Marcelo E. Heinig
A. Ahsan Ejaz
Main Points
· The prevalence of acute renal failure continues to increase.
· Mortality of acute renal failure, despite improvements, remains high.
· Loss of autoregulation of renal blood flow, vasoconstriction, and subsequent downstream effects potentiate the inflammatory cascade in acute renal failure.
· Pre-existing organ dysfunction affects the prognosis of critically ill patients.
· The decrease in physiologic reserve in acute renal failure influences drug dosing, treatment modality, and response to interventions in patients in the intensive care unit.
· The treatment of acute renal failure is based on the principle that the preservation of renal blood flow and optimal perfusion pressure improves outcomes.
Acute renal failure (ARF) is a common finding in hospitalized patients. The accurate incidence of acute renal failure is undetermined due to the differences in definitions used in databases at different time points. However, data from hospitalized Medicare beneficiaries—using an acute renal failure definition from ICD-9-CM as “the sudden, severe onset of inadequate kidney function”—yields an overall incidence rate of 23.8 cases per 1,000 discharges (1). Others have reported incidences of hospital-acquired acute renal failure of 4.9% to 7%. Patients who develop ARF have an in-hospital mortality of 15.2%, which increases to 32.9% if dialysis is also required. In the intensive care unit (ICU), the incidence of ARF—again, depending on the definitional criteria used—has been reported to range between 5% and 17%. Nonetheless, the mortality rates of ICU patients with ARF were high in every study, ranging between 24% and 53%; when renal replacement therapy was required, the mortality was even higher, ranging between 45% and 79% (2,3).
Knowledge of risk factors for ARF in the ICU can be helpful in the determination of clinical outcome and contribute to more defined risk assessment and management in this cohort (Table 47.1). This chapter reviews pertinent renal physiology, the mechanisms and outcomes of ischemic acute renal failure, the progression of chronic kidney disease and its impact on acute critical illness, and the principles of management of acute renal failure. Specifics on the treatment of ARF and a detailed description of the various treatment modalities are to be found in later chapters of this textbook.
Physiology of the Kidney
General Anatomy
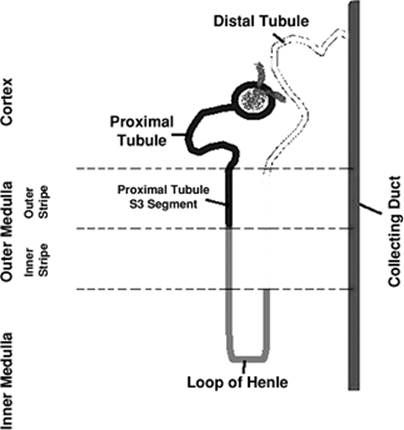
The ability of the kidney to maintain the equilibrium of the corporal fluids and electrolytes depends on three essential processes: (i) filtration of the circulating blood by the glomerulus to form an ultrafiltrate, (ii) reabsorption of specific solutes from the tubular fluid to the blood, and (iii) secretion from the peritubular capillary blood system to the tubular space. The functions of the kidney are dependent on the unique anatomic arrangement of its structures.
The afferent arteriole divides into several branches after entering the glomerular tuft and forms the capillary network present in the glomerulus (Fig. 47.1). The confluence of several capillaries forms the efferent arteriole, which drains the blood from the glomerulus. During the ultrafiltration process, water and solutes pass through the endothelium, the glomerular basement membrane, and the slit-diaphragm between the podocytes. The determinant of the filtration of a substance is its size and charge. Substances with molecular radius of less than 2 nm are filtered freely, whereas ionic charges of substances measuring between 2 and 4 nm determine the amount of their filtration. Substances with molecular radius greater than 4 nm are not filtered.
The medullary region of the kidneys is characterized by low oxygen tension (10 to 15 mm Hg) under normal conditions. The tubular segments located in this region—pars recta or S3 segment of proximal tubule and medullary thick ascending limb—are characterized by active transport of Na+, which is dependent on oxidative phosphorylation for energy. The high rates of oxygen consumption associated with the precarious blood flow in the medullary region are responsible for the vulnerability of this area to ischemia.
General Renal Physiology
Glomerular Filtration Rate and Renal Plasma Flow
The glomerular filtration rate (GFR) and the renal plasma flow (RPF) are rate measurements that help to characterize the status of renal function. The total rate at which fluid is filtered into all the glomeruli constitutes the glomerular filtration rate (GFR). The normal GFR varies between 100 and 120 mL/min/1.73 m2, depending on various factors including gender, age, and body weight. Changes in the GFR can result from changes in the glomerular permeability or capillary surface area or from changes in the net ultrafiltration. In a single glomerulus, the driving pressure for the glomerular filtration is determined by the difference of the gradient of the hydrostatic and oncotic pressures between the capillaries and the Bowman space.
|
Table 47.1 Predictors of acute renal failure in the intensive care unit |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The rate at which plasma flows through the kidney is called renal plasma flow. Renal blood flow (RBF) is the volume of blood delivered to the kidney per unit time (1 to 1.2 L/min). Renal blood flow calculations are based on renal plasma flow and hematocrit:
RBF = RPF/1 - hematocrit
It is possible to measure the RPF using para-aminohippurate as a tracer in humans, with a normal value about 625 mL/min, but the test is not commonly used in clinical practice due to labor intensity and cost.
Autoregulatory Control of Renal Blood Flow
Despite the significant variations in mean arterial pressure, renal blood flow and GFR remain constant, a phenomenon known as autoregulation. Autoregulation is affected via changes in diameter of the afferent arterioles in response to a combination of two mechanisms:
1. The myogenic reflex: When the renal perfusion pressure increases, the afferent arteriole constricts automatically.
2. Tubuloglomerular feedback: Situations associated with an increased delivery of NaCl to the macula densa result in vasoconstrictive response of the afferent arteriole. The increased uptake of chloride ions by the macula densa cells leads to ATP release into the surrounding extracellular space. ATP is then converted to adenosine which binds to adenosine A1 receptors causing afferent arteriolar vasoconstriction.
Basic Principles of Tubular Transport
The kidneys filter about 180 L of plasma, and all but 2 L are reabsorbed. This massive reabsorption is accomplished through several modifications of the glomerular ultrafiltrate, consisting of absorption and secretion of water and solutes before becoming the final urine. In general, three different tubular segments are involved in this process and can be recognized based on the differences in the function of their cells.
|
|
|
Figure 47.1. Nephron—the functional unit of the kidney. |
1. The proximal tubule reabsorbs most of the filtered glucose, amino acids, low-molecular-weight proteins, and water (approximately 65%). Other solutes, such as Na+, K+, Cl-, HCO3-, Ca2+, phosphate, and urea are also absorbed in this nephron segment. The terminal segment of the proximal tubule—the pars recta or S3—is responsible for the secretion of numerous drugs and toxins.
2. The straight portion of the proximal tubule, the thin ascending and descending limbs, and the thick ascending limb constitute the region known as the loop of Henle. This region is responsible for the continuing reabsorption of the solutes that escaped the proximal tubules (Na+, Cl-, K+, Ca2+, Mg2+). It is the major area responsible for the ability of the kidneys to concentrate or dilute the final urine. The principal luminal transporter expressed in the thick ascending limb is the Na-K2Cl cotransporter, which is the target of diuretics such as furosemide.
3. The distal nephron is responsible for the final adjustments in the urine. Critical regulatory hormones such as vasopressin and aldosterone regulate the acid and potassium excretion and the urinary concentration at this segment. Thiazide diuretics act at the distal convoluted tubule through an apical cotransporter of Na+.
The Glomerulotubular Balance
The fact that the tubules tend to reabsorb a constant proportion of a glomerular filtrate rather than a constant amount is called glomerular balance. As an example, if the filtered load of Na+ was increased by 10%, total Na+ reabsorption in the tubules would also increase by 10%, keeping the final amount of Na+ in the urine—100 to 250 mEq/day—stable. In the absence of this mechanism, even small changes in the GFR would cause major changes in the final amount excreted of any solute. The mechanisms responsible for this balance are not fully understood, but changes in the oncotic pressure in the peritubular capillaries and in the delivery of certain solutes (glucose and amino acids) to the proximal tubule are probably involved.
Control of Effective Circulating Volume via Integrated Mechanisms
Most volume-regulatory mechanisms in the kidney use the effective circulating volume, or the degree of fullness of the vasculature, as the final target. Under normal conditions, the effective circulating volume varies in direct proportion to the extracellular fluid volume. As Na+ is the most abundant extracellular solute, the kidney excretion or retention of Na+ is a crucial step to control of effective circulatory fluid volume. Osmoregulation is under the control of a single hormonal system, the antidiuretic hormone (ADH), but volume regulation requires a complex set of redundant and overlapping mechanisms.
The kidneys are able to conserve water by excreting the solute load in concentrated urine in conditions of excess water loss. Similarly, in high water intake states, the urinary volume may increase to as high as 14 L/day, with an osmolality significantly lower than that of the plasma. Vasopressin or ADH regulates the water permeability in the distal nephron and is the principal hormone responsible for the determination of the urinary concentration and volume. Normally, the major stimulus to secretion of ADH is the plasma osmolality, but in situations of extracellular volume depletion, the set point to release ADH is shifted, and higher levels of this hormone are common even in hypotonic states.
The renin-angiotensin system plays a central role in the control of effective circulatory fluid volume. The afferent arteriolar cells that form part of the juxtaglomerular apparatus release renin in response to increased sympathetic nervous stimulation, reduced arterial blood pressure, or reduced delivery of NaCl to the macula densa region. Renin cleaves angiotensinogen into angiotensin I and is then converted to angiotensin II by the angiotensin-converting enzyme. Angiotensin plays important roles in the control of blood pressure and the effective circulatory fluid volume:
· Angiotensin II has the direct effect of increasing the sodium reabsorption in the proximal tubule (stimulation of Na+/H+ exchange).
· The aldosterone secreted by the adrenal glands in response to the angiotensin II stimulates sodium reabsorption in the distal nephron.
· Angiotensin II causes general arteriolar vasoconstriction, thereby increasing arterial pressure.
Increased renal sympathetic tone enhances renal salt reabsorption and can decrease renal blood flow at higher frequencies. In addition to its direct effects on renal function, increased sympathetic outflow promotes the activation of the renin-angiotensin system.
Pathophysiology of Acute Renal Failure
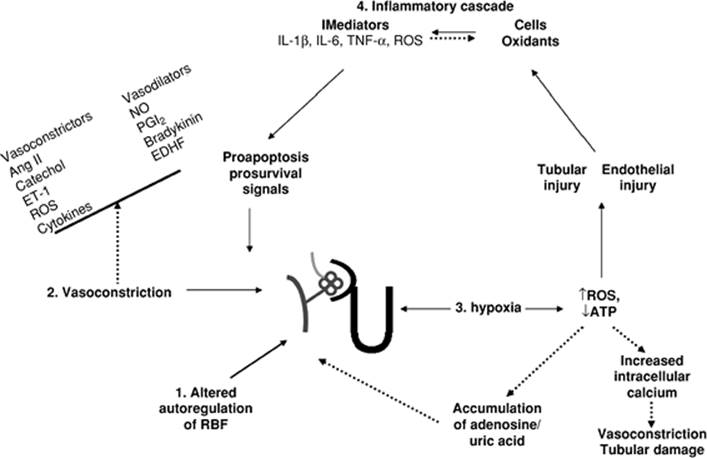
Ischemic acute renal failure is a syndrome that develops following a sudden transient drop in total or regional blood flow to the kidney. Classically, a decrease in mean arterial pressure has been associated with a reduction in renal blood flow with subsequent tissue hypoxia, tubular and vascular injury, and loss of renal structure and function. However, renal blood flow and glomerular filtration rate can decrease by as much as 50% despite the maintenance of mean arterial pressure (4), suggesting the presence of renal vasoconstriction. Mechanisms for these observations in ischemic acute renal failure are discussed below (Fig. 47.2).
|
|
|
Figure 47.2. Theories of the mechanism of acute renal failure. |
Theories of the Mechanism of Acute Renal Failure
Loss of Autoregulation of Renal Blood Flow
Renal blood flow is dependent on systemic blood pressure and intrarenal vascular resistance. The autoregulatory mechanisms, through changes in vascular resistance, ensure that over a wide range of perfusion pressures, renal blood flow remains stable and glomerular filtration can be maintained. However, in ischemic acute renal failure, autoregulation of renal blood flow is lost. Consequently, renal blood flow diminishes with decreased mean arterial pressure over the autoregulatory range (5). The loss of autoregulation of renal blood flow is related to an increase in renovascular resistance.
Imbalance of Mediators of Vascular Tone
The paradoxical rise in renovascular resistance seen with decreasing renal perfusion in ischemic acute renal failure is due to the loss of the usual balance of vasoconstrictors and the vasodilators required to maintain the normal tone of the renal vasculature. The aberrant responses to neurohormonal stimuli and the persistent vasoconstriction worsen renal perfusion and impair oxygen and nutrient delivery to the areas supplied by the postglomerular vessels.
Tissue Hypoxia
The partial pressure of oxygen in the outer medulla is about 10 to 15 mm Hg. Even a mild decrease in renal perfusion can lead to a hypoxic insult (oxidative stress) to the vulnerable medullary nephron segments. Tissue hypoxia can result in depletion of cellular ATP stores, increased intracellular calcium, and subsequent disruption of actin cytoskeleton in the endothelial and vascular smooth muscle cells, with resultant hemodynamic impairment and tubular injury. Adenosine nucleotide metabolic products are not reused for the regeneration of ATP and are, instead, diverted through the degradatory pathways to generate xanthine and uric acid. Accumulation of adenosine and uric acid worsens vasoconstriction and renal perfusion via their effects on adenosine receptors and afferent arterioles, respectively. Cellular activation also leads to reactive oxygen species generation, phospholipase activation, and membrane lipid alterations.
The Inflammatory Cascade
Hypoxia, with subsequent reperfusion, leads to acute inflammatory changes. Inflammation is one of the major pathophysiologic pathways contributing to ischemic acute renal failure. Ischemic injury to the vasa recta results in enhanced adherence of leukocytes to the vascular endothelial cells, sequestration of leukocytes, vascular congestion—that is, a no-flow phenomenon—cellular infiltration, production of inflammatory mediators, and generation of reactive oxygen species. Cytokines and chemokines, released from the injured cells, attract and activate inflammatory cells to the site of injury and potentiate the inflammatory cascade. A similar inflammatory response is also seen with tubular cell injury, which is also capable of producing inflammatory mediators. The inflammatory changes are most pronounced in the outer medullary stripe, the region that is most susceptible to hypoxic insult.
Prosurvival and Proapoptotic Signaling Pathways
Numerous stress response mechanisms are rapidly activated in response to oxidative insults. Some of the pathways are preferentially linked to cell survival whereas others are proapoptotic. These pathways intersect and modulate each other's activities. Whether a particular insult leads to cell repair and survival—or death—depends on the nature and severity of the insult, the balance between the proapoptotic and antiapoptotic signals, and the basal state of the cells. Ongoing efforts to elucidate factors that play a role in microvascular endothelial injury and dysfunction, expression of adhesion molecules that facilitate leukocyte-endothelial interactions, the cytokine network, the cellular response to oxidative stress, and the gene activation patterns that regulate tissue injury and repair will result in a better understanding of the complex mechanisms involved in the pathogenesis of ischemic acute renal failure.
Role of Uric Acid in Acute Renal Failure
Serum uric acid is frequently elevated during cardiovascular surgery and has been shown to correlate with the risk of developing acute renal failure (6). Hyperuricemia worsens cisplatin-induced acute renal failure via a crystal-independent mechanism. Uric acid decreases the bioavailability of nitric oxide, increases afferent arteriolar vasoconstriction, and decreases the glomerular filtration rate. Uric acid causes preglomerular arteriolar smooth muscle proliferation and may interfere with autoregulation of the renal blood flow. The proinflammatory and prooxidative properties of uric acid can potentiate the inflammatory cascade, mediating acute renal failure.
Outcome of Acute Renal Failure of Critical Illness
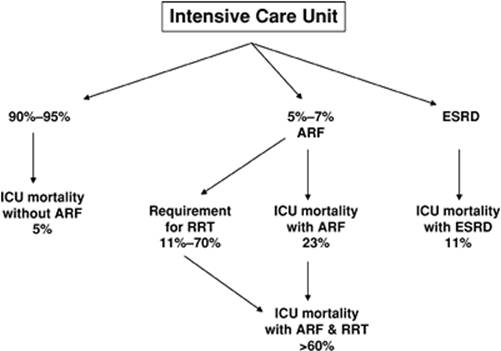
The outcome of acute renal failure of critical illness is of immense importance. The mean duration of in-hospital acute renal failure is 14 days. Most episodes resolve in the first month of evolution (7), and only 11% of the patients require renal replacement therapy (2). However, the requirement for renal replacement therapy increases to over 70% when acute renal failure is severe, that is, in the presence of oliguria or severe azotemia (3,8). The usual ICU mortality approximates 5% without acute renal failure, 23% with acute renal failure, and over 60% with acute renal failure requiring renal replacement therapy (Fig. 47.3). Of the patients with acute renal failure who expire, 78% do so within 2 weeks after the renal insult. The 90-day and 1-year survival of those who are discharged from the hospital are 64% and 50%, respectively (8). Interestingly, the ICU mortality of patients with end-stage renal disease is 11%, much lower than for acute renal failure patients who do not need dialysis support (2). The increased mortality associated with the acute decline in renal function is not explained simply by loss of organ function.
|
|
|
Figure 47.3. Outcome of acute renal failure of critical illness. |
The recovery of renal function is influenced by many factors, including pre-existing chronic illness. In one review, only 41% of the patients were reported to be in good health 3 months before entry into the intensive care unit (9); chronic kidney disease has been reported in 30% of all patients admitted to the intensive care unit (3,8,9). Most of the survivors of acute renal failure recover their renal function within 2 weeks, and 65% to 94% of them have independent renal function at discharge from the hospital (3,8,10).
Pathophysiology of Chronic Kidney Disease
Epidemiology
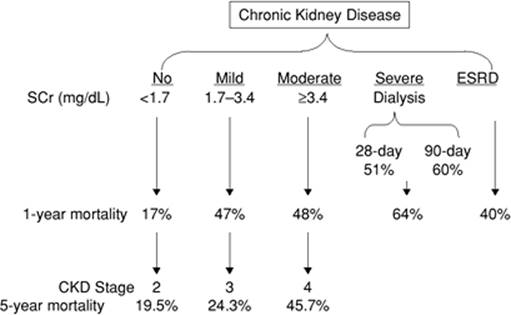
An increasingly elderly population with pre-existing renal dysfunction is treated in our intensive care units. The presence of chronic renal disease on admission to the intensive care unit is associated with an incremental increase in long-term mortality in survivors of acute renal failure (Fig. 47.4). Furthermore, recovery from acute renal failure is often accompanied by residual renal dysfunction and the perils associated with chronic kidney disease. Chronic kidney disease affects 12% of the U.S. population (11). The prevalence of chronic kidney disease is relatively stable, but the number of end-stage renal disease patients who require maintenance dialysis treatments continues to grow. It is estimated that the number of end-stage renal disease treatments will increase 60% by the year 2010. One reason for the discrepancy between the size of the chronic kidney disease pool and the incidence of end-stage renal disease may be the premature cardiovascular death in many patients before progression to end-stage renal disease. In fact, chronic kidney disease has a five to ten times higher risk of death than end-stage renal disease (12). The 5-year mortality of patients with chronic kidney disease stages 2, 3, and 4 are 19.5%, 24.3%, and 45.7%, respectively; 1.1%, 1.3%, and 19.9% progress to renal replacement therapy, respectively (13). The above data underscore the impact of pre-existing organ dysfunction on the prognosis of critically ill patients.
|
|
|
Figure 47.4. Effect of the stage of chronic kidney disease on ICU mortality. |
Theories of Chronic Kidney Disease
The U.S. National Kidney Foundation's Kidney Disease Outcomes Quality Initiative classification of chronic kidney disease (Table 47.2) facilitates the development of appropriate management plans but does not provide information on the future risk of decline in renal function. Once renal damage reaches a certain threshold, the progression of renal damage is consistent, irreversible, and independent of initial insult. The characteristic histologic findings of tubular atrophy, interstitial fibrosis, and glomerulosclerosis in chronic kidney disease of diverse causes suggest that multifactorial and complex interactions between numerous pathways of cellular damage by both cellular and humoral pathways contribute to their progression to a common final pathway. Brief overviews of the proposed mechanisms that are involved in the progression of chronic kidney disease are provided below.
|
Table 47.2 Definition and classification of chronic kidney disease (CKD) |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
The Hyperfiltration Theory
The hyperfiltration theory (14) emphasizes glomerular hemodynamic changes as the final common pathway in progressive chronic kidney disease. Accordingly, renal injury from diverse causes results in hyperfunction of the remaining glomeruli. The sustained elevations in glomerular pressures and flows favor hyperfiltration and the loss of selectivity of the permeability of the glomerulus. The ensuing proteinuria causes tubule cell injury by “misdirected filtration”—the accumulation of glomerular filtrate outside of the Bowman space and into periglomerular space, creation of a cellular cover around the focus of misdirected filtration by interstitial fibroblasts, extension over the entire glomerulus leading to global sclerosis (15), luminal obstruction, and overwhelming the tubular mechanism by excessive uptake and degradation of filtered protein by the proximal tubule cells. The subsequent extravasation of the accumulated filtered plasma protein into the interstitium causes inflammatory reaction and tubulointerstitial fibrosis (16).
The Complement Activation Theory
The complement activation theory maintains that the proteinuria-induced intraluminal activation of the terminal complement cascade, leading to the formation of C5b-9 membrane attack complex, is the principal mediator of chronic progressive interstitial damage and progressive renal failure in proteinuric renal disease, irrespective of primary glomerular injury. This is supported by the demonstration of increased urinary excretion of C5b-9 in nonimmunologic glomerular injury, correlation of tubulointerstitial deposition of the C5b-9 with interstitial myofibroblast accumulation and proteinuria, and the observation that, in experimental models, progressive interstitial injury was maintained only in hypocomplementemic animals.
The Chronic Hypoxia Theory
The chronic hypoxia theory emphasizes chronic ischemic damage in the tubulointerstitium as a final common pathway in end-stage renal injury. The extent of tubulointerstitial damage is better correlated with impaired renal function than the degree of glomerular injury. The countercurrent arrangement of the descending postglomerular vessels and ascending vasa recta vessels, elevated vasa recta permeability to oxygen, and high metabolic requirement results in a graded fall in oxygen tension from the outer cortex to the inner medulla. In extensive tubulointerstitial injury, the loss and distortion of peritubular capillaries, increased transposition of extracellular matrix, vasoconstriction, glomerular damage, anemia, and oxidative stress impair oxygen supply to the corresponding regions. Hypoxia leads to apoptosis or epithelial-mesenchymal transdifferentiation and exacerbates fibrosis of the kidney and subsequent hypoxia, setting in motion the vicious cycle to end-stage renal disease (17).
Elevated Asymmetric Dimethylarginine
The endothelium plays a crucial role in the maintenance of vascular tone and structure. Endothelium produces nitric oxide, a crucial mediator of vasodilation, inhibition of vasoconstrictor influences, antithrombosis, anti-inflammation, and antiproliferation. The generation of nitric oxide by nitric oxide synthase is inhibited by asymmetric dimethylarginine (ADMA). Elevated plasma ADMA levels are inversely related to GFR and significantly associated with progression of chronic kidney disease (18,19). Elevated ADMA in chronic kidney disease is not due to impaired urinary clearance, but to increased ADMA generation (synthesized by protein methyltransferase) and decreased degradation (mainly by dimethylarginine dimethylaminohydrolase). It is speculated that uremic oxidative stress is involved in the dysregulation of protein methyltransferase and dimethylarginine dimethylaminohydrolase (20).
Anemia
Erythrocytes represent a major antioxidant component of blood. The generation of oxidants is amplified in anemia; it is also enhanced in chronic kidney disease due to increased oxygen consumption by the remnant hyperfunctioning nephrons. Hypoxia of the tubular cells due to decreased delivery of oxygen may be the main link between interstitial fibrosis and tubular destruction. Hypoxia stimulates the production of extracellular matrix by tubular cells and renal interstitial fibroblasts and the release of profibrotic cytokines. Treatment with erythropoetin increases red blood cell mass, improves red blood cell survival by antiapoptotic effects, and decreases oxidative stress. Whether the correction of anemia can decrease the progression of chronic kidney disease remains to be seen (21).
Microvascular Endothelium
Renal microvasculature is maintained by the balance of angiogenic growth factors, alteration of which impairs capillary repair, causes loss of microvasculature, and leads to a decrease in glomerular filtration rate, and oxygen and nutrient supply to the tubules and interstitial cells. The progressive loss of the endothelium results in capillary collapse and development of glomerulosclerosis, impaired blood flow, and tubulointerstitial fibrosis. Increased expression of thrombospondin-1 and other antiangiogenic factors, and decreased expression of vascular endothelial growth factor (VEGF) and other proangiogenic factors, influence the renal microvasculature in progressive chronic kidney disease. VEGF expression correlates with the severity of peritubular capillary loss and inversely correlates with the degree of tubulointerstitial inflammation; it is inhibited by macrophage-derived inflammatory cytokines (IL-10, IL-6, TNF-α) and angiotensin II, and modulated by nitric oxide—factors involved in the pathogenesis of renal injury (22).
Impact of Chronic Kidney Disease on Critical Illness
Chronic kidney disease is associated with a decrease in structural and functional reserve. The renal capacity to autoregulate renal blood flow; maintain normal systemic blood pressure; excrete solutes, fluids, electrolytes, and the daily acid load; and metabolize drugs is diminished (Table 47.3). The reduction in physiologic reserves influence drug dosing, treatment modality, and response to interventions in patients in the intensive care unit.
Blood Pressure
Some degree of autonomic dysfunction occurs in most patients with moderate to severe chronic kidney disease. The presence of autonomic dysfunction impairs the patient's ability to maintain the systemic blood pressure, which may complicate dialysis treatment of patients in the ICU who require removal of large quantities of fluid. Despite the significant down-regulation of alpha and beta adrenergic receptors, plasma catecholamine levels are often elevated in these patients and may increase the risk of cardiac complications. Uremic pericardial effusion may also cause vasopressor-resistant hypotension that, if unrecognized, may have dire consequences for the patient, as the onset of cardiac tamponade can be rapid without premonitory signs.
|
Table 47.3 Impact of Chronic Kidney Disease on other organ systems |
||
|
Cardiac Dysrhythmias
Cardiac dysrhythmias are common in patients with chronic renal failure due to underlying left ventricular hypertrophy, calcific cardiomyopathy that involves the conducting tissues, a “disturbed” metabolic milieu, and chronic tissue hypoxia. An increased incidence of cardiac dysrhythmias is seen in patients with postoperative acute renal failure that may be exacerbated with rapid fluctuations in hemodynamics and electrolyte concentrations in those requiring dialysis support.
Chest Pain
The presence of renal dysfunction can influence the symptoms, manifestations, and progression of coronary syndromes. Chronic kidney disease affects outcome in patients with acute coronary syndrome and is an independent risk factor for the development of coronary artery disease and for more severe coronary heart disease. Chronic kidney disease is also associated with an adverse effect on prognosis from cardiovascular disease. Many dialysis patients with angina often have a fairly typical history of exercise-induced chest discomfort that is similar to those with normal renal function. However, silent myocardial ischemia is also common among patients with severe kidney disease. It has been speculated that the extremely poor prognosis among dialysis patients with an acute myocardial infarction may be due in part to a relatively increased number of atypical clinical presentations, resulting in both underdiagnosis and undertreatment. The presence of dyspnea alone due to an acute myocardial infarction in an individual scheduled to undergo a regular chronic dialysis procedure may be mistakenly attributed to volume overload. In addition, baseline abnormalities on the electrocardiogram, such as left ventricular hypertrophy, may mask characteristic changes with ischemia.
Respiratory Failure
Most patients with acute respiratory failure on mechanical ventilation require some form of renal replacement therapy. Conversely, alterations in respiratory drive, mechanics, muscle function, and gas exchange are frequent consequences of uremia. The development of acute renal failure predisposes patients to overall fluid overload, decreased plasma oncotic pressure, and leakage of fluid from pulmonary capillaries. The restrictive effects of pulmonary interstitial and alveolar edema, pleural effusion, and chest wall edema increase the work of spontaneous breathing and may contribute to the development of acute ventilatory failure. In addition, the metabolic acidosis present in most instances of acute renal failure increases the demand for ventilation through compensatory respiratory alkalosis, further disrupting the relationship between the patient's ventilatory needs and capabilities. Pulmonary edema and ventilation at low lung volumes can cause or worsen hypoxemia.
Acute renal failure can necessitate several modifications in the management of mechanical ventilation. Higher airway pressure is required to maintain the same level of ventilation in the presence of pulmonary edema, pleural effusion, or total-body fluid overload. Airway mucosal edema can reduce effective airway diameter, predisposing to air trapping and intrinsic positive end-expiratory pressure, which can reduce venous return, further compromising cardiac function and increasing the risk of alveolar rupture. The management of acute lung injury and acute respiratory distress syndrome using lung-protective ventilation is made more difficult in the presence of metabolic acidosis, which increases ventilatory drive and worsens acidemia related to permissive hypercapnia.
Disordered Consciousness
Uremic encephalopathy is an organic brain disorder that develops in patients with acute or chronic renal failure, usually when creatinine clearance falls, and remains, below 15 mL/min. Accumulation of toxins, increases in intracellular concentration of calcium in brain cells, and imbalances of neurotransmitter amino acids within the brain are thought to be responsible, although urea itself is not thought to be causative. Clinical manifestations vary with worsening uremia, but prompt identification and initiation of dialysis treatment can readily reverse the symptoms. Initiation of dialysis treatment can also lead to disordered consciousness, especially when advanced states of uremia are dialyzed for excessive lengths of time during their first treatment sessions—the dialysis disequilibrium syndrome.
Decreased Metabolism of Drugs
The pharmacokinetics of most drugs are altered in renal dysfunction; their clearances are impaired or they accumulate in tissues and continue to exert their effects long after their administration. Some are broken down into their metabolites with deleterious consequences. Most of the drugs are not removable by dialysis due to their high protein binding. Many effective drug therapies cannot be used because of the risk of accumulation and toxicity. Some drugs are removed by dialysis and require postdialysis supplementation. The varying clearances of the different continuous renal replacement therapies mandate the knowledge of the clearance of the particular modality used to effectively dose a particular drug.
General Principles of Management
Despite the remarkable progress achieved in understanding the pathophysiology of acute renal failure, no specific pharmacologic agent has yet been approved for its treatment and prevention remains the principle element in its management.
The treatment of established acute renal failure is based on the following principles:
· The preservation of renal blood flow and optimal perfusion pressure favorably influences the deterioration of renal function.
· Correction of uremia, electrolyte, acid-base, endocrine, hematologic and nutritional disorders, and hypervolemia can favorably affect outcome
· The pharmacokinetics and clearance of drugs are altered in renal failure, and the appropriate dosage adjustment requires knowledge of the pharmacokinetic parameters of the drugs and clearance characteristics of the different renal replacement techniques.
· The treatment and complications of secondary causes of acute renal failure may determine its outcome.
· The appropriate integration of care provided by intensivists and organ specialists can favorably affect outcome.
In certain clinical situations, such as in cardiovascular surgery, ischemic acute renal failure is strongly associated with occult renal ischemia—associated with poor cardiac performance, fixed atherosclerotic disease of the renal arteries and/or prolonged hypoxemia, and reduced renal functional reserve. Due to the silent nature of renal ischemia, prognostic stratification using reliable surrogates can guide clinical decision making (23,24,25,26). Recently, the use of atrial natriuretic peptides has been shown to improve dialysis-free survival in thoracic aortic aneurysm surgery patients with impaired renal function. However, the use of atrial peptides in acute renal failure remains controversial—two major clinical trials have reported unfavorable outcomes whereas two smaller clinical trials have shown favorable outcomes.
The prevalence of acute renal failure continues to rise; however, there are indications that the mortality of patients with acute renal failure may be declining. This decline in mortality is not due to the effects of newer drugs in the treatment of acute renal failure, but rather, it is due to the increased cooperation between the intensivists and subspecialists, which has led to a concerted approach to treatment. This has resulted in increased awareness of disease states, early initiation and higher doses of dialysis treatments, maintenance of euglycemia, and other interventions that play important roles in reversing mortality.
References
1. Xue JL, Daniels F, Star RA, et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol. 2006;17:1135–1142.
2. Clermont G, Acker CG, Angus DC, et al. Renal failure in the ICU: comparison of the impact of acute renal failure and end-stage renal disease on ICU outcomes. Kidney Int. 2002;62:986–996.
3. Uchino S, Kellum JA, Bellomo R, et al; Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–818.
4. Lundberg S. Renal function during anaesthesia and open-heart surgery in man. Acta Anaesthesiol Scand Suppl. 1967;27:1–81.
5. Kelleher SP, Robinette JB, Conger JD. Sympathetic nervous system in the loss of autoregulation in acute renal failure. Am J Physiol. 1984;246:F379–386.
6. Ejaz AA, Mu W, Kang DH, et al. Could uric acid have a role in acute renal failure? Clin J Am Soc Nephrol. 2007;2(1):16–21.
7. Liano I, Liaño F, Pascual J; the Madrid ARF Study Group. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Kidney Int. 1996;50:811–818.
8. Hegarty J, Middleton RJ, Krebs M, et al. Severe acute renal failure in adults: place of care, incidence and outcomes. QJM. 2005;98:661–666.
9. Silvester W, Bellomo R, Cole L. Epidemiology, management, and outcome of severe acute renal failure of critical illness in Australia. Crit Care Med. 2001;29:1910–1915.
10. Bagshaw SM, Laupland KB, Doig CJ, et al. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care. 2005;9:R700–709.
11. Coresh J, Byrd-Holt D, Astor BC, et al. Chronic kidney disease awareness, prevalence, and trends among U.S. adults, 1999 to 2000. J Am Soc Nephrol. 2005;16:180–188.
12. Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305.
13. Keith DS, Nichols GA, Gullion CM, et al. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–663.
14. Brenner BM, Meyer TW, Hostetter TH. Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med. 1982;307:652–659.
15. Kriz W, Elger M, Hosser H, et al. How does podocyte damage result in tubular damage? Kidney Blood Press Res. 1999;22:26–36.
16. Zandi-Nejad K, Eddy AA, Glassock RJ, et al. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int. 2004;92:S76–89.
17. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25.
18. Fliser D, Kronenberg F, Kielstein JT, et al. Asymmetric dimethylarginine and progression of chronic kidney disease: the mild to moderate kidney disease study. J Am Soc Nephrol. 2005;16:2456–2461.
19. Ravani P, Tripepi G, Malberti F, et al. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: a competing risks modeling approach. J Am Soc Nephrol. 2005;16:2449–2455.
20. Matsuguma K, Ueda S, Yamagishi S, et al. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J Am Soc Nephrol. 2006;17:2176–2183.
21. Rossert J, Fouqueray B, Boffa JJ. Anemia management and the delay of chronic renal failure progression. J Am Soc Nephrol. 2003;14:S173–177.
22. Kang DH, Kanellis J, Hugo C, et al. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 2002;13:806–816.
23. Chertow GM, Lazarus JM, Christiansen CL, et al. Preoperative renal risk stratification. Circulation. 1997;95:878–884.
24. Chawla LS, Abell L, Mazhari R, et al. Identifying critically ill patients at high risk for developing acute renal failure: a pilot study. Kidney Int. 2005;68:2274–2280.
25. Yegenaga I, Hoste E, Van Biesen W, et al. Clinical characteristics of patients developing ARF due to sepsis/systemic inflammatory response. Am J Kidney Dis. 2004;43:817–824.
26. Vivino G, Antonelli M, Moro ML, et al. Risk factors for acute renal failure in trauma patients. Intensive Care Med. 1998;24:808–814.