy intentionally decreases blood glucose levels, reverses ketone body formation, and improves insulin sensitivity, it also places the patient at significant risk for hypoglycemia and its associated serious complications, including significant cognitive dysfunction, coma, and death. The incidence of serious hypoglycemic episodes associated with DKA treatment can be substantially decreased with the institution of low-dose insulin protocols, the addition of dextrose-containing solutions to intravenous fluid management when the blood glucose concentration reaches 250 to 300 mg/dL, and the institution of frequent blood glucose monitoring with frequent insulin infusion rate titration (4,34). Additionally, hyperglycemia—with the potential for DKA to recur—can be seen following the resolution of DKA, due in large part to abrupt termination of the intravenous insulin infusion without adequate overlap of nonintravenous therapies, including subcutaneous insulin.
Hypokalemia may be regularly encountered in the treatment phase of DKA due to intracellular translocation of potassium during insulin treatment with inadequate replacement. This electrolyte abnormality may be responsible for associated dysrhythmias, skeletal muscle weakness, and ileus. Treatment of the acidemic state with bicarbonate and inadequate potassium replacement predisposes patients to hypokalemia.
Cerebral edema is a rare, but devastating, complication of DKA therapy. It occurs more commonly in pediatric populations, and may be seen in up to 1% of the patients treated for DKA (4). The exact reason for the development of cerebral edema during DKA treatment remains unproven; however, it is felt to be related to an overly rapid correction of the hyperosmolar state. For this reason, current recommendations state that the change in serum osmolality resultant from therapy should not exceed 3 mOsm/kg/hour, and, in patients with concomitant cardiac and renal compromise, serum osmolality should be monitored frequently (22,37,38,39,40); we check serum osmolality every 4 to 6 hours, depending on the severity of the episode of DKA and underlying disease.
With worsening cerebral edema, intracranial pressure may significantly increase—occasionally to the point of brainstem herniation with associated respiratory arrest and cardiovascular derangements. For this reason, careful monitoring should be done during DKA treatment for signs of cerebral edema and brainstem herniation such as the acute onset of headache, changes in the level of consciousness, the development of papilledema, or the onset of seizures. Additionally, sodium and water deficits are slowly corrected, and rapid decreases in blood glucose levels are avoided. Treatment of this condition is largely supportive, and may be improved with the use of mannitol as an osmotic diuretic to decrease the amount of cerebral edema.
Acute respiratory distress syndrome is also a rare but potentially fatal complication that may occur at any time during the process of DKA. The disorder may be linked to the underlying cause of DKA as a source of infection, or the treatment phase with associated fluid resuscitation, fluid shifts, and changing osmotic gradients. Pulmonary aspiration of gastric secretions may also be responsible for the respiratory involvement. Patients with hypoxemia, widened alveolar-to-arterial oxygen gradients, or other pre-existing cardiorespiratory conditions warrant close monitoring and possibly more judicious fluid resuscitation (14,41).
Hyperosmolar Hyperglycemic Nonketotic Syndrome
HHS is a medical emergency that develops in response to one of many precipitating conditions (Table 163.1) in patients with type 2 DM. Among adults in the United States, the incidence of HHS is approximately 17.5 cases per 100,000 persons per year, and results in significant morbidity and mortality (42). The mortality rate from HHS is related directly to patient age, considering that mortality is, for example, less than 10% in patients younger than 75 years of age compared to 35% in patients older than 84 years of age (43,44).
In approximately 20% of patients presenting with HHS, this diagnosis is their initial presentation with type 2 diabetes (42). Additionally, the diagnosis is usually made after significant delay, and is made more complex because HHS can coexist with DKA in approximately 30% of patients (44).
Pathophysiology
The basic pathophysiologic abnormality in HHS is a relative insulin deficiency caused by both an increase in peripheral insulin resistance and an increase in blood levels of counterregulatory hormones (Fig. 163.1) (44,45,46). These hormones—glucagon, cortisol, and growth hormone—and various catecholamines increase hepatic and renal glucose production, and further worsen peripheral tissue glucose utilization (44,45). Together, these defects cause an insidious but dramatic rise in serum glucose concentration, typically over days to weeks (45).
With increasing serum glucose concentration, an osmotic gradient develops between the intravascular and extravascular compartment (47). Because water moves from the extravascular compartment down this osmotic gradient, both intracellular dehydration and a transiently increased intravascular volume with relative serum hyponatremia can occur. As the serum glucose concentration continues to rise, osmotic diuresis causes profound decreases in intravascular volume, coupled with losses of vital electrolytes such as sodium, potassium, phosphate, and magnesium (43). This large intravascular volume loss can result in life-threatening end-organ hypoperfusion and nonketotic metabolic acidosis.
Compared to DKA, ketones are minimally produced in patients with HHS likely due to the ability of the pancreas to secrete insulin. The amount of insulin, while not sufficient to prevent hyperglycemia in these patients, does prevent fatty acid lipolysis and the formation of ketone bodies and development of ketoacidosis.
Presentation and Diagnosis
Because patients with HHS typically fail to develop ketoacidosis, the time from onset to diagnosis and treatment can be significantly longer than in patients with DKA (45). The clinical diagnosis of these patients, therefore, requires a high clinical suspicion to promptly recognize the signs and symptoms of HHS and institute appropriate diagnostic and treatment modalities.
Patients with suspected HHS may initially exhibit nausea/ vomiting, visual disturbances, muscle weakness, and leg cramps (48). Left untreated, these patients eventually develop confusion, lethargy, hemiparesis, seizures, and coma (49). Physical examination may reveal both signs of profound dehydration—such as decreased skin turgor and dry mucous membranes—as well as abdominal distension from gastroparesis (50).
Initial laboratory evaluation in patients with suspected HHS should include serum glucose, ketones, electrolytes, and creatinine concentration; serum measured and calculated osmolality; urinalysis; and appropriate empiric bacterial and fungal cultures (Table 163.2) (45). Because these patients can have significantly elevated serum glucose concentrations—as high as or higher than 1,000 mg/dL, the serum osmolality can be quite high and seems to correlate with neurologic symptoms (51).
Management
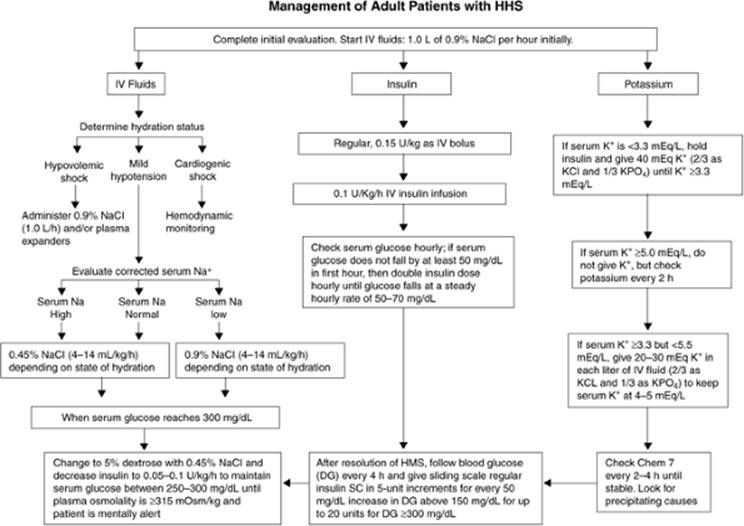
The treatment goals for HHS include aggressive intravascular fluid replacement, insulin administration to correct hyperglycemia, appropriate electrolyte replacement, and, if indicated, respiratory system support (Fig. 163.4). Ultimately, effective patient education and long-term patient support are also important.
HHS treatment is typically undertaken in two phases. The first is the acute—emergency—phase and consists of rapid restoration of circulatory volume and electrolyte deficits with concomitant insulin administration to correct serum hyperglycemia and hyperosmolality. Additionally, treatment of the triggering disorder should be started. This phase ends with near-correction of hyperglycemia and metabolic acidosis. The second phase is a transitional phase centered on changing insulin replacement to appropriate chronic diabetes therapy (i.e., subcutaneous or oral hypoglycemic regimen), as well as patient education and support.
|
|
|
Figure 163.4. Management of adult patients with hyperosmolar hyperglycemic nonketotic syndrome (HHS). DG,; HMS,. (Copyright 2001, American Diabetes Association, from Diabetes Care 2001;24:131–153. Reprinted with permission from the American Diabetes Association.) |
In patients with HHS, the total body water deficit can be as high as 100 to 200 mL/kg in adults; thus, fluid replacement is the mainstay therapy for intravascular collapse and poor organ perfusion, and can lower glucose concentration independent of insulin administration. Initially, 0.9% saline solution should be infused at 15 to 20 mL/kg total body weight per hour to restore extracellular fluid volume deficit. Normal saline infusion should continue until blood pressure and end-organ perfusion have been normalized. The intravenous solution should then be changed to 0.45% saline solution at a reduced rate to restore the intracellular fluid deficit. The overall fluid resuscitation goal should be replacement of one half of the estimated fluid deficit over the first 8 hours, and the other half of the estimated fluid deficit over the next 16 hours. Care should be taken to ensure that the serum osmolality does not decrease more than 3 mOsm/kg/hour to reduce the risk of acute cerebral edema.
The cornerstone of therapy for HHS is intravenous insulin given to restore normal peripheral glucose uptake, suppress lipolysis, and decrease hepatic gluconeogenesis. Complications of insulin therapy include hypoglycemia, hypokalemia (insulin infusion should not begin with serum potassium less than 3.5 mEq/dL), and hypophosphatemia. Insulin should be initially given as an intravenous bolus of 0.15 units/kg, followed by a continuous infusion of 0.1 unit/kg/hour with a goal glucose decrease of 50 to 75 mg/dL/hour. While the patient is receiving intravenous insulin, the glucose should be monitored every 1 to 2 hours via either capillary or serum samples. Once the serum glucose decreases to 250 mg/dL, dextrose should be added to the intravenous fluid administration, and the insulin infusion should be decreased to 0.05 to 0.1 unit/kg/hour.
Electrolyte replacement is also an important component to the management of HHS. Hypokalemia can develop during HHS treatment because insulin administration causes an intracellular shift of potassium ions from the extracellular compartment. Additionally, the ongoing osmotic diuresis can cause a dramatic depletion of total body potassium stores; this loss can exceed 400 mEq in severe HHS. For this reason, electrocardiographic monitoring should be utilized during this phase of therapy, and aggressive potassium replacement—with up to 40 mEq/hour—may be necessary. Hypophosphatemia may also develop secondary to the ongoing osmotic diuresis. Replacement of phosphate during HHS treatment seems prudent; however, several prospective randomized studies have failed to show a definitive benefit to phosphate replacement in the absence of decreased cardiac or respiratory function and anemia (36,52). If phosphate replacement is necessary, potassium phosphate may be an ideal replacement infusion to correct both hypokalemia and hypophosphatemia.
Complications
In addition to the electrolyte abnormalities discussed above, complications from HHS include pancreatitis, rhabdomyolysis, thromboembolism, hyperchloremic metabolic acidosis, cerebral edema, acute gastric dilatation, and ARDS (Table 163.3). Patients with HHS are at increased risk for thromboembolism, and thus, subcutaneous heparin administration may be warranted to prevent thromboembolic complications (43,53). Complications of HHS treatment include intravascular volume overload and acute cerebral edema from overaggressive intravenous fluid administration. Intravascular volume overload is seen as hypoxemic respiratory failure—often with pulmonary edema—and lower extremity pitting edema. Acute cerebral edema is manifested by headache, lethargy, and depressed levels of consciousness that can rapidly progress to brainstem herniation. Treatment of this potentially devastating complication consists of administering an osmotic diuretic such as mannitol and supportive care.
Summary
Future directions in DKA and HHS therapy involve further refinements in protocols and therapies aimed to improve the hyperglycemia, acidosis, and electrolyte imbalances while minimizing risks such as volume overload, ARDS, and cerebral edema. Additionally, ongoing efforts to provide patients and caregivers with the tools for early identification and the need for aggressive treatment of these hyperglycemic emergencies may ultimately have the largest impact on long-term morbidity and mortality.
Glycemic Control and Insulin Therapy in the Intensive Care Unit
Over the last decade, multiple published studies (Table 163.4) (54,55,56,57,58) suggested that intensified glycemic control of ICU patients by using intravenous insulin infusion results in improved outcomes, including decreased mortality rate. While these observations led to dramatic changes in the management of ICU blood glucose control in both Europe and North America, concerns have been raised about the optimal range of glycemic control for ICU patients and the clinical implication of the increased risk of iatrogenic hypoglycemia (59,60).
The recommendation that glucose levels be maintained no higher than 4.4 to 6.1 mmol/L (1 mmol/L of glucose = 18 mg/dL) has its origins in a key study conducted in Leuven, Belgium, by Van den Berghe et al. and published in 2001 (54). In this large randomized, single-center trial of predominantly cardiac surgery ICU patients, there was a reduction in ICU mortality with intensive intravenous insulin protocol targeting blood glucose to 4.4 to 6.1 mmol/L (for all patients, a 43% relative and 3.4% absolute mortality reduction; for those with >5 days in the ICU, a 48% relative and 9.6% absolute mortality reduction). In addition, the study demonstrated a reduction in organ dysfunction and ICU length of stay (LOS) in the subset with ICU LOS >5 days from a median of 15 to 12 days.
Although there were significant differences in design, blood sugar target, and patient population among all studies listed in Table 163.4, the results were, for the most part, fairly similar. Intensive therapy with intravenous insulin produced a consistent decrease in mortality rate in a variety of patient populations. However, several important differences should be noted. In some cases, all the participants had diabetes, whereas in others, most participants were nondiabetic. In addition, there were remarkable differences in the range of blood glucose levels achieved. For instance, the glucose levels of the control group of the Leuven studies were similar to—and in some cases, lower than—the levels in the intensive treatment groups of the other studies. However, the differences in blood glucose levels between the control and intensive treatment groups were similar in all five studies. Furthermore, there was a comparable improvement in mortality rate in the critically ill patients targeted to reach a serum glucose goal of 6.1 mmol/L and 7.3 to 9.8 mmol/L.
|
Table 163.4 Relation Between Intensive Insulin Therapy and Outcome in Critically Ill Patients |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Another important observation is that the two studies that achieved by far the lowest glucose levels in the intensively treated patients were also the only two studies in which parenteral nutrition (PN) was often associated with hyperglycemia and hypertriglyceridemia (62). Furthermore, there is considerable evidence that PN can be responsible for other adverse outcomes in critically ill patients. In a meta-analysis of 27 studies comparing PN with enteral nutrition and involving >1,800 patients, Braunschweig et al. found a >50% higher incidence of infection in participants receiving PN, but no difference in mortality rate (62). The implications of these findings in interpreting the Leuven I study are obvious. Patients in the Leuven I study received PN, a treatment known to increase infection rates, and when glucose level was lowered with insulin infusion to an average of 5.7 mmol/L, an improvement in mortality rate, based mostly on a reduction in infection rate, was observed. Hence, the administration of intravenous insulin in this study merely reduced infectious complications to a level that would have been observed if PN had not been otherwise given.
Furthermore, one of the mechanisms that may explain an increased rate of infection in diabetic individuals and hospitalized patients with hyperglycemia is impaired immune function secondary to a defect in the adherence properties of polymorphonuclear leukocytes (63,64). An analysis of data from the 363 patients who remained in intensive care for >7 days in the Leuven I study revealed a strong relationship between serum triglyceride (TG) and ICU mortality rate by univariate analysis (an approximate 400% increase in mortality rate in patients with TAG >3.4 mmol/L compared with individuals with TAG <1.1 mmol/L). Accordingly, an alternative explanation for the benefits of insulin infusion is that they were not a direct consequence of lowering glucose, but rather due to effects of insulin on lipid control, especially in patients who received PN (65).
Most recently, two additional multicenter randomized control trials (RCTs) of intensive insulin therapy—one focused on patients with severe sepsis (VISEP) and the second on medical and surgical ICU patients—failed to demonstrate improvement in mortality, though they are not yet fully published (66,67). Nevertheless, both were stopped earlier than planned because of high rates of hypoglycemia and adverse events in the intensive insulin groups. This finding is particularly important for ICU patients who are commonly sedated and mechanically ventilated since aggressive lowering of serum glucose levels may carry the risk of hypoglycemia, which is potentially more difficult to detect (59).
|
Table 163.5 The Surviving Sepsis Campaign 2008 Consensus Recommendation on Glucose Control during Severe Sepsis |
||
|
To address the above concerns, a large RCT in 20 ICUs in Australia and New Zealand is currently under way to compare target serum glucose levels of 4.5 to 6.0 mmol/L (80–110 mg/dL) versus 8 to 10 mmol/L (140–180 mg/dL) (68). This trial will provide information about the effect of normoglycemia in a heterogeneous group of critically ill patients. In addition, it will recruit >6,000 patients and most likely will produce ≥500,000 glucose measurements that subsequently can be used to reveal some of the many unknown dimensions of glycemic control and its consequences in ICU patients.
In the meantime, several recommendations for glycemic control in the ICU have been suggested in the most current Surviving Sepsis Campaign (Table 163.5) (69). These recommendations are intended to provide some direction for the clinician caring for ICU patients with severe sepsis while awaiting results from the ongoing studies on potential differences in outcomes with different glucose targets and various insulin protocols.
Until the full results of the ongoing studies become available, intensive insulin therapy to all ICU patients remains unsupported, and should be viewed with a healthy degree of scientific skepticism.
Summary
The weight of available data indicates that intensified insulin treatment of the critically ill is associated with impressive reductions in mortality rate. Therefore, the value of intensive insulin treatment should not be in doubt. However, the question remains if the available data justify a glycemic target of 6.1 mmol/L. As pointed out above, the reduction in mortality reported in Leuven I can be secondary to lipid, rather than glucose, control. Since these are mere associations, additional research will be required in which lipid levels are maneuvered more directly in order to assess whether nonglucose effects of insulin are, in fact, responsible for the apparent benefits of insulin. In addition, since the use of PN in the Leuven I study may impose additional constraints on its subjects, intensive insulin treatment in this study may merely have counteracted the adverse effects of PN. Finally, considering that the Leuven I study was not designed to compare different glycemic targets with intensive insulin treatment, judgments concerning whether a goal of 6.1 mmol/L will produce better outcomes than a higher goal must await the results of ongoing studies.
Furthermore, since hypoglycemia is likely to be more frequent and more severe with lower glucose targets, it seems prudent to adjust the dose of intravenous insulin to target glucose levels ≥5.0 and <8.3 mmol/L.
References
1. American Diabetes Association. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care. 2003;26:S5.
2. Burke JP, Williams K, Gaskill SP, et al. Rapid rise in the incidence of type 2 diabetes from 1987 to 1996: results from the San Antonio heart study. Arch Intern Med. 1999;159:1450–1456.
3. Currie CJ, Morgan CL, Peters JR. The epidemiology and cost of inpatient care for peripheral vascular disease, infection, neuropathy, and ulceration in diabetes. Diabetes Care. 1998;21:42–48.
4. Kitabchi AE, Umpierrez GE, Murphy MB, et al. Hyperglycemic crises in diabetes. Diabetes Care. 2004;27:S94–102.
5. Fishbein H, Palumbo PJ. Acute Metabolic Complications in Diabetes. Bethesda, MD: National Institutes of Health; 1995 #NIH 95–1468. 9. Centers for Disease Control, Division of Diabetes Translations. Diabetes Surveillance, 1991. Washington, DC: US Government Printing Office; 1992:635–1150.
6. White NH. Diabetic ketoacidosis in children. Endocrinol Metab Clin North Am. 2000;29:657–682.
7. Kitabchi AE, Wall BM. Diabetic ketoacidosis. Med Clin North Am. 1995; 79:9–37.
8. Stentz FB, Umpierrez GE, Cuervo R, et al. Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes. 2004;53:2079–2086.
9. Merimee TJ, Tyson JE. Stabilization of plasma glucose during fasting: normal variations in two separate studies. N Engl J Med. 1974;291:1275– 1278.
10. Gerich JE, Lorenzi M, Bier DM, et al. Effects of physiologic levels of glucagon and growth hormone on human carbohydrate and lipid metabolism: studies involving administration of exogenous hormone during suppression of endogenous hormone secretion with somatostatin. J Clin Invest. 1976;57:875–884.
11. Felig P, Sherwin RS, Soman V, et al. Hormonal interactions in the regulation of blood glucose. Recent Prog Horm Res. 1979;35:501–532.
12. McGarry JD, Woeltje KF, Kuwajima M, et al. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab Rev. 1989;5:271–284.
13. DeFronzo RA, Matzuda M, Barret E. Diabetic ketoacidosis: a combined metabolic nephrologic approach to therapy. Diabetes Rev. 1994;2:209–238.
14. Kitabchi AE, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. 2001;24:131–153.
15. Faich GA, Fishbein HA, Ellis SE. The epidemiology of diabetic acidosis: a population-based study. Am J Epidemiol. 1983;117:551–558.
16. Katz MA. Hyperglycemia-induced hyponatremia: calculation of expected sodium depression. N Engl J Med. 1973;289:843–844.
17. Adrogue HJ, Wilson H, Boyd AE, et al. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med. 1982;307:1603–1610.
18. Stephens JM, Sulway MJ, Watkins PJ. Relationship of blood acetoacetate and 3-hydroxybutyrate in diabetes. Diabetes. 1971;20:485–489.
19. Wallace TM, Matthews DR. Recent advances in the monitoring and management of diabetic ketoacidosis. Q J Med. 2004;97:773–780.
20. Waldhausl W, Kleinberger G, Korn A, et al. Severe hyperglycemia: effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes. 1979;28:577–584.
21. Owen OE, Licht JH, Sapir DG. Renal function and effects of partial rehydration during diabetic ketoacidosis. Diabetes. 1981;30:510–518.
22. Kitabchi AE, Fisher JN, Murphy MB, et al. Diabetic ketoacidosis and hyperglycemic, hyperosmolar nonketotic state. In: Kahn CR, Weir GC, eds. Joslin's Diabetes Mellitus Textbook. Philadelphia: Lea & Febiger; 1993: 753–760.
23. Foster DW, McGarry JD. The metabolic derangements and treatment of diabetic ketoacidosis. N Engl J Med. 1983;309:159–169.
24. Fleckman AM. Diabetic ketoacidosis. Endocrinol Metab Clin North Am. 1993;22:181–207.
25. Kitabchi AE. Low-dose insulin therapy in diabetic ketoacidosis: fact or fiction? Diabetes Metab Rev.1989;5:337–363.
26. Burghen GA, Etteldorf JN, Fisher JN, et al. Comparison of high dose and low-dose insulin by continuous intravenous infusion in the treatment of diabetic ketoacidosis in children. Diabetes Care. 1980;3:15–20.
27. Magee MF, Bankim AB. Management of decompensated diabetes. Diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome. Crit Care Clin. 2001;17:75–106.
28. Naunheim R, Jang TJ, Banet G, et al. Point-of-care test identifies diabetic ketoacidosis at triage. Acad Emerg Med. 2006;13:683–685.
29. Wallace TM, Meston NM, Gardner SG, et al. The hospital and home use of a 30-second hand-held blood ketone meter: guidelines for clinical practice. Diabetes Med. 2001;18:640–645.
30. Foster KJ, Alberti KG, Hinks L, et al. Blood intermediary metabolite and insulin concentrations after an overnight fast: reference ranges for adults, and interrelations. Clin Chem. 1978;24:1568–1572.
31. Umpierrez GE, Watts NB, Phillips LS. Clinical utility of b-hydroxybutyrate determined by reflectance meter in the management of diabetic ketoacidosis (Letter). Diabetes Care. 1995;18:137–138.
32. Luzi L, Barrett EJ, Groop LC, et al. Metabolic effects of low-dose insulin therapy on glucose metabolism in diabetic ketoacidosis. Diabetes. 1988; 37:1470–1477.
33. Sulway MJ, Malins JM. Acetone in diabetic ketoacidosis. Lancet. 1970; 2:736–740.
34. Umpierrez GE, Kitabchi AE. Diabetic ketoacidosis: risk factors and management strategies. Treat Endocrinol. 2003;2:95–108.
35. Abramson E, Arky R. Diabetic acidosis with initial hypokalemia: therapeutic implications. JAMA. 1966;196:401–403.
36. Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983;57:177–180.
37. Ennis ED, Stahl E, Kreisberg RA. The hyperosmolar hyperglycemic syndrome. Diabetes Rev. 1994;2:115–126.
38. Hillman K. Fluid resuscitation in diabetic emergencies—a reappraisal. Intensive Care Med. 1987;13:4–8.
39. Marshall SM, Walker M, Alberti KGM. Diabetic ketoacidosis and hyperglycemic non-ketotic coma. In: Alberti KGM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. New York: John Wiley; 1997:1215–1229.
40. Ennis ED, Stahl EJ, Kreisberg RA. Diabetic ketoacidosis. In: Porte D Jr, Sherwin RS, eds. Diabetes Mellitus: Theory and Practice. Amsterdam: Elsevier; 1997:827–844.
41. Carroll P, Matz R. Adult respiratory distress syndrome complicating severely uncontrolled diabetes mellitus: report of nine cases and a review of the literature. Diabetes Care. 1982;5(6):574–580.
42. Yared Z, Chiasson JL. Ketoacidosis and the hyperosmolar hyperglycemic state in adult diabetic patients. Minerva Med. 2003;94:909–913.
43. Venkatraman R, Singhi SC. Hyperglycemic hyperosmolar nonketotic syndrome. Ind J Pediatr. 2006;73(1):55–60.
44. Gaglia JL, Wyckoff J, Abrahamson MJ. Acute hyperglycemic crisis in the elderly. Med Clin N Am. 2004;88(4):1063–1084.
45. American Diabetes Association. Hyperglycemic crises in patients with diabetes mellitus. Diabetes Care. 2001;24:1988–1996.
46. Chiasson JL, Aris-Jilwan N, Belanger R, et al. Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ. 2003;168:859–866.
47. Brenner ZR. Management of hyperglycemic emergencies. AACN Clin Issues. 2006;17:56–65.
48. Stoner GD. Hyperosmolar hyperglycemic state. Am Fam Physician. 2005; 71:1723–1730.
49. Ting JY. Hyperosmolar diabetic non-ketotic coma, hyperkalaemia and an unusual near death experience. Eur J Emerg Med. 2001;8:57–63.
50. Delaney MF, Zisman A, Kettyle WM. Diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Endocrinol Metab Clin North Am. 2000;29:683–705.
51. Siperstein MD. Diabetic ketoacidosis and hyperosmolar coma. Endocrinol Metab Clin North Am. 1992;21:415–432.
52. Wilson HK, Keuer SP, Lea AS, et al. Phospate therapy in diabetic ketoacidosis. Arch Intern Med. 1982;142:517–520.
53. Mather HM. Management of hyperosmolar coma. J Royal Soc Med. 1980; 73:134–138.
54. Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359–1367.
55. Malmberg K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. BMJ. 1997;314:1512–1515.
56. Furnary AP, Gao G, Grunkemeier GL, et al. Continuous insulin infusion reduces mortality in patients with diabetes undergoing coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2003;125:1007–1021.
57. Krinsley JS. Effect of an intensive glucose management protocol on the mortality of critically ill adult patients. Mayo Clin Proc. 2004;79:992–1000.
58. Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449–461.
59. Cryer PE. Hypoglycaemia: the limiting factor in the glycaemic management of the critically ill?. Diabetologia. 2006;49:1722–1725.
60. Bhatia A, Cadman B, Mackenzie I. Hypoglycemia and cardiac arrest in a critically ill patient on strict glycemic control. Anesth Analg. 2006;102:549–551.
61. Klein CJ, Stanek GS, Wiles CE 3rd. Overfeeding macronutrients to critically ill adults: metabolic complications. J Am Diet Assoc. 1998;98:795–806.
62. Braunschweig CL, Levy P, Sheean PM, et al. Enteral compared with parenteral nutrition: a meta-analysis. Am J Clin Nutr. 2001;74:534–542.
63. Bagdade JD, Root RK, Bulger RJ. Impaired leukocyte function in patients with poorly controlled diabetes. Diabetes. 1974;23:9–15.
64. McMahon MM, Bistrian BR. Host defenses and susceptibility to infection in patients with diabetes mellitus. Infect Dis Clin North Am. 1995;9:1–9.
65. Mesotten D, Swinnen JV, Vanderhoydonc F, et al. Contribution of circulating lipids to the improved outcome of critical illness by glycemic control with intensive insulin therapy. J Clin Endocrinol Metab. 2004;89:219– 226.
66. Brunkhorst FM, Kuhnt E, Engel C, et al. Intensive insulin therapy in patient with severe sepsis and septic shock is associated with an increased rate of hypoglycemia—results from a randomized multicenter study (VISEP). Abstr Infect. 2005;33:19–20.
67. Preiser JC. Intensive glycemic control in medsurg patients (European Glucontrol trial). Program and abstracts of the Society of Critical Care Medicine 36th Critical Care Congress, February 17–21, 2007, Orlando, FL.
68. Normoglycemia in Intensive Care Evaluation and Survival Using Glucose Algorithm Regulation (NICE-SUGAR). Current controlled trials: a multi-centre, open label, randomised controlled trial of two target ranges for glycaemic control in intensive care unit (ICU) patients. http://controlled-trials.com/isrctn/trial/ISRCTN04968275/0/04968275. Accessed June 10, 2008.
69. Dellinger P, Levy MM, Jean M, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296–327.