Anand Kumar
Aseem Kumar
Septic shock (shock due to infection) and sepsis-associated multiple organ failure are the dominant cause of death in intensive care units of the industrialized world. As many as 800,000 cases of sepsis are admitted every year to American hospitals (comparable to the incidence of first myocardial infarctions), with half of those developing septic shock (1). Historically, the mortality associated with sepsis and septic shock has been approximately 50% to 75% (2,3,4). The major advance in the therapy of septic shock was the development of antibiotic therapy 50 years ago, which resulted in a reduction in sepsis-associated mortality in the 30% to 50% range (2,3). However, the past 40 years have seen a gradual year-to-year increase in the incidence of sepsis (5). As a result, total deaths in the United States have increased even though the overall mortality rate has fallen from 27.8% to 17.9% during that period (5). At present, the total death toll from sepsis is comparable to that from myocardial infarction and far exceeds the impact of illnesses such as acquired immune deficiency syndrome (AIDS) or breast cancer (1,6).
The total number of cases continues to gradually increase due to a burgeoning population of patients with a chronic and high degree of susceptibility to infection (age, AIDS, organ failure with transplant, and other chronic illness); an increased use of invasive medical devices; and increased use of cytotoxic agents for autoimmune disease, transplants, and malignancy for patients at high risk for sepsis. Current estimates suggest a doubling of total United States cases by 2050 but with only a projected increase in population of 33% (1). Until recently, despite major advances in technology and constant refinement of our understanding of sepsis pathophysiology, numerous clinical trials have failed to produce any new drugs with consistent beneficial effects on this patient population. Nonetheless, the last 50 years have seen a gradual improvement in mortality, perhaps related to improvements in supportive care (5,7).
Definitions
Derived from the Greek word “sepo,” meaning “I rot,” the first introduction of the term sepsis occurs in the poems of Homer (circa eighth century B.C.) (8). Over the intervening 2,700 years, through Homer, Hippocrates, Aristotle, and Galen to current-day physicians, the term has continued to be used virtually unchanged in meaning. Hugo Schottmüller modernized the term with his 1914 definition, “Septicemia is a state of microbial invasion from a portal of entry into the blood stream which causes signs of illness” (9). From the time of Schottmüller's definition of septicemia until recent years, terms such as septicemia, sepsis, toxemia, and bacteremia were all used interchangeably to indicate patients exhibiting systemic responses to infection.
A significant problem with the term septicemia (as defined by Schottmüller) is that most patients with a septic response cannot be documented to have bacteremia/fungemia, and many with bacteremia/fungemia (e.g., endocarditis, catheter-related infection) do not exhibit overt sepsis. Recognizing that future large-scale clinical trials of novel sepsis therapies will require more consistent and precise definitions of the septic response, consensus definitions were developed in 1991 (10). These criteria were developed primarily as a tool to enhance the ability to perform clinical sepsis research. However, the terminology soon entered the clinical lexicon. These consensus definitions were revised in 2001 to accommodate the clinician's perspective (11). Current and previous definitions follow.
Infection:
A microbial phenomenon characterized by an inflammatory response to the presence of micro-organisms or the invasion of normally sterile host tissue by these organisms.
Bacteremia:
The presence of viable bacteria in the blood. The presence of other organisms in the blood should be described in like manner—viremia, fungemia, and so on. Bacteremia can either be transient, sustained, or intermittent.
Systemic Inflammatory Response Syndrome (SIRS):
The systemic inflammatory response to various severe clinical insults, including but not limited to infection. Various other clinical insults include pancreatitis, ischemia, multiple trauma and tissue injury, hemorrhagic shock, immune-mediated organ injury, and exogenous administration of inflammatory mediators such as tumor necrosis factor or other cytokines. Previous criteria for SIRS are enumerated in Table 57.1. The more recent revision to sepsis definitions removed these SIRS criteria while retaining the concept. However, some understanding of these criteria remains crucial for the intensivist/clinical researcher, as most trials in the last 15 years have been predicated on patients having three or more of these criteria.
Sepsis:
The systemic response to infection. This response is similar to SIRS, except that it is considered to result from an infection. The previously accepted definition required at least two of the four SIRS criteria in the presence of documented or suspected infection. The recent revision of the criteria enumerates multiple potential diagnostic criteria for sepsis (Table 57.2) and no longer specifically requires the discarded elements of the SIRS criteria.
Severe Sepsis:
Sepsis associated with organ dysfunction, perfusion abnormalities, or hypotension. Organ system dysfunction can be described by organ failure scoring systems (12,13).
Septic Shock:
Sepsis with hypotension despite adequate fluid resuscitation, in conjunction with perfusion abnormalities.
Standard abnormalities in an adult include mean arterial pressure (MAP) <60 mm Hg, systolic blood pressure <90 mm Hg, or a drop in systolic blood pressure >40 mm Hg from baseline.
|
Table 57.1 Definition of Systemic Inflammatory Response Syndrome (SIRS) |
||
|
Multiorgan Dysfunction Syndrome (MODS):
The presence of altered organ function in an acutely ill patient, such that homeostasis cannot be maintained without intervention. Primary MODS is the direct result of a well-defined insult in which organ dysfunction occurs early and can be directly attributable to the insult itself. Secondary MODS develops as a consequence of a host response and is identified within the context of SIRS.
The relationship of many of these conditions to each other is demonstrated in Figure 57.1. An understanding of sepsis definitions has become increasingly important since most clinical trials in the last two decades have used the modified version of the 1991 sepsis definitions (usually requiring three rather than two SIRS criteria) in their entry criteria. The concept of a compensatory anti-inflammatory response has also been introduced after the demonstration that traditional anti-inflammatory mediators were also elevated during sepsis (14).
Epidemiology
Although the sepsis syndromes (from sepsis to septic shock) have been a major burden on human health in both the developed and undeveloped world, there has been a surprising dearth of epidemiologic information. In North America, this has been caused by the earlier lack of consensus definitions of these syndromes and, more recently, the absence of syndrome-specific diagnostic codes for sepsis within the International Classification of Disease (ICD) coding system. In the last 20 years, the development of consensus definitions and application of computerized hospital and government administrative databases has allowed substantial insight into the problem.
Martin et al. (5) have estimated 660,000 annual cases of sepsis in the United States during 2000 (adjusted rate 240/100,000 population) using an analysis of ICD-9 codes associated with National Hospital Discharge Survey data. With the exception of a single major study with much higher values (1), estimates for severe sepsis from sites across North America and Europe have been fairly consistent at 50 to 80/100,000 population (15,16,17,18,19). These cases account for approximately 10% to 15% of all intensive care unit (ICU) admissions (16,17,19,20,21). Approximately 25% of cases of sepsis (22) and 50% to 75% of cases of severe sepsis progress to septic shock (20). Septic shock represents between 5% and 8% of all ICU admissions (21,23). In the United States, the cost of sepsis and severe sepsis ranges from $22,000 to $60,000 per episode at a total cost of approximately $17 billion annually (1,24). Sepsis and related conditions are the tenth leading cause of death in the United States (6).
|
Table 57.2 Revised Diagnostic Criteria for Sepsis |
|||||||
|
|||||||
|
|
|||||||
|
Figure 57.1. Venn diagram showing the relationship between infection and other sepsis-associated terms. The intersection of systemic inflammatory response syndrome (SIRS) and infection defines sepsis. Severe sepsis is a subset of sepsis defined by the presence of organ failure. Septic shock is a subset of severe sepsis in which the organ failure is cardiovascular (i.e., shock). Patients with certain inflammatory conditions (e.g., extensive burn injury, pancreatitis, major trauma, postpump syndrome, and so on) may demonstrate a “septic” appearance (i.e. SIRS) without the presence of infection required for a diagnosis of sepsis. (Adapted from Bone R. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–874.) |
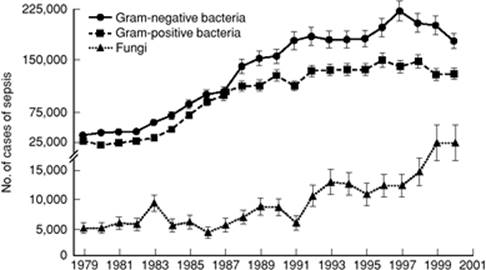
The incidence of sepsis appears to be increasing at a rate of about 9% per year in the United States (5) (Fig. 57.2). Reasons for this increase include the following: (a) An aging population with increased predisposition to illness; (b) increased proportion and longevity of the subpopulation with conditions that predispose to systemic infection including chronic organ failure (e.g., cirrhosis, renal failure, cardiomyopathy, chronic obstructive pulmonary disease [COPD]), and other conditions (e.g., diabetes, cancer, AIDS, etc.); (c) extensive use of invasive diagnostic and therapeutic modalities (indwelling catheters and devices), which lead to breakdown of native resistance to infection; and (d) widespread use of immunosuppressive chemotherapies for a wide range of diseases (asthma, inflammatory bowel disease, rheumatoid arthritis, systemic lupus erythematosus, and other autoimmune diseases).
|
|
|
Figure 57.2. Incidence of sepsis in the United States stratified by organism group. The incidence of sepsis increased approximately 9% per year between 1979 and 2001 with the greatest relative increase in fungal infections. In addition, as of the late 1980s, Gram-positive pathogens became numerically dominant over Gram-negative organisms. (From Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–1554. Copyright © 2003 Massachusetts Medical Society. All rights reserved.) |
Age is a substantial risk factor for sepsis, severe sepsis, and septic shock (1,5,25). Patients older than the age of 65 years are approximately 13-fold more likely to develop sepsis compared to others (5). Similarly, septic shock is 18 times more likely in the >80-year age group compared to those in the 20- to 29-year age group (23). Given that the average age of the North American population is increasing, and that the incidence of all the sepsis-related syndromes is markedly elevated in the elderly (23), the fact that the average age of patients with sepsis has climbed over the last few decades can be no surprise (1,5). That septic shock is substantially a geriatric illness is reflected in the median age of 67 years (25). The persistent 60:40 male:female preponderance in sepsis, severe sepsis, and septic shock may have its origins in men's increased predisposition to smoking-associated cases of pneumonia and peptic ulcer disease/gastrointestinal malignancy-associated gastric and bowel perforation (1,5,17,20,22,23). Nonwhite racial groups are also at substantially increased risk, particularly African Americans (5). However, low socioeconomic status is a substantial risk factor for septic shock (a fourfold increased risk in the lowest quintile of income compared to any other quintile) (23). In this context, it is unclear whether race may be relevant only as a marker of socioeconomic status. Comorbidities are common in patients with sepsis, as might be expected given an average age of 55 to 65 years for sepsis and perhaps higher for septic shock (5,19,25,26,27,28,29). Diabetes, COPD, renal failure, congestive heart failure, and malignancy can each be found in 10% to 20% of patients with sepsis or septic shock. At least 50% of patients with severe sepsis have at least one major medical comorbidity (5). Patients with septic shock have an even higher incidence (>90%) of major comorbidities. Alcoholism and substance abuse also substantially increases the risk of sepsis, as well as death from sepsis and septic shock (30).
As might be expected, mortality increases with the severity of the septic syndrome. Mortality is <15% for sepsis, 25% to 50% for severe sepsis, and >50% for septic shock (1,5,15,16,17,20,21,22,25,31). This mortality rate for septic shock, while staggering, nevertheless represents an improvement in survival from 35 years ago when mortality rates frequently exceeded 80% (32,33). Early septic mortality (<3 days) appears to be associated most closely with shock with other deaths within the first week due to multiple organ failure. Later deaths tend to be most closely associated with pre-existing comorbidities (34). Of those succumbing to septic shock, approximately 75% are early deaths (within 1 week of shock), primarily due to hyperdynamic circulatory failure (35).
Throughout recorded history, there has been an evolution of the organisms that cause infectious diseases and the associated clinical syndromes. This phenomenon has become particularly pronounced since the advent of antibiotics in the last half of the previous century. By the 1960s and 70s, Gram-negative organisms had become the dominant pathogens over Staphylococcus aureus and streptococci. During the 1980s, resistant Gram-positive organisms (methicillin-resistant S. aureus, coagulase-negative staphylococci, penicillin-resistant S. pneumoniae, and enterococci) again re-emerged as major pathogens. Gram-positive cocci account for approximately 40% to 50% of single isolates (excluding fungi) in sepsis and septic shock (20,25,31,36,37,38).
Most recently, yeast and other fungi have demonstrated a remarkable increase in their contribution to sepsis (5% of total) and septic shock (8.2% of total), with an increase of about 10% per year (5,25,37,38). Candida albicans remains numerically dominant (about 60% of total fungal infections), but fluconazole-resistant yeasts are the most rapidly increasing species (39,40,41). Other major concerns in recent years include the emergence of vancomycin-resistant enterococci (42), extended spectrum β-lactamase (ESBL) resistance in Gram-negative organisms (reliably sensitive only to carbapenems) (43), and an endemic strain of virulent, methicillin-resistant S. aureus in the community (44). In addition, concerns regarding sporadic cases of vancomycin-resistant S. aureus (VRSA) are growing (45).
Pathogenesis of Sepsis, Severe Sepsis, and Septic Shock
Sepsis and septic shock or sepsis-associated multiple organ failure typically begin with a nidus of infection within the body (e.g., pneumonia, peritonitis, urinary tract infection, abscess). Within that nidus, the organism replicates. Eventually, the infection at the inciting focus releases sufficient microbial antigens to elicit a systemic inflammatory response designed to eliminate the invading microbes (Fig. 57.3). Many constitutive and/or inducible elements of invasive microorganisms are capable of inciting the systemic inflammatory responses that result in sepsis and septic shock (Fig. 57.3, Table 57.3). Beyond endotoxin (lipopolysaccharide; LPS) of Gram-negative bacteria, other major triggers of the systemic inflammatory response characteristic of sepsis include various exotoxins (bacteria), peptidoglycans (streptococci), and teichoic acid (S. aureus); lipoarabinomannan of mycobacteria; and mannoproteins and β-glucan of fungi (46). Bacterial DNA may possess sufficient antigenic properties (based on unique CG repetitions and lack of deoxyribonucleic acid [DNA] methylation) to initiate a substantial inflammatory response independent of other bacterial elements (47,48,49). Bacterial ribonucleic acid (RNA) may be able to do the same (50). Recent investigations suggest a surprising commonality of signaling mechanisms in septic shock via Toll-like receptors from a broad range of etiologic agents (48,51,52,53,54).
Despite the large number of potential elements of pathogenic microorganisms that can drive the septic response, endotoxin of Gram-negative bacteria remains the prototype of such factors and the model for subsequent research. This antigen is thought to be central in initiating the powerful host response to infection with these organisms (55). LPS and other antigens interact with immune cells (particularly macrophages), resulting in the induction of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) secreted by monocytes, macrophages, and other cells (Fig. 57.3) (56). These cytokines initiate a complex signaling sequence involving the release of secondary mediators (platelet-activating factor, leukotrienes, prostaglandins) and monocytes, as well as endothelial tissue factor expression, inducible nitric oxide synthetase induction, microvascular coagulation, cell-adhesion molecule up-regulation, and apoptosis (57,58,59,60). To maintain homeostasis (and likely as part of a feedback mechanism), several anti-inflammatory mediators are also released, including interleukin-10 (IL-10), transforming growth factor-β (TGFβ), and interleukin-1 receptor antagonist (IL-1ra). If homeostasis cannot be maintained, progressive and sequential dysfunction of various organ systems (i.e., MODS) may occur. If the inflammatory stimulus is particularly intense, or if there is limited cardiovascular reserve, effects on the cardiovascular system as manifested by septic shock may dominate the clinical presentation.
Microbial Antigen Signaling
As the prototypical and best-studied microbial antigen, an understanding of the signaling cascade of endotoxin is instructive. Endotoxin is an amphiphilic macromolecule located on the outer cell wall membrane of Gram-negative bacteria. It is composed of lipid A (a diglucosamine-based acylated phospholipid), and a polysaccharide side chain (61,62) (Fig. 57.4). The polysaccharide chain is composed of a short, highly conserved, proximal section (core polysaccharide) and a highly variable, longer distal oligosaccharide side chain. The core polysaccharide and lipid A are sometimes referred to as the core glycolipid. The highly conserved lipid A moiety is the toxic element of endotoxin and can reproduce the manifestations of endotoxic shock when administered alone (62,63,64,65,66,67). As a circulating form in the plasma, endotoxin exists in a multimeric aggregate form.
Lipopolysaccharide-binding protein (LBP) is an acute phase reactant protein present in plasma (61,68,69). The levels increase with inflammatory stimulation. LBP catalyzes the transfer of endotoxin from serum aggregates to either serum lipoproteins, such as high-density lipoprotein (HDL), leading to endotoxin neutralization or to CD14 receptors (either membrane-bound [mCD14] or soluble [sCD14]), the putative primary LPS receptor (Fig. 57.5). The degree to which endotoxin is shunted through either pathway appears to play a significant role in the phenotypic physiologic response (46). LBP, by forming a complex with endotoxin monomers, appears to enhance the ability of endotoxin to bind CD14 and allows cellular activation at relatively low endotoxin concentrations (61,69).
Although LBP appears to be a specific carrier molecule for endotoxin, available data suggest that other microorganism toxins associated with sepsis may use similar carrier proteins (70,71).
|
|
||||||||||||||||||||||||||||||||||||||||
|
Figure 57.3. Pathogenesis of sepsis and septic shock. ATIII, antithrombin III; DNA, deoxyribonucleic acid; HMGB1, high mobility group box 1 protein; LPS, lipopolysaccharide; MIF, macrophage migration inhibitory factor; TFPI, tissue factor pathway inhibitor; TGF, transforming growth factor; Toxin A, Pseudomonas toxin A; TSST-1, toxic shock syndrome toxin 1. (Adapted from Parrillo JE. Pathogenic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477.) |
||||||||||||||||||||||||||||||||||||||||
|
Table 57.3 Elements of Microorganisms Capable of Inducing a Septic Response |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
CD14, a glycoprotein receptor, is found primarily in the cells of the myelomonocytic lineage (monocytes, macrophages, polymorphonuclear leukocytes) (72). Although there appear to be several other membrane-associated LPS receptors, membrane-associated CD14 (mCD14) represents the only receptor that is clearly involved in LPS binding and activation of cellular inflammatory responses. In contrast to the low endotoxin concentrations required to activate CD14 (an effect mediated by the LBP-LPS interaction [73]), other receptors such as CD18 appear to require exceptionally high concentrations of LPS to elicit a cellular effect, suggesting a lack of physiologic relevance (74).
|
|
|
Figure 57.4. Endotoxin (lipopolysaccharide). Endotoxin is a component of the cell wall of Gram-negative bacilli. (From Young LS, Martin WJ, Meyer RD, et al. Gram-negative rod bacteremia: microbiologic, immunologic, and therapeutic considerations. Ann Intern Med. 1977;86:456–471, with permission.) |
Recent data suggest that CD14, far from being uniquely a receptor for LPS, may also bind ligands from various pathogens, including peptidoglycan and lipoteichoic acid of Gram-positive bacteria, lipoarabinomannan of mycobacteria, and chitin of fungi (Table 57.4) (46,75). In several of these, binding is serum dependent, suggesting the possibility of serum carrier/binding proteins similar to LBP (70). This convergence of receptor-signaling mechanisms may explain why downstream intracellular signaling events (activation of NF-κB, MAP kinases, etc.) and cellular responses (cytotoxicity, cytokine generation, etc.) appear to be so highly conserved in sepsis due to different etiologic agents. Although elements of different microorganisms bind and activate CD14, limited data suggest that the precise binding sites vary.
Despite the importance of CD14, the receptor lacks the ability to initiate intracellular signaling on its own because of the lack of an intracytoplasmic-signaling domain. CD14 signaling requires the involvement of the most recently discovered (and most central) element of microbial antigen-mediated signal transduction, the Toll-like receptors (TLRs) (52,76,77,78,79). The original Toll receptor was initially described as an essential component of embryogenesis of Drosophila (80). In mammals, various TLRs have been shown to play a crucial role in the recognition of microbial antigens and initiation of the immune response. TLR4, and to a lesser extent TLR2, have been implicated in signaling associated with endotoxin (53,77,78,79,81). TLR4 appears to be coexpressed and forms a plasma membrane complex with mCD14. mCD14 appears to bind with the LPS/LBP complex to enable transfer to TLR4 and an accessory protein, MD-2 (82). mCD14, acting as a receptor for other non-LPS microbial antigens, also appears to have a role in TLR2 signaling (83). The exact nature of the CD14-TLR interaction is as yet undetermined. However, interaction of CD14 and TLR4 stimulates downstream activity of the intracellular domain of TLR to generate NF-kB and other intracellular mediators that drive the response to LPS (Fig. 57.5). Notably, the intracellular domain of the TLRs is shared with the IL-1 receptor. Several other TLR receptors are known to be involved in microbial antigen signaling from various pathogens, including Gram-positive and Gram-negative bacteria, fungi, mycobacteria, and viruses (Table 57.5).
|
|
|
Figure 57.5. Endotoxin signaling pathway related to CD14 and TLR4 (Toll-like receptor 4). IκB, inhibitory κB; IKK, IκB kinase; IRAK, IL-1R–associated kinase; LBP, lipopolysaccharide-binding protein; LPS, lipopolysaccharide; MYD88, myeloid differentiation factor; NFκB, nuclear factor-κB; NIK, nuclear factor κB–inducing kinase; TRAF 6, tumor necrosis factor receptor associated factor. |
Besides the Toll-like receptor pathways, other important routes of microbial antigen signaling exist. In particular, some Gram-positive organisms produce potent exotoxins that are implicated in the pathogenesis of toxic shock syndromes. These include the toxic shock syndrome toxin-1 associated with staphylococcal toxic shock and pyrogenic toxins predominantly associated with group A streptococci. These exotoxins appear to be superantigens in that they are able to activate broad polyclonal groups of lymphocytes, resulting in massive cytokine generation and toxic shock (84,85).
Cytokines
The concept of a systemic inflammatory response syndrome (SIRS) has already been discussed in the context of sepsis. The notion of an innate anti-inflammatory response, termed compensatory anti-inflammatory response syndrome (CARS), during sepsis also exists (14). This model suggests that a clinical insult (such as infection or injury) initiates a proinflammatory response that is countered by an endogenous anti-inflammatory reaction. The aggregate responses produce endogenous circulating mediators (cytokines, soluble receptors, adhesion molecules, growth factors, eicosanoids, etc.), generating systemic phenomena such as septic shock or immunosuppression. Clinical manifestations and patient outcome are dependent on the balance between proinflammatory and anti-inflammatory elements. The predominance of the inflammatory response corresponds to SIRS and may lead to cardiovascular compromise, shock, and organ dysfunction. However, a predominance of anti-inflammatory mediators produces a state of immune paralysis associated with a propensity to infection and inability to fight infection. Both may ultimately lead to death. In patients with sepsis, the duration of monocyte inactivation (a potential manifestation of CARS) correlates with mortality (86). If the counterinflammatory response is able to balance the inflammatory stimuli (while the infecting micro-organism is effectively cleared), homeostasis is achieved and clinical recovery will occur. In this model, sepsis has a dynamic nature based on the development and balance of the above-described responses (Fig. 57.6). This interplay is influenced by the nature of the inflammatory injury and the genetically determined variability of the host immune response (87,88).
|
Table 57.4 CD14 Binding-Capable Microbial Products |
||||||||||||||||||||
|
Proinflammatory cytokines have multiple effects, including the stimulation of production and release of other proinflammatory mediators. TNF-α, interleukin-1β (IL-1β), and interleukin-6 (IL-6) are the best known proinflammatory cytokines and have overlapping and synergistic effects in stimulating the inflammatory cascade. The next phase in the cytokine response to infection is the endogenous counterinflammatory cascade in response to the systemic activity of proinflammatory cytokines. Cytokine inhibitors (e.g., IL-1 receptor antagonist [IL-1ra], soluble TNF receptor) and anti-inflammatory cytokines (e.g., TGFβ, IL-4, IL-10, and IL-13) are involved in this phase of the response. Other cytokines like HMGB1 may be involved even later in the syndrome. Thus, the cytokine network in sepsis involves proinflammatory cytokines, anti-inflammatory cytokines, and cytokine inhibitors (Table 57.6). It is the balance between these cytokines at different time points that determine the clinical manifestations and outcome of sepsis.
|
Table 57.5 Microbial Ligands of the Toll-like Receptors (TLRs) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Nitric Oxide
Another important mediator, nitric oxide (NO), has a vital role in normal intracellular signal transduction (89). NO is synthesized by a family of enzymes called NO synthases (NOS) that incorporate nitrogen from one of the guanidine terminals of L-arginine with molecular oxygen to form NO and L-citrulline. Three distinct nitric oxide synthases have been purified, cloned, and characterized: (i) Neuronal NOS or nNOS, (ii) inducible NOS or iNOS, and (iii) endothelial NOS or eNOS, reflecting the cell types from which they were originally identified.
NO has several important roles in infection, sepsis, and septic shock. The iNOS gene is induced in immunoactivated cells. NO formed by these cells plays a role in host defense against bacterial, viral, and protozoan infections. Of particular importance in relation to septic shock, nitric oxide is the mediator through which endothelial cells normally cause relaxation of adjacent smooth muscle (89). Endothelial cells, through eNOS, produce picomolar quantities of nitric oxide in response to several vasodilatory stimuli such as shear stress, acetylcholine, and bradykinin. This nitric oxide diffuses to adjacent smooth muscle and activates guanylate cyclase to produce cyclic GMP, which effects vascular relaxation. Activity of endothelial NOS is regulated and is calcium and calmodulin dependent.
|
|
|
Figure 57.6. The dynamic cytokine inflammatory response. Sepsis is associated with an early transient dominance of proinflammatory cytokines corresponding to the systemic inflammatory response syndrome (SIRS) and the onset of organ damage. After this initial phase, the anti-inflammatory pathways of CARS (compensatory anti-inflammatory response syndrome) become active with the development of a refractory state characterized by a decreased capacity of mononuclear cells to produce proinflammatory cytokines. Recovery occurs if homeostasis is re-established. (Adapted from van der Poll T, van Deventer SJ. Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am. 1999;13(2):413–426.). |
During septic shock, an iNOS capable of producing nanomolar quantities of nitric oxide is generated in endothelium and vascular smooth muscle (89,90). Following this generation, the activity of this iNOS is unregulated and constant. Nitric oxide–mediated generation of cyclic guanosine monophosphate (cGMP) explains the profound loss of arterial vascular tone and venodilatation seen in septic shock (90,91) and may, in part, explain the irreversible vascular collapse seen late in hemorrhagic shock (92) (Fig. 57.7). A potential role for NO in inflammation-associated edema and third-spacing during shock has also been suggested (93). The in vitro myocardial depressant effects of TNF-α, IL-1β, and serum from septic humans may be mediated by a similar NO- and cGMP-dependent pathway (94,95). TNF-α, IL-1β, and IFN-γ have been identified as key mediators of iNOS activation. An alternative pathway by which NO may play a role in the cardiovascular pathophysiology of shock and sepsis involves the production of peroxynitrite (ONOO-), a highly reactive oxidant, from the interaction of superoxide (OH-) and nitric oxide (NO-) (96).
|
Table 57.6 Major Proinflammatory and Anti-inflammatory Cytokines and Receptors in Sepsis |
|||||||||||||||||||||||||||||||||
|
Hemostasis
The coagulation cascade represents a highly conserved antimicrobial defense mechanism common to even the most primitive complex organisms, such as the Limulus horseshoe crab. The hemolymph of the horseshoe crab, one of the oldest complex organisms still in existence, clots rapidly in response to minute quantities of endotoxin or beta-(1,3) glucan, a component of fungi. Pathogens are immobilized in the clot, allowing subsequent elimination (97,98). This commonality of purpose and function of the coagulation and inflammatory systems in eliminating invading microbes has persisted in evolution to present-day mammals including humans (99). These systems, in sharing common activation pathways, are inextricably linked.
Although both these systems are normally highly adaptive in nature, excessive activity of the coagulation and inflammation pathways can result in vascular injury, aberrant tissue blood flow, tissue damage, and, ultimately, organ dysfunction. Recent clinical and laboratory investigations have established that, in conjunction with the cytokine cascade, the coagulation system plays a key role in inflammatory states such as sepsis (100,101,102) (Fig. 57.8). A critical process in sepsis-induced coagulopathy is the activation of the extrinsic pathway (100).
During the normal hemostatic response, exposure of blood to nonvascular cell-bound tissue factor in the subendothelial layer initiates the extrinsic pathway through the binding of tissue factor to activated factor VII. The resulting enzyme complex, in turn, activates factor IX of the intrinsic pathway and factor X of the common pathway. With factor V as a cofactor, activated factor X cleaves prothrombin to form thrombin. Thrombin then converts fibrinogen to fibrin, which results in clot formation (103).
|
|
|
Figure 57.7. Physiologic and pathophysiologic vasodilatory factors relevant in sepsis and septic shock. cGMP, cyclic GMP; eNOS, endothelial nitric oxide synthetase; IL-1, interleukin-1β; iNOS, inducible nitric oxide synthetase; NO, nitric oxide; ONOO-, peroxynitrite; PAF, platelet-activating factor; PGE2, prostaglandin E2; PGI1, prostacyclin; TNF, tumor necrosis factor-α. (Adapted from Kumar A, Parrillo JE. Shock: pathophysiology, classification and approach to management. In: Parrillo JE, Dellinger RP, eds. Critical Care Medicine: Principles of Diagnosis and Management in the Adult. 3rd ed. St. Louis, MO: Mosby; 2007:379–422.) |
In sepsis, however, the expression of tissue factor is either directly or indirectly induced by inflammatory cytokines. Overexpression of proinflammatory cytokines, such as TNF-α, IL-1β, and interleukin-8, are thought to upset the balance toward a procoagulant state (60,101,104) (Fig. 57.8). TNF-α and IL-1β, for example, can induce the expression of tissue factor in circulating monocytes and endothelial cells (101). The vascular endothelial injury resulting from inflammation can also further expose tissue factor in subendothelial tissue and perivascular cells. Endothelial injury also inhibits the production and activity of anticoagulants such as proteins C and S, the heparin–antithrombin complex, and thrombomodulin. Loss of native anticoagulant function is indicated by decreased activity and circulating levels of protein C (105,106), antithrombin III, (ATIII) (101,106), and tissue factor pathway inhibitor (TFPI) (107,108) in patients with severe sepsis and septic shock.
Current evidence suggests that the pathogenesis of sepsis is associated with (a) systemic activation of coagulation resulting in consumption of coagulant factors, (b) suppression of the anticoagulant system by the same proinflammatory mediators that activate coagulation, and (c) early activation followed by later suppression of fibrinolysis (60,101) (Fig. 57.8). Whereas the coagulation cascade is clearly activated in sepsis, the specific inciting events and the molecular linkages between inflammation and coagulation remain to be elucidated (60,101,102,103). Given observational studies demonstrating the depletion of anticoagulant factors (decreased activity levels of protein C [60,102], ATIII [101,103], and TFPI [28]) in patients with severe sepsis and septic shock, such markers may be useful to indicate the presence or severity of sepsis in the future.
|
|
|
Figure 57.8. Cytokines induce the endothelial cell to shift from an antithrombotic to a prothrombotic phenotype. Expression of tissue factor by monocytes, and perhaps a subset of endothelial cells, initiates coagulation through the extrinsic system in patients with severe sepsis and septic shock. At the same time, fibrinolysis is inhibited through the release of thrombin-activatable fibrinolysis inhibitor (TAFI) and plasminogen activator inhibitor-1 (PAI-1). IL-1, interleukin-1β; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α. (Copyright © 2002 Eli Lilly and Company. All rights reserved. Printed with permission. Permission to reproduce the copyrighted material must be obtained from Lilly prior to reproducing or using the image.) |
Host Genetic Factors
Although the characteristics of the pathogen have much to do with the occurrence of clinical infection and progression to sepsis and septic shock, a growing body of data suggests that genomic variations between patients are equally important. These genomic variations in microbial and cell signaling, innate immunity, and coagulation and inflammatory stress cytokine responses appear to explain individual variations in susceptibility to infection, sepsis/septic shock, and septic death. They likely explain why identical organisms cause fulminant disease with septic shock in some but only minimal clinical illness in others. The importance of inheritable elements in susceptibility and mortality risk of life-threatening infections is demonstrated by adopted twin studies which demonstrated remarkable convergence in the causes of death (including sepsis/infection) of such individuals (109).
|
Table 57.7 Human Genetic Markers Associated with Risk of Infection and Sepsis/Septic Shock |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The advent of complete gene mapping via high throughput analysis techniques (e.g., microarray gene chips, etc.) have resulted in a rapid expansion of the list of human genetic markers associated with risk of infection, sepsis/septic shock, and death. These markers fall into several broad groups, including those involved with microbial ligand binding, intracellular signaling, cytokine generation, and coagulation factor generation/activity as described in Table 57.7. It should be noted that some genetic polymorphisms may be linked to other genetic loci. An association between a given polymorphism and susceptibility to infection, sepsis, septic shock, or septic death does not always imply a direct causal relationship.
Bioenergetic Failure
The underlying metabolic defect in sepsis and septic shock has been the source of substantial controversy over the last 30 years. Most forms of shock are associated with low cardiac output (CO) and tissue hypoperfusion leading to overt tissue ischemia. This results in anaerobic glycolysis with intracellular acidosis, increased lactate, and high-energy phosphate depletion in the affected tissues. Blood oxygen extraction ratio (the ratio of oxygen consumed divided by the oxygen delivered) is increased as tissues maximize oxygen extraction in order to maintain oxygen consumption. During septic shock, the same tissue metabolic phenomenon of intracellular acidosis and increased lactate production is noted. However, cardiac output and total tissue perfusion is typically increased, and the oxygen extraction ratio falls. The explanation for tissue acidosis and lactate production in septic shock in the presence of tissue hyperperfusion is unknown.
|
|
|
Figure 57.9. Microanatomic shunting in sepsis and septic shock. One explanation of the increased lactatic acidosis and MvO2 found in septic shock is the potential presence of opening of nonnutrient blood vessels between the arterial and venous vascular beds. MvO2, mixed venous oxygen saturation. |
Loss of vascular autoregulatory control may explain some of the typical metabolic findings of sepsis and septic shock. An early theory postulated the existence of microanatomic shunts between the arterial and venous circulations (110) (Fig. 57.9). During sepsis, these shunts were said to result in decreased systemic vascular resistance (SVR) and increased mixed venous oxygen saturation (MvO2) (111). The resultant decrease in perfusion to tissue beds with normal or even increased metabolic demand could generate tissue ischemia and lactic acid. However, whereas microanatomic shunting has been noted in localized areas of inflammation, systemic evidence of this phenomenon in sepsis and septic shock is lacking (111,112,113,114,115). Another theory involving “functional” shunting due to defects of microcirculatory regulation in sepsis has also been proposed (Fig. 57.10) (116,117). Overperfusion of tissues with low metabolic requirements would result in increased MvO2 and narrowing of the arteriovenous oxygen content difference. The relative vasoconstriction of vessels supplying more metabolically active tissues would result in tissue hypoxia and lactate production due to anaerobic metabolism. Observations that some capillary beds may be occluded by platelet microaggregates, leukocytes, fibrin deposits, and endothelial damage support this theory (112,116,118). Additional support comes from studies that demonstrate evidence of supply-dependent oxygen consumption in sepsis (119,120,121,122,123). Both of these theories of the metabolic defect of energy metabolism in sepsis and septic shock fall within the category of “stagnant” hypoxia as described by Barcroft in 1920 (124).
|
|
|
Figure 57.10. Functional shunting in sepsis and septic shock. Loss of ability to appropriately regulate microvascular flow according to tissue metabolic demand can lead to overperfusion of low-metabolic-demand tissue beds resulting in increased MvO2 (mixed venous oxygen saturation). Underperfusion of high-metabolic-demand beds can result in tissue ischemia, anaerobic metabolism, and lactic acidosis. |
A third theory of the metabolic presentation of sepsis and septic shock suggests that circulating mediators cause an intracellular metabolic defect involving substrate use. This results in bioenergetic failure with high-energy phosphate (adenosine triphosphate [ATP] and phosphocreatine) depletion and lactate production (125,126,127). Increased mixed venous oxygen saturation could then be explained by perfusion, which is maintained in excess of tissue oxygen use capability. This phenomenon has been termed histotoxic (124) or cytopathic (127) hypoxia. Potential mechanisms to explain this form of hypoxia include impairment/ inactivation of pyruvate dehydrogenase; nitric oxide or peroxynitrite-mediated inhibition of mitochondrial respiration; uncoupling of oxidative phosphorylation or activation of poly-(ADP-ribosyl)-polymerase (PARP) (127). Observations demonstrating preservation of tissue PO2 (128), absence of tissue hypoxia (129), and impairment of mitochondrial function (127,130,131,132) during sepsis and septic shock support this possibility.
In particular, near-infrared spectroscopy (NIRS) has been used to examine mitochondrial function in a primate model of septic shock using live Escherichia coli infusion. NIRS demonstrated the presence of mitochondrial dysfunction in skeletal muscle in animals with experimentally induced sepsis. This was manifested by the impairment of oxidation of cytochrome a,a3 with reperfusion after transient ischemia in septic animals compared to controls (131). Another primate study demonstrated early disturbance of mitochondrial redox state in skeletal muscle and brain in the presence of live E. coli bacteremia. Of note, these changes occurred before the onset of overt hemodynamic alterations (133). In a limited observational study, uncoupling of tissue oxyhemoglobin levels and mitochondrial oxygen consumption, as indicated by cytochrome a,a3 redox state (indicating mitochondrial oxidative stress), predicted the development of multiple organ failure in patients with major trauma (134). These data particularly support the possibility of a decreased ability of mitochondria to use oxygen as a potential cause of decreased tissue high-energy phosphate in sepsis.
All these theories of septic bioenergetic metabolism would be expected to result in a deficit of tissue high-energy phosphates during septic shock. A series of studies using biochemical analysis of harvested tissues and nuclear magnetic resonance (NMR) spectroscopy of septic animals have suggested that high-energy phosphate reserves are decreased in animal models of septic or endotoxic shock (125,135,136). It can be argued that in many of these studies, animals were inadequately fluid resuscitated, which resulted in tissue hypoperfusion. However, animals in at least one study (125) were clearly adequately resuscitated (cardiac output and tissue oxygen tension were maintained comparable to shams) and demonstrated similar evidence of high-energy phosphate depletion (skeletal muscle biopsy) along with an increased lactate/pyruvate ratio during rat peritonitis induced by cecal ligation and perforation (125). Little human data exist. In one study of critically ill patients (most of whom were septic), the acetoacetate/β-hydroxybutyrate ratio (a marker of mitochondrial redox state) rose significantly in nonsurvivors compared to survivors (137). Evidence of increased acetoacetate/β-hydroxybutyrate ratio, along with an increase in ATP degradation products in critically ill patients with sepsis, also exists (138,139). In addition, independent studies using skeletal muscle biopsies in patients with sepsis/septic shock observed decreased ATP and phosphocreatine but variable changes in lactate levels (140,141).
In contrast, other animal studies using NMR spectroscopy demonstrate that high-energy phosphates are not depleted in septic animals as would be expected in these theories of septic bioenergetic failure (142,143,144). According to these and other studies, cellular ischemia is not the dominant factor in metabolic dysfunction in sepsis (129,142,143,144,145,146,147). Rather, circulating mediators may result in cellular dysfunction, aerobic glycolysis, and lactate production in the absence of global ischemia (143). This position is weakened by data suggesting that increased lactate in septic shock is also associated with decreased pH (which would not be expected in aerobic glycolysis) (143). Nonetheless, ongoing controversy of this issue remains.
Cardiac and Vascular Responses
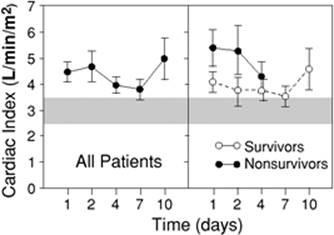
Prior to the introduction of the balloon-tipped pulmonary artery catheter (PAC) and echocardiography to assess cardiovascular performance, much of our understanding of septic hemodynamics was based on clinical findings. Two distinct clinical presentations of septic shock were proposed: Warm shock characterized with high CO, warm dry skin, bounding pulses and hypotension; and cold shock characterized with low CO, cold clammy skin, and diminished pulses (148). These two presentations were thought to represent a progressive continuum, starting with warm shock (in the initial hemodynamically well-compensated phase) and progressing to cold shock (indicating decompensation), culminating in death. This notion was supported by studies showing a correlation between survival and a high cardiac index (CI) (148,149). A major problem with this interpretation was that these studies used central venous pressure (CVP) as a reflection of left ventricular end-diastolic volume (LVEDV) and adequacy of fluid resuscitation. The central role of adequacy of intravascular volume status to CI and survival was suggested in a handful of studies at that time (150,151). Based on evidence collected over the past four decades, CVP is now accepted to be a poor measure of preload in critically ill patients, particularly those with sepsis and septic shock (152). Studies in recent years have clearly shown that adequately resuscitated septic shock patients typically exhibit a persistent hyperdynamic state, high CO, and low SVR (153,154). In nonsurvivors, this hyperdynamic state usually persists until death (Fig. 57.11) (35,155).
More than any other form of shock, distributive and, particularly, septic shock involves substantial elements of the hemodynamic characteristics of other shock categories. All forms of distributive shock involve decreased mean peripheral vascular resistance. Before fluid resuscitation, distributive shock also involves a hypovolemic component with decreased central venous and pulmonary artery occlusion pressures. The primary cause of this relative hypovolemia is an increase of the vascular capacitance due to venodilatation. This phenomenon has been directly supported in animal models of sepsis (156,157,158,159,160) and is reinforced by the fact that clinical hypodynamic septic shock (low CO) can usually be converted to hyperdynamic shock (high CO) with adequate fluid resuscitation (35,148,161). Relaxation of vascular smooth muscle is attributed to several of the mediators known to circulate during sepsis. These same mediators also contribute to the second cause of hypovolemia in sepsis: Third-spacing of fluid to the interstitium due to loss of endothelial integrity. Further, decreased oral fluid and salt intake during the course of the illness may play a role. As a consequence, CO and central/mixed venous oxygen saturation in unresuscitated and poorly resuscitated septic shock patients is usually decreased (161,162). Septic shock also involves a cardiogenic element. Myocardial depression is common in human sepsis and septic shock (163,164). Circulating substances such as TNF-α, IL-1β, platelet-activating factor (PAF), leukotrienes, and most recently, IL-6 and macrophage migration inhibitory factor have been implicated in this process (95,165,166,167,168,169,170,171,172).
|
|
|
Figure 57.11. Cardiac index in resuscitated septic shock. The mean (standard error of the mean [SEM]) cardiac index plotted against time for all patients, survivors, and nonsurvivors. The hatched areas show the normal range. All groups maintained an elevated cardiac index throughout the study period. The difference between the survivors and nonsurvivors was not statistically significant. Open circles, survivors; closed circles, nonsurvivors. (Adapted from Parker MM, Shelhamer JH, Bacharach SL, et al. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100:483–490.) |
Organ System Dysfunction Due To Sepsis And Septic Shock
Table 57.8 summarizes organ system dysfunction in sepsis and septic shock.
Central Nervous System
Septic encephalopathy is the most common neurologic manifestation of sepsis and septic shock, encompassing between 8% and 80% of patients with sepsis (173,174,175,176). The likely reason for the divergent frequencies of the syndrome in studies is the difficulty of identifying the condition in patients with superimposed hypotension, sedation, hypoxemia, acidosis, electrolyte disturbances, hypoglycemia/hyperglycemia, hypothermia/hyperthermia, and/or concurrent hepatic/renal failure/encephalopathy. The diagnosis, requiring the presence of altered mentation with an extracranial source of infection, is often one of exclusion. Although deficits can range from impairment of higher cognitive functions to delirium or coma, asterixis, myoclonus, and seizure activity are highly atypical (173,176). The diagnosis is best made by electroencephalography (EEG) (177). The occurrence and severity of septic encephalopathy (graded by EEG or Glasgow coma scale) appears to be associated with increased mortality (as high as 70%) (173,178).
|
Table 57.8 Organ System Dysfunction in Sepsis and Septic Shock |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
Critical illness-associated neuromuscular syndromes (inclusive of critical illness polyneuropathy and myopathy) are the most common cause of neuromuscular problems in the ICU (179). The primary clinical manifestation of this condition is muscle weakness. Since many patients who are in the ICU with sepsis and septic shock require ventilatory support, the initial overt manifestation may be either respiratory failure or failure to wean from ventilation. Studies have suggested an incidence between 35% and 50% based on clinical criteria and 40% to 80% based on electromyography (EMG)/nerve conduction studies (180,181,182). Although the disorder is commonly noted later in the recovery phase of sepsis and septic shock, EMG/nerve conduction data suggest that the onset is much earlier (concurrent or within days of the onset of septic shock) (183,184). The condition is a predominantly peripheral motor neuropathy in association with the presence of the systemic inflammatory response. Physical findings may include difficulty in weaning from the ventilator, symmetric paresis greater in the lower extremities, reduced deep tendon reflexes, and ataxia (180). A distal sensory neuropathy is also common. Approximately 25% of patients who are awake after a week on mechanical ventilation have significant weakness that lasts at least a week (185). The condition is considered to be an element of and is closely associated with the occurrence of MODS.
Cardiovascular System
The major clinically apparent manifestations of shock on the heart are due to sympathoadrenal stimulation. Heart rate is almost universally increased in the absence of disturbances of cardiac conduction; the degree of increase is predictive of outcome (35). In addition, catecholamine-driven supraventricular tachycardias and ventricular ectopy with ischemic electrocardiography (ECG) changes, particularly in patients predisposed to myocardial ischemia, may be found.
Like the brain, the blood supply to the heart is autoregulated, rendering it resistant to sympathetically driven vasoconstriction and shock-related hypoperfusion. Perfusion of the heart is unchanged or even increased during sepsis and septic shock (186,187). The occurrence of septic myocardial depression has already been addressed. Circulating myocardial depressant substances contribute to myocardial depression in sepsis and septic shock (188,189). This has been linked to decreased beta-adrenoreceptor affinity and density (190,191,192), as well as potential defects of intracellular signal transduction involving nitric oxide, G proteins, cyclic adenosine monophosphate (cAMP), and cGMP (95,193,194,195,196,197).
Although septic myocardial depression is a transient phenomenon in survivors, myocardial cell injury as evidenced by increased troponin levels does occur (198,199). Serum troponin is elevated in almost half of patients with septic shock (without myocardial creatine kinase [CK-MB] elevation or ischemic ECG changes) (200). A correlation between left ventricular (LV) dysfunction and troponin I (TnI) positivity has been shown (199). Serum TnI correlated with left ventricular dysfunction and was an independent predictor of the need for inotropic/vasopressor support, adverse outcome, and mortality in septic shock patients (200). Whether the clinically inapparent myocardial cell injury that is the source of elevated troponin contributes to, or is a consequence of, septic shock is yet to be determined. Although troponin is used as a marker of myocardial injury (particularly in the context of myocardial ischemia), it does not specifically suggest myocardial infarction in other contexts.
Respiratory System
Early respiratory responses to sepsis include tachypnea and hyperventilation. Gas exchange may be mildly abnormal. Later in the course of sepsis, patients may develop diffuse alveolar damage consistent with the acute lung injury (ALI) or adult respiratory distress syndrome (ARDS). Infections account for about one half of all cases of ARDS. These infections can involve local pneumonia or distant foci of infection associated with sepsis or septic shock. The risk of ARDS in association with sepsis increases with the severity of the syndrome (sepsis to septic shock) (201). From 40% to 60% of patients with Gram-negative septic shock develop ARDS. Sepsis is the single condition most closely associated with progression to acute lung injury or ARDS, with an incidence of 40% (202). Several comorbid factors increase the risk of ARDS, including chronic alcohol abuse, chronic lung disease, and severe acidemia (202). Most patients with septic ARDS also have other organ failure, i.e., MODS. Death is more commonly due to MODS or the underlying sepsis, although the impact of low tidal volume ventilation in ARDS studies suggest that the lung injury may still play a significant role (perhaps as a source of persistent inflammatory stimulation) (202,203,204). The mortality of ARDS/MODS is approximately 40%, although some recent reports suggest that it may be decreasing (202,205). Failure to improve in the first week is associated with progression of the syndrome and poor prognosis, as are MODS, chronic liver disease, and age; interestingly, indices of oxygenation and ventilation are not predictive (202).
Renal
Acute renal failure (ARF) is a major complication of sepsis and septic shock and occurs with increasing frequency in relation to the severity of the syndrome, from 16% to 19% with sepsis to 51% with septic shock (31,201,206). Sepsis has been the leading cause of acute tubular necrosis (ATN) in some ICU studies, accounting for almost 50% of cases (207,208,209). Sepsis-associated acute renal failure is associated with a substantially higher mortality risk (75%) than nonseptic ARF (45%); within this group, septic shock mortality is higher (80%) than in those with severe sepsis (70%) (201,208). Compared with nonsepsis-associated ARF, sepsis-related ARF patients are significantly older, sicker, require mechanical ventilation more often, and present later in the hospital course more frequently (208).
Gastrointestinal
The gut is relatively sensitive to circulatory failure due to the responsiveness of the splanchnic vasculature to vasoconstrictive stimulation by extrinsic factors. In addition, gut tissues may have increased sensitivity to proinflammatory cytokine-driven inflammatory injury. Typical clinical gut manifestations of hypoperfusion, sympathetic stimulation, and inflammatory injury associated with sepsis and septic shock include ileus, erosive gastritis, pancreatitis, acalculous cholecystitis, and colonic submucosal hemorrhage (210). In addition, enteric ischemia produced by circulatory shock and free radical injury with resuscitation may breach gut barrier integrity (211,212). Some theories propose that enteric bacteria and antigens (notably endotoxin) may translocate from the gut lumen to the systemic circulation during gut ischemia, resulting in irreversible shock (213) and MODS (214).
Hepatobiliary
Two major forms of organ injury can be seen in the liver with sepsis and septic shock (215,216). “Shock liver” (ischemic hepatitis) is associated with massive ischemic necrosis and major elevations of transaminases, which can occur with septic shock and is atypical in the absence of extensive hepatocellular disease (217). When it does occur, it can contribute substantially to lactic acidosis since the liver accounts for most serum lactate clearance. Hypoglycemia may also be seen. Centrilobular injury with mild increases of transaminases and lactate dehydrogenase is much more common. Transaminases usually peak within 1 to 3 days of the insult and resolve over 3 to 10 days. In both cases, there are only mild increases in bilirubin and alkaline phosphatase in the early phase. Despite the production of acute-phase reactants in early sepsis and septic shock, synthetic functions may be impaired, with decreased generation of prealbumin, albumin, and hepatic coagulation factors (increased prothrombin time [PT]). After, or independent of, the occurrence of septic shock, evidence of biliary stasis with increased bilirubin and alkaline phosphatase may be present (216). Increases in transaminases are modest.
Hematologic
Sepsis and septic shock are associated with a range of hematologic disorders including overt disseminated intravascular coagulation (DIC), thrombocytopenia, and coagulopathy. Thrombocytopenia and coagulopathy are multifactorial in nature. Bone marrow suppression, consumption, and medications can contribute to thrombocytopenia, whereas consumption and decreased liver production of coagulant factors, as well as malnutrition (leading to depleted vitamin K stores), contribute to coagulopathy. Nonetheless, whenever these findings are present, early disseminated intravascular coagulation (DIC) is possible.
Septic shock is the single most common cause of DIC, characterized by microangiopathic hemolysis, consumptive thrombocytopenia, consumptive coagulopathy, and microthrombi with tissue injury. Overt DIC occurs in one quarter to one half of cases of Gram-negative sepsis (218). Although Gram-positive sepsis has been thought to be less closely associated with DIC, the frequency of occurrence is quite similar (218,219). The occurrence of DIC in sepsis is associated with a doubling of projected mortality (218,220). DIC may also represent both a driver and manifestation of MODS. The deposition of microvascular thrombi can cause significant endothelial injury and inflammatory responses, leading to ischemic and inflammatory tissue injury, the basis of MODS.
A prolonged prothrombin time and partial thromboplastin time, hypofibrinogenemia, elevated level of fibrin split products, and the presence of the D-dimer herald the onset of disseminated intravascular coagulation. Since it is due to simultaneous systemic activation of coagulation and fibrinolysis cascades, it can be differentiated from the coagulopathy of liver failure by determination of endothelial cell-produced factor 8 (normal or increased with hepatic dysfunction). The pathogenesis of this disorder is linked to activation of tissue factor on endothelial cells and macrophages, probably by proinflammatory cytokines induced by exogenous bacterial toxins (220,221).
Metabolic
Specific, predictable, and overlapping metabolic alterations occur in both sepsis and shock. Foremost among these is hyperglycemia. There are two reasons for hyperglycemia in sepsis and states of shock. Early in sepsis, when hemodynamic stress initiates compensatory responses, endogenous catecholamines are released as a consequence of enhanced sympathoadrenal stimulation. In addition, increased release of adrenocorticotropic hormone (ACTH), glucocorticoids, and glucagon with a concomitant decreased release of insulin results in glycogenolysis and gluconeogenesis (222,223). Increased epinephrine also results in skeletal muscle insulin resistance, sparing glucose for use by glucose-dependent organs such as the heart and brain (224). In addition, proinflammatory, stress-related cytokines such as TNF-α, IL-1β, and IL-6 contribute to insulin resistance in peripheral tissues (225). Pharmacologic therapies of sepsis and shock, including catecholamine vasopressors/inotropes, steroids, and total parenteral nutrition, can add to these effects. It is notable that, despite insulin resistance, the increased metabolic demands of sepsis also result in increased overall glucose uptake and utilization (226).
With the evolution of sepsis to septic shock, metabolic responses progress. Late in shock, hypoglycemia may develop, possibly due to glycogen depletion or failure of hepatic glucose synthesis (227). Fatty acids are increased early in sepsis but fall later with hypoperfusion of adipose-containing peripheral tissue (226,228). Hypertriglyceridemia is often seen during shock as a consequence of catecholamine stimulation and reduced lipoprotein lipase expression induced by circulating TNF-α (223,226,229). Increased catecholamines, glucocorticoids, and glucagon also increase protein catabolism, resulting in a negative nitrogen balance (223,228).
Endocrine
Endocrine abnormalities are frequently underappreciated in sepsis and septic shock. Notable alterations in levels of pituitary, adrenal, thyroid, growth, and sex hormones are known to occur (225,230,231,232,233,234,235,236). In recent years, “relative” adrenal insufficiency in septic shock has received substantial attention. Few septic patients exhibit overt adrenal insufficiency. Relative bradycardia and a nontoxic appearance in a patient with septic shock is suggestive of this possibility. These are often elderly patients who have survived an initial episode of septic shock and either fail to fully recover or suffer a relapse. However, a considerable body of literature suggests that a suboptimal cortisol response (within the normal range) to sepsis and septic shock can have deleterious effects, including prolonged pressor dependence and increased mortality. Estimates of the frequency of adrenal insufficiency in septic shock vary wildly from 0% to 95% (237,238). In great part, this is due to the use of varying definitions based on baseline or cosyntropin-stimulated cortisol levels or changes in levels from baseline in response to cosyntropin. Common definitions in septic shock patients include random cortisol of <700 nmol/L (25 µg/dL), peak postcosyntropin level of <500 to 550 nmol/L (1–20 µg/dL), or postcosyntropin change in cortisol of <200 to 250 nmol/L (7–9 µg/dL) (230,237,239,240). Interestingly, pituitary dysfunction may play a role in many patients with adrenal insufficiency, as 85% of critically ill patients have decreased levels of adrenocorticotropic hormone (ACTH) (241).
Abnormalities of thyroid hormones are also present in sepsis and septic shock, although the clinical significance is less certain. In humans, serum T4 and T3 levels fall shortly after the onset of severe clinical infection. Euthyroid sick syndrome is manifested by low serum levels of thyroid hormones in clinically euthyroid patients with severe nonthyroidal systemic illness. Decreased T3 levels are most common. Patients with more severe or prolonged illness also have decreased T4 levels. Serum reverse T3 (rT3) is increased. Patients are clinically euthyroid and do not have clinically significant thyroid-stimulating hormone (TSH) elevations.
Sepsis and septic shock are clearly associated with perturbations of various hormones including insulin, growth hormone, TSH, thyroxin, ACTH, cortisol, growth hormone (242), and sex hormones. Perturbations of hormones of the posterior pituitary should be expected. In addition to abnormal prolactin levels (243), sepsis and septic shock are accompanied by relative deficiencies of vasopressin/antidiuretic hormone (ADH) levels. Vasopressin, produced in the hypothalamus and stored in the posterior pituitary gland, is released in response to hyperosmolarity. Hypotension as seen in shock states is an even more powerful stimulus for release. Recent human studies have suggested a relative deficit of circulating vasopressin in patients with septic shock (relative to those with cardiogenic or hypovolemic shock). This deficiency may be related to depletion of neurohypophyseal stores combined with NO-mediated inhibition of production (225,235). Clinically, vasopressin exerts powerful vasopressor effects in hypotensive patients, particularly those with septic shock. To some extent, this effect appears to be mediated through reestablishment of reduced sensitivity to catecholamine (244).
Diagnosis Of Sepsis
Under ideal circumstances, each patient with evidence of sepsis would undergo a thorough evaluation at presentation prior to the initiation of therapy. In the context of sepsis and septic shock, circumstances are rarely ideal, so an abbreviated initial assessment focusing on critical diagnostic and management planning elements is frequently necessary.
To ensure maximally rapid implementation of effective therapy, an initial presumptive diagnosis of severe sepsis and septic shock is mandated. The criteria for this presumptive diagnosis should be highly inclusive and based primarily on clinical criteria.
The initial presumptive diagnosis of sepsis with organ dysfunction (severe sepsis) may be made in the presence of the following elements:
· Suspected infection based on a minimal clinical constellation of localizing (e.g., dyspnea, cough, purulent sputum production, dysuria, pyuria, focal pain, local erythema, etc.) and systemic signs and/or symptoms of infection and sepsis (Table 57.9)
· Clinical evidence of organ dysfunction (e.g., hypotension with peripheral hypoperfusion, oliguria, hypoxemia, obtundation, etc.)
|
Table 57.9 Clinical Symptoms/Signs for Presumptive Diagnosis of Severe Sepsis/Septic Shock |
|
|
Similarly, an initial diagnosis of septic shock is established in the presence of suspected infection with sustained hypotension without a definitive alternate explanation.
The initial presumptive diagnosis of severe sepsis or septic shock is based on clinical criteria and does not require microbiologic, radiographic, or other laboratory evidence of specific infection or organ injury. Only clinical evidence of infection and organ failure is necessary. For the most part, available laboratory tests or imaging studies represent supportive, not diagnostic, elements. This clinical approach allows a parallel, rapid initiation of empiric antimicrobials and supportive measures.
Although a suggestive clinical examination is sufficient for the presumptive diagnosis of severe sepsis and septic shock, more authoritative investigations (both laboratory and radiologic) are generally required for confirmation. For this reason, the definitive diagnosis of severe sepsis and septic shock involves a broader range of clinical and laboratory evidence of sepsis (Table 57.10) and organ dysfunction (arterial hypotension, lactic acidosis, or any organ dysfunction variables in Table 57.2). Establishment of a definitive diagnosis can help to more specifically target antimicrobial therapy and trigger specific therapies such as surgical source control and activated protein C.
|
Table 57.10 Supportive/Confirmatory Findings for Severe Sepsis/Septic Shock |
||
|
History
The initial history should focus on two major areas: The key symptoms with respect to diagnosis of sepsis and of the specific site of infection, and key factors that would modify initial empiric therapies such as antimicrobials, fluid resuscitation, and possibly, vasopressors/inotropes.
With respect to symptoms, constitutional complaints are entirely nonspecific. The classic pattern of fever, rigors, and chills is common but far from universal. Fatigue, malaise, anxiety, or confusion may be observed, particularly in the elderly. Occasionally, the elderly, the immunocompromised (nonspecific immune dysfunction due to chronic organ failure), and the immunosuppressed (specific immune defects) may present without classic signs and symptoms.
Fever is a common feature of infection and/or sepsis. Fever is caused by a direct effect of inflammatory mediators, such as IL-1β, on the hypothalamus. The fever response may be suppressed in septic shock and may be absent in the elderly, immunocompromised, or immunosuppressed patient. Hypothermia in septic shock is associated with reduced cardiac output and portends a poor prognosis (245). Septic encephalopathy manifested by disorientation or confusion is especially common in elderly individuals. Apprehension, anxiety, and agitation may all occur early in the course. With severe disease (i.e., septic shock) or progression of sepsis, overt encephalopathy with a decreased level of consciousness and coma can occur. Hyperventilation with respiratory alkalosis can manifest even before the onset of metabolic acidosis as a consequence of cytokine-mediated stimulation of the respiratory center in the medulla.
Localizing symptoms as described in Table 57.11 may be more helpful in determining the septic cause of the constitutional manifestations of sepsis. The key historical factors used to modify initial therapies include antimicrobial sensitivities/allergies, recent infections/antimicrobial use, the locale of infection acquisition (i.e., nosocomial vs. community), and major comorbidities. The existence of comorbidities (e.g., AIDS; chemotherapy; hematologic malignancy; neutropenia resulting in immunosuppression or chronic renal, heart, liver, or other organ failure; COPD; dementia; inflammatory bowel diseases; diabetes; or via invasive catheters/devices) resulting in immunocompromise mandate the use of extended-spectrum antimicrobial therapy. Chronic renal, liver, or heart failure may also influence the choice and volume/dose of antimicrobials, resuscitation fluids, and vasopressors. Recent antimicrobial use and nosocomial or institutional acquisition of infection may also mandate consideration of extended-spectrum antimicrobial therapy to adequately cover nosocomial pathogens.
|
Table 57.11 Localizing Clinical Symptoms and Signs in Severe Infections |
|||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||
Physical Examination
The physical examination should focus on ensuring that the patient is stable and on rapid localization of the site of infection. The physical examination should first ensure that the airway is patent, the patient is breathing satisfactorily, and vital signs and peripheral perfusion are acceptable.
Tachypnea and tachycardia are almost universal. Normothermia and fever are consistent with sepsis, but hypothermia should be of concern due to its association with shock/hypoperfusion. All patients with sepsis should be observed for signs of hypoperfusion (mottling, pallor, diaphoresis, impaired capillary refill in nail beds). An acutely ill, flushed, and toxic appearance is common in the septic patient, particularly early in the course. In the early stages of sepsis, CO is well maintained or even increased, skin and extremities are warm, and capillary refill is normal. As sepsis progresses, venodilation results in reduced central venous pressure and venous return. Hypovolemic manifestations with hypotension, reduced stroke volume, and CO with signs of tissue hypoperfusion develop. As patients are aggressively fluid resuscitated, a hyperdynamic circulatory state (albeit with distributive shock) again dominates the clinical picture and will usually persist until recovery or death.
The most common sites of infection causing sepsis and septic shock in order of frequency are respiratory, abdominal, urinary, and soft tissue. Abdominal infections are more closely associated with septic shock whereas urinary infections are more common in sepsis. Intravascular catheters are a frequently overlooked source of infection and sepsis. A recent study suggested that central venous catheters might account for as much as 3.7% of cases of septic shock (25). Similarly, cases of Clostridium difficile–related septic shock are often overlooked in the absence of overt toxic megacolon. Adding to the difficulty of managing the ICU patient with sepsis and/or septic shock is that many patients have simultaneous infection at more than one site.
Laboratory Studies
Patients with sepsis require urgent lab testing to help make a firm diagnosis and to evaluate the severity of the illness. Sepsis and septic shock typically present with somewhat different, though naturally overlapping, laboratory parameters (see Table 57.12). Lab tests usually start with a complete blood count (CBC). Hemoglobin is often decreased, although this is usually due to the presence of chronic disease. Hemoglobin can occasionally be increased in patients with substantial interstitial third-spacing and relative hypovolemia. The white cell count is increased in sepsis but may transiently normalize or even drop below normal range, with progression to septic shock. Although this phenomenon has been linked to Gram-negative septic shock, it can be seen in septic shock due to any pathogen. Leukopenia in this setting has been linked to poor outcome. Toxic granulation and the presence of Dohle bodies are also seen more frequently, with progression to more severe disease. Similarly, a marked left shift with increasing immature forms (bands) is more common in septic shock. Platelets often respond as an acute-phase reactant, with increases early in infection/sepsis. However, platelet counts drop, with septic shock reaching a nadir around day 5 in survivors.
|
Table 57.12 Key Laboratory Values in Infection/Sepsis versus Septic Shock |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In contrast, the international normalized ratio (INR) may be mildly abnormal at the onset of sepsis (due to malnourishment) and is usually most abnormal at onset of septic shock. Fibrinogen is an acute-phase reactant and is usually elevated with onset of infection/sepsis. However, levels will drop with septic shock, especially if DIC intervenes. Fibrin split products and D-dimers are very sensitive markers of progression of sepsis and are almost universally elevated with septic shock.
Serum creatinine and blood urea nitrogen (BUN) may actually be decreased due to increased renal blood flow in the early hyperdynamic phase of sepsis but will increase with the onset of septic shock. An increase in serum creatinine denotes an increased mortality risk even within a few hours of the onset of septic shock. Similarly, elevated serum lactate is closely correlated with increased mortality risk in septic shock.
Septic patients should have both site-specific and blood cultures drawn prior to initiation of antimicrobial therapy. In the case of septic shock, however, antimicrobial therapy should never be delayed to accommodate these cultures because of the antimicrobial delay-dependent increase in mortality risk (25). Gram stain should be performed on all site samples. Although there are some data to suggest that Gram stain is not useful in the initial management of certain infections (nosocomial pneumonia, peritonitis due to bowel perforation), a good specimen, appropriately interpreted, can provide invaluable information.
Imaging Studies
Although in most cases the clinical examination will localize the source of infection with a reasonable degree of confidence, basic radiographic imaging can be very useful in cases where an obvious site of infection is not apparent. Advanced imaging studies (computerized axial tomography [CAT], magnetic resonance imaging [MRI], ultrasound) rarely yield information regarding localization of the infection that has not been provided by the clinical examination and basic imaging studies. However, these techniques may be highly useful when definitive or precise localization and/or delineation of extent of disease are required.
A chest radiograph should be obtained in most patients admitted to the hospital with sepsis. Elderly, immunocompromised, and immunosuppressed patients with occult sepsis will often be found to have a pulmonary source on radiographic examination. Supine and upright or lateral decubitus abdominal films are useful if bowel perforation is of concern. In the appropriate clinical context of crepitus, bullae, hemorrhage, or foul-smelling exudate with intense local pain, evidence of gas in soft tissues on plain extremity radiographs is almost pathognomonic of necrotizing soft tissue infection with clostridia or facultatively anaerobic Gram-negative bacilli.
CT scan with contrast is the preferred imaging modality to rule out intra-abdominal, intracranial, epidural, perinephric, and soft tissue abscesses, as well as retroperitoneal abscess or mediastinal infection. They can also be useful for localizing bowel wall injury and assessing necrotizing soft tissue infections (although MRI is preferred for the latter). Ultrasound is the initial imaging modality of choice for biliary sepsis and obstructive uropathy, although CT scan is also sensitive and specific.
Management of Severe Sepsis and Septic Shock (i.e., The Sepsis Six-Pack)
To optimize outcome in sepsis with organ dysfunction (severe sepsis), the initiating triggers, amplification cascade, and downstream organ dysfunction must be addressed; this requires monitoring and therapeutic elements. With respect to the initiating triggers, antimicrobials and, where possible, surgical and nonsurgical source control are mandated. With respect to the amplification cascade, one new agent (activated protein C) has been developed that directly dampens septic response by exerting both anti-inflammatory and antithrombotic effects, such that mortality is improved. Organ dysfunction is addressed through direct supportive measures. The most immediate of these—fluid and vasopressor/inotropic resuscitation—support the circulatory system. However, mechanical ventilation and dialysis have also been shown to improve outcome in severe sepsis and septic shock.
Six major areas in the evaluation and treatment of severe sepsis can be identified. These include the following:
1. Fluid resuscitation
2. Antimicrobial therapy
3. Vasopressors and inotropes
4. Invasive and noninvasive monitoring
5. Specific therapy
6. Miscellaneous supportive therapy
Fluid Resuscitation
The development of shock in patients with sepsis involves disturbances of global and regional perfusion. Initially, ventricular filling pressures as reflected by CVP and pulmonary wedge pressure (PWP) are decreased. As a consequence, venous return falls, resulting in limitation of CO. Although an increase in insensible losses and decreased fluid intake may contribute to this effect, nitric oxide–mediated venular dilatation and loss of endothelial barrier integrity (resulting in a drop in colloid oncotic pressure from loss of albumin into the interstitium) probably play a dominant role (246,247). A significant degree of hypovolemia is almost universal in early, untreated severe sepsis or septic shock. Available data suggest that initial isotonic fluid deficits can exceed 10 L (248).
Management of sepsis requires consideration of both global and regional perfusion defects, making the establishment of goals for therapy more complex than for other forms of shock. Support of global perfusion takes initial precedence. Since hypovolemia is a major factor in the hypotension and hypoperfusion of early septic shock, foremost among the appropriate initial therapeutic considerations is infusion of intravascular fluids. Fluid infusion should be implemented rapidly by large-bore peripheral intravenous catheters. Infusion of fluids can improve global perfusion indices (blood pressure, CO, and MvO2/central venous oxygen saturation [ScvO2]) and may reveal the presence of regional perfusion disturbances and/or myocardial depression that may require therapy with vasopressors/inotropes.
The three issues to consider in optimizing fluid resuscitation are the type of fluid used, the rapidity of infusion, and the amount of fluid administered.
Initial resuscitation of septic patients should be aimed at rapid intravascular volume expansion
The view that intravascular fluid depletion plays a central role in the pathogenesis of early septic shock has been recognized since the past midcentury. Several studies suggested that septic shock is associated with reduced total circulating blood volume (149,150). Since almost all untreated patients with severe sepsis or septic shock have a significant element of hypovolemia, a hypodynamic circulation with decreased cardiac output is typical prior to fluid resuscitation. This hypovolemia is probably the basis of early observations that death in sepsis is associated with decreased cardiac output. The patients in those studies were clearly inadequately resuscitated by current standards (149,150). Additional support for the central importance of functional hypovolemia in early septic shock comes from a more recent demonstration that the venous oxygen saturation is decreased in early preresuscitation septic shock (consistent with the findings in other forms of hypodynamic shock) (161).
Aggressive fluid loading is the standard early therapy of septic shock and results in the generation of a hyperdynamic circulatory state in over 90% of patients (249). Rapid fluid resuscitation may reveal severe sepsis without shock in a significant subset of patients with apparent septic shock (248). Increased total blood volume has been associated with higher cardiac output and increased survival in human septic shock (150). Intravascular volume dependence of the hyperdynamic circulatory state in sepsis has been confirmed in animal models (158). Although the demonstration that resuscitation from hypovolemia improves outcome in traumatic shock dates back to the early work of Cannon (250) and Cournand et al. (251), clear evidence that early aggressive fluid resuscitation improves outcome in septic shock is limited to a small series of pediatric septic shock (252) and a recent randomized study of goal-directed resuscitation (253).
Initial fluid resuscitation should be titrated to specific clinical end points
Aggressive fluid loading in patients with septic shock can increase total blood volume, cardiac output, oxygen delivery, and consumption while reducing lactic acidosis (119). Older studies have suggested that an increased blood volume associated with normalization of cardiac output is associated with improved survival (149,150).
In the absence of early invasive or echocardiographic monitoring, clinical end points can be used for titration of fluid resuscitation. Since both initial heart rate and blood pressure have been shown to be associated with outcome in septic shock as well as hypovolemic shock (35,254,255,256), standard goals may include the following:
· Heart rate ≤100 beats/minute
· Systolic blood pressure (≥90 mm Hg)
· Mean arterial pressure (≥60–65 mm Hg)
· Urine output (≥0.5 mL/kg/hour)
It should be noted that these clinical parameters can underestimate initial resuscitative requirements in critically ill subjects including those with septic shock (257,258,259).
Mortality in both septic and other forms of shock has also been associated with increased arterial lactate and base deficit levels (260). Normalization of these parameters can be used to augment clinical end points for titration of fluid resuscitation (261). However, both parameters represent relatively late responses to cellular stress, and resolution may similarly lag following the implementation of effective resuscitation (262).
Initial fluid resuscitation should be achieved using isotonic crystalloid solutions
Effective fluid resuscitation can be delivered with either isotonic crystalloid (e.g., normal saline, lactated Ringer solution) or colloid solutions (e.g., hydroxyethyl starch, human albumin). All of these solutions are equally effective if titrated to the same clinical end points. Given the difference in distribution of such compounds, it typically requires approximately four times more crystalloid to achieve the same hemodynamic effect as a given amount of colloid (263). Several animal and human studies have pointed out theoretical advantages to colloids in limiting interstitial fluid accumulation (which may benefit ARDS) in sepsis and septic shock (264,265,266). However, no clinical study has suggested improved clinical outcomes (morbidity or mortality) with colloid solutions (267,268). Although the severe sepsis subset of one recent randomized controlled trial (RCT) trended toward a more favorable outcome with albumin resuscitation (269), another (meta-analytic) study suggested an opposite trend toward increased mortality with albumin use (268,270). In addition, colloids are substantially more expensive than crystalloid solutions. For these reasons, isotonic crystalloids are recommended as the initial resuscitative solution for severe sepsis and septic shock. The development of a hyperchloremic acidosis can be anticipated with use of large volumes of normal saline. Use of lactated Ringer solution may limit this effect. Hypertonic saline is not recommended for the routine resuscitation of septic shock.
Rapid volume expansion (500 mL isotonic crystalloid every 10–30 minutes) should be continued until clinical and physiologic treatment targets are met. Vasopressor/inotropic support is required if fluid infusion alone fails to achieve physiologic response targets
Early aggressive resuscitation to achieve physiologically normal hemodynamic goals reduces subsequent morbidity and mortality in patients with septic shock. In a pediatric population with septic shock, rapid fluid resuscitation in the first hour of presentation to hospital improved survival (252). In an adult study, the effect of early goal-directed resuscitation to normal physiologic values in patients presenting to an emergency department with severe sepsis or septic shock was examined (253). All patients (both conventional and goal-directed therapy groups) were resuscitated in the emergency room for the first 6 hours to standard hemodynamic end points of CVP ≥8 mm Hg, MAP ≥65 mm Hg, and urine output ≥0.5 mL/kg per hour. The experimental early goal-directed therapy group, in addition, was managed using an experimental protocol to achieve both the standard goals and a central venous oxygen saturation ≥70% (as measured by an oximetric central venous catheter). During the 6 hours of their protocolized emergency room support, the experimental group received 1.5 L more fluid than the control group, and a substantially larger fraction of the patients in the experimental group achieved the physiologic resuscitative goals (99.2% vs. 86.1%). Overall mortality was significantly lower in the early goal-directed therapy group.
Antimicrobial Therapy and Source Control
Historically, critically ill patients with overwhelming infection have not been considered a unique subgroup comparable to neutropenic patients for purposes of selection of antimicrobial therapy. However, critically ill patients with severe sepsis and septic shock, similar to neutropenic patients, are characterized by distinct differences from the typical infected patient that impact on the optimal management strategy. These differences include the following:
· Marked alterations in antibiotic pharmacokinetics
· Increased frequency of hepatic and renal dysfunction
· High prevalence of unrecognized immune dysfunction
· Predisposition to infection with resistant organisms
· Marked increase in frequency of adverse outcome if there is a failure of rapid initiation of effective antibiotic therapy
Critical management decisions in this patient group must often be made emergently in the absence of definitive data regarding the infecting organism and its sensitivity pattern, patient immune status, and organ function. Since outcomes in severe sepsis and septic shock are strongly influenced by the rapidity of administration of an appropriate antimicrobial regimen at first presentation, a particularly thoughtful and judicious approach to initial empiric antimicrobial therapy is required (271,272,273).
Empiric antibiotic regimens should approach 100% coverage of pathogens for the suspected source of infection
Initial administration of inappropriate antimicrobials increases morbidity in a wide range of infections. The occurrence of initiation of inadequate antimicrobial therapy may occur as frequently as 17.1% in community-acquired and 34.3% in nosocomial bacteremia in patients admitted to the ICU (273). Similarly, 18.8% and 28.4% of septic shock cases were initially treated with inadequate antimicrobial therapy in another large study (274). Retrospective studies have shown that the risk of death increases from 30% to 60% in ICU bacteremia (4,272) to 70% to 100% in Gram-negative shock (4) when the initial empiric regimen fails to cover the inciting pathogen. More recent data suggest that the survival of septic shock with inappropriate initial antimicrobial therapy is reduced approximately 5-fold (range 2.5 to 10-fold in selected subgroups) to about 10% (274). These findings of a sharply increased mortality risk with initial inadequate antimicrobial therapy apply to serious infections caused by Gram-negative and Gram-positive bacteria as well as Candida species (4,274,275,276,277,278).
As a consequence, empiric regimens should err on the side of overinclusiveness. The most common cause of initiation of inappropriate antimicrobial therapy is a failure of the clinician to appreciate the risk of infection with antibiotic-resistant organisms (either uncommon organisms with increased native resistance or antibiotic-resistant isolates of common organisms). Selection of an optimal antimicrobial regimen requires knowledge of the probable anatomic site of infection; the patient's immune status, risk factors, and physical environment; and the local microbiologic flora and organism resistance patterns. Risk factors for infection with resistant organisms include a prolonged hospital stay, prior hospitalization, and prior colonization or infection with multiresistant organisms.
|
Table 57.13 Indication for Extended Empiric Antibiotic Therapy of Severe Sepsis/Septic Shock |
||||||||
|
||||||||
Superior empiric coverage can be obtained through the use of a local antibiogram or via consultation with an infectious disease specialist (279). Although not routinely required, extended-spectrum Gram-negative regimens, vancomycin, and/or antifungal therapy may be appropriate in specific high-risk cases with severe sepsis (Table 57.13). In addition, given that 90% to 95% of patients with septic shock have comorbidities or other factors that make them high risk for resistant organisms, it may be appropriate to initially treat all patients with septic shock using a combination of antimicrobials that result in a broadly expanded spectrum of coverage for the first few days. This approach should improve the adequacy of antimicrobial coverage initially, while ensuring that high-risk patients are not inappropriately categorized as low risk.
Intravenous administration of broad-spectrum antimicrobials should be initiated immediately (preferably <30 minutes) following the clinical diagnosis of septic shock
Appropriate intravenous, empiric broad-spectrum therapy should be initiated as rapidly as possible in response to clinical suspicion of infection in the presence of hypotension, i.e., presumptive septic shock. An assumption that hypotension is caused by anything other than sepsis in the setting of documented or suspected infection should be avoided, unless there is very strong data indicating a specific alternate cause. Retrospective studies of human bacteremia, pneumonia, and meningitis with sepsis suggest that mortality in sepsis increases with delays in antimicrobial administration (271,278,280,281,282). One major retrospective analysis of septic shock has suggested that a delay in the initial administration of effective antimicrobial therapy is the single strongest predictor of survival (25). Initiation of effective antimicrobial therapy within the first hour following the onset of septic shock-related hypotension was associated with 79.9% survival to hospital discharge. For every additional hour to effective antimicrobial initiation in the first 6 hours post onset of hypotension, survival dropped an average of 7.6%. With effective antimicrobial initiation between the first and second hour post hypotension onset, survival had already dropped to 70.5%. With effective antimicrobial therapy delay to 5 to 6 hours after hypotension onset, survival was just 42.0%, and by 9 to 12 hours, 25.4%. The adjusted odds ratio of death was already significantly increased by the second hour post hypotension onset, and the ratio continued to climb with longer delays.
Substantial delays before initiation of effective therapy have been shown in several studies of serious infections (271,282,283,284). In septic shock, the median time to delivery of effective antimicrobial therapy following initial onset of recurrent/persistent hypotension was 6 hours (25).
A potential survival advantage may exist if a pathogenic organism can be isolated in severe infections, including septic shock. Every effort should be made to obtain appropriate site-specific cultures to allow identification and susceptibility testing of the pathogenic organism; however, such efforts should not delay antimicrobial therapy.
Antimicrobial therapy should be initiated with dosing at the high end of the therapeutic range in all patients with life-threatening infection
Early optimization of antimicrobial pharmacokinetics can improve the outcome of patients with severe infection, including septic shock. This is most easily achieved by initiating antibiotic therapy with high-end dosing regimens.
Early in sepsis, before the onset of hepatic or renal dysfunction, cardiac output is increased in many patients. In association with increased free drug levels due to decreased albumin levels, drug clearance can be transiently increased (285). As the illness progresses, ICU patients with sepsis or septic shock exhibit substantially increased volumes of distribution and decreased clearance rates. Consequently, suboptimal dosing of antibiotics is common in these conditions (286,287,288,289,290,291). Data is most well developed in reference to aminoglycosides but also exists for fluoroquinolones, β-lactams, and carbapenems (286,287,288,289,290,291). Failure to achieve targets on initial dosing has been associated with clinical failure with aminoglycosides (292,293). Similarly, clinical success rate for treatment of serious infections tracks with higher peak blood levels of fluoroquinolones (nosocomial pneumonia and other serious infections) (294,295,296) and aminoglycosides (Gram-negative nosocomial pneumonia and other serious infections) (297,298). Although there are extensive data in experimental animals and less serious human infections, data for optimization of outcomes using β-lactams in critically ill, infected patients is relatively limited (299,300). A single recent paper has shown improved survival in patients with Pseudomonas bacteremia when treated with extended infusions rather than standard intermittent dosing of piperacillin/tazobactam (301).
Achievement of optimal serum concentrations of aminoglycosides (peak antibiotic serum concentration:pathogen minimal inhibitory concentration [MIC] ratio of ≥12) and longer periods of bactericidal β-lactam and carbapenem serum concentrations (minimum time above MIC in serum of 60% of dosing interval) are appropriate goals (294,302,303). This can most easily be attained with once-daily dosing of aminoglycosides (304). For β-lactams and related antibiotics, increased frequency of dosing (given identical total daily dose) is recommended. For example, piperacillin/tazobactam can be dosed at either 4.5 g every 8 hours or 3.375 g every 6 hours for serious infections; all things being equal, the latter would achieve a higher time above MIC and should be the preferred dosing option. A similar dosing approach should be used for other β-lactams in critically ill patients with life-threatening infections. Limited data suggest that continuous infusion of β-lactams and related drugs may be even more effective, particularly for relatively resistant organisms (305,306,307,308,309).
Multidrug antimicrobial therapy is preferred for the initial empiric therapy of septic shock
Probable pathogens should be covered by at least two antimicrobials with different bactericidal mechanisms. Given that highly resistant organisms are endemic in the critical care environment, multidrug antimicrobial therapy will reduce the probability of failure to cover these organisms. In addition, most patients with septic shock (even those without specific pre-existing immune defects) exhibit significant deficits of neutrophil and monocyte function during the course of their illness (310,311,312,313,314,315,316). Furthermore, malnutrition and organ dysfunction (e.g., renal or hepatic failure), which are common in ICU patients, suppress cell-mediated immunity. Based on these data, septic shock patients likely have a reduced ability to clear infection and may be best managed with multidrug therapy similar to that recommended for patients with neutropenic sepsis (317,318).
No prospective controlled study has specifically compared multiple versus single antimicrobial therapy in a broad range of severe sepsis or septic shock patients. Most infectious diseases physicians and other experts suggest no advantage to multidrug therapy in serious infections, including bacteremia (319,320). However, a subgroup analysis of the sickest subset of patients with Gram-negative bacteremia, with or without shock, has tended to suggest improved survival with the use of two or more antibiotics to which the causative organism is sensitive (321,322,323,324). Similarly, at least two retrospective and one prospective analyses of the most severe, critically ill patients with bacteremic pneumococcal pneumonia suggested improvement in outcome if two or more effective agents were used (325,326,327). This occurred even as patients with pneumococcal bacteremia with a lower severity of illness demonstrated no such benefit (325). A recent secondary analysis of a prospective study of community-acquired pneumonia has shown benefit with multidrug therapy compared to monotherapy but only in the subset of septic shock (328).
Empiric antimicrobial therapy should be adjusted to a narrower regimen within 48 to 72 hours if a plausible pathogen is identified or if the patient stabilizes clinically (i.e., resolution of shock)
Although several retrospective studies have demonstrated that inappropriate therapy of bacteremic septic shock yields increased mortality (4,272,276,277,278), none have suggested that early narrowing of antibiotic therapy is detrimental if the organism is identified or if the patient is responding well clinically. This approach will maximize appropriate antibiotic coverage of inciting pathogens in septic shock while minimizing selection pressure toward resistant organisms. Although it is tempting to continue a broad-spectrum regimen in the 15% of improving patients who are culture-negative for a potential pathogen, intensivists must recognize that a strategy of broad-spectrum initial antimicrobial therapy will be sustainable only if overuse of these agents can be avoided. Aggressive de-escalation of antimicrobial therapy within 48 to 72 hours after initiation is required.
Where possible, early source control should be implemented in patients with severe sepsis, septic shock, and other life-threatening infections
Source control is a critical issue in the management of infection associated with severe sepsis. Infections found in ICU patients frequently require source control for optimal management. The need for such source control may initially be overlooked in many infections commonly found in the ICU (e.g., pneumonia-associated bacterial empyema, decubitus ulcers, C. difficile colitis). Causes of septic shock where source control may be required are noted in Table 57.14.
|
Table 57.14 Common Sources of Severe Sepsis/Septic Shock Requiring Urgent Source Control |
|
|
Source control may include removal of implanted or tunneled devices, open surgical/percutaneous drainage of infected fluids or abscesses, and surgical resection of infected tissues. In a broader sense, it is inclusive of elimination of inciting chemotherapies (e.g., antibiotics driving C. difficile colitis or chemotherapy causing gut injury). Efforts to identify infections requiring invasive forms of source control frequently require rapid (<2 hours) radiographic imaging (often CT scan) or, if clinical status and findings are supportive, direct and immediate surgical intervention without an imaging effort. With rare exceptions, surgical source control should follow aggressive resuscitative efforts to minimize intraoperative morbidity and mortality. In some cases (e.g., rapidly progressive necrotizing soft tissue infections, bowel infarction), optimal management mandates simultaneous aggressive resuscitation and surgical intervention. Subgroup analysis in at least one large prospective, severe sepsis study has suggested that failure to implement adequate source control is associated with increased mortality (329). Earlier surgical intervention has been shown to have a significant impact on outcome in certain rapidly progressive infections such as necrotizing fasciitis (330,331). In a large retrospective study of septic shock, time from hypotension to implementation of source control was found to be highly correlated with outcome (332).
The necessity for or efficacy of source control efforts should be reassessed within 12 to 36 hours following admission and/or source control efforts should be based on clinical response.
Vasopressors and Inotropes
Following fluid resuscitation, patients with severe sepsis or septic shock may demonstrate persistent vasomotor dysfunction characterized by regional perfusion deficits with or without systemic hypotension despite normal or increased CO. Clinical manifestations may include lactic acidosis and ongoing progression of organ failure.
Until recently, the only available approach to correction of regional perfusion defects was vasopressor therapy. Unfortunately, vasopressors do not represent a specific therapy for this problem. Their primary use is to increase systemic arterial pressure to a range that potentially sustains the ability of the vasculature to autoregulate flow on a tissue and organ level (333,334). This allows vital organ perfusion to be supported (potentially at the expense of peripheral perfusion) until definitive therapy (infection source control and antibiotics) can be implemented.
The aim of vasopressor/inotropic therapy in septic shock is simply the optimization of critical organ and tissue perfusion. However, the specific global and/or regional perfusion goals required to achieve this result are complex and controversial. Although specific targets can be suggested, therapy for each patient must be highly individualized and dynamic. Appropriate goals will change over time and should be re-evaluated on a continuing basis.
If hypotension and/or clinical evidence of tissue hypoperfusion persist after adequate fluid resuscitation of septic shock, vasopressor therapy is indicated. Norepinephrine and dopamine are both effective as initial therapy
Initiation of vasopressor support is dependent on the patient's clinical status following fluid resuscitation. If systemic hypotension in association with evidence of tissue/organ hypoperfusion (oliguria, obtundation, lactic acidosis) persists, vasopressor support is indicated. Selection of a vasopressor agent is based on an individualized assessment of the patient's needs. The patient's hemodynamic presentation, the anticipated cardiovascular effect of each vasoactive agent (based on the distribution of receptor activity), and the physician's experience and comfort with each drug should be considered. As a consequence of the variety of factors that may play a role in vasopressor selection, septic shock patients with a predominantly distributive hemodynamic pattern can be appropriately and effectively managed with one of several vasopressors including dopamine, norepinephrine, or phenylephrine.
Ideally, patients should have achieved the targeted intravascular volume status prior to initiation of vasopressors. Although vasopressors can be used to maintain blood pressure for brief periods while intravascular volume is repleted, the infusion of high-dose vasopressors to volume-depleted patients may substantially aggravate ischemic organ injury.
Studies suggesting that norepinephrine is superior to dopamine are less than definitive (335,336,337,338,339,340). No controlled study has directly assessed norepinephrine and dopamine in terms of survival, and few have compared the two agents with respect to markers of organ dysfunction. Studies assessing the effects of these agents on renal and splanchnic perfusion have been mixed, with neither agent demonstrating conclusive superiority (336,337,340,341,342,343,344,345,346,347). Norepinephrine may have more powerful vasopressor activity than dopamine (348). In addition, its inotropic effects are mediated by direct activity on myocardial β-adrenoreceptors. Dopamine pressor effects are weaker than those of norepinephrine, and inotropic effects are substantially indirect (through stimulation of release of myocardial catecholamine stores); excessive tachycardia may be more common. In addition, dopamine may exert significant immunosuppressive effects through suppression of prolactin production from the hypothalamus (349). Phenylephrine, a relatively pure β-adrenergic agonist, has minimal or absent inotropic effects and tends to cause reflex bradycardia. For that reason, it can be very useful in the context of excessive tachycardia or concurrent tachyarrhythmias. However, phenylephrine consistently decreases cardiac output and has an increased propensity to cause ischemic complications. Despite potent inotropic and vasopressor activity, epinephrine is not commonly used as the initial pressor therapy in septic shock because it can generate profound tachycardia, tissue ischemia, and metabolic disturbances.
Dobutamine is indicated for patients with low cardiac index or other evidence of hypoperfusion following achievement of adequate blood pressure. Milrinone can be used as an alternate agent if the response to dobutamine is suboptimal
In some cases of septic shock, clinical or laboratory evidence of hypoperfusion (e.g., oliguria, altered mentation, decreased mixed venous oxygen saturation, increased lactic acidosis) persists despite an adequate blood pressure. In this circumstance, the patient may require a higher blood pressure or assessment of cardiac output (via PAC or echocardiography) to determine the need for inotropic support. In the small proportion of septic shock patients who manifest overt myocardial depression following fluid resuscitation, dobutamine or milrinone may be indicated. Dobutamine can increase cardiac index in septic shock, although the inotropic response is frequently blunted relative to normal subjects (350,351). If catecholamine responsiveness is inadequate, low-dose milrinone may be effective since its inotropic activity is mediated through an alternate mechanism (352). When using either agent, patients must be adequately fluid resuscitated. Severe hypotension can result if intravascular volume is deficient when either dobutamine or milrinone is initiated (350,352).
Although the aim of inotropic therapy in severe sepsis/septic shock is to improve cardiac output and tissue perfusion, specific goals for cardiac index have been controversial; the currently recommended target is a CO within the normal range (approximately 2.5–4 L/minute per m2). The utility of MvO2/ScvO2 as global indices of tissue perfusion adequacy in severe sepsis and septic shock is also uncertain. Limited studies suggest that an MvO2/ScvO2 below the normal range (65%–70%) may indicate inadequacy of resuscitation and/or total perfusion in early septic shock (161). If other hemodynamic targets have been achieved, an MvO2 below 65% may represent an appropriate indication to increase oxygen delivery by starting inotropic agents. Recommendations to increase MvO2 are based on mixed evidence. No benefit was noted in a randomized trial of goal-directed therapy using MvO2 in critically ill patients after the onset of organ dysfunction (353,354). On the other hand, early goal-directed therapy targeting a ScvO2 of ≥70% was associated with improved outcome in another study (253).
Supranormal hemodynamic goals are not indicated in the management of septic shock. Observational studies of medical and surgical critical care patients have demonstrated lower values of physiologic variables such as oxygen consumption (VO2), oxygen delivery (DO2), and CI in nonsurvivors relative to survivors of septic shock (355,356). These observations formed the basis of efforts to implement goal-directed therapy in septic shock to achieve supranormal physiologic parameters consistent with levels observed in survivors (i.e., CI ≥4.5 L/min/m2, DO2 ≥600 mL/minute per m2, and VO2 ≥170 mL/minute per m2). Although a single clinical trial and at least one meta-analysis have suggested some promise with this approach (357,358), several large randomized trials have failed to demonstrate an overall significant benefit of supranormal oxygen delivery in patients with severe sepsis and septic shock (353,354,359,360,361). One has suggested increased mortality when supranormal oxygen delivery was generated with dobutamine (354). The absence of a beneficial effect with supranormal oxygen delivery in patients with severe sepsis and septic shock has been supported in recent meta-analytic reviews (362).
Continuous infusion of vasopressin (0.01–0.04 U/minute) exerts a strong pressor effect and may be beneficial in catecholamine-resistant septic shock following adequate volume resuscitation
Recently, vasopressin levels in septic shock patients have been shown to be decreased (363). Further studies have demonstrated that intravenous infusion of vasopressin in patients with septic shock results in a profound pressor response (236,364,365), an effect that is absent with even larger amounts of vasopressin in normotensive patients (235). A randomized, controlled, double-blind trial of 4-hour infusion of norepinephrine and vasopressin in high-dose, pressor-dependent shock has demonstrated significant improvement in urine output and creatinine clearance, along with a concomitant reduction in conventional vasopressor requirements in the vasopressin group (365). Another RCT has recently demonstrated that, while vasopressin can spare the need for high doses of sympathomimetic agents, outcome is not affected (366).
Because of the limited experience with this compound and the relatively prolonged pharmacologic effect of the drug, vasopressin should be used only after hemodynamic stabilization with standard agents (catecholamines) has been attempted.
At high dose (>0.04 U/minute), vasopressin may produce increased blood pressure, bradycardia, arrhythmias (premature atrial contractions, heart block), severe peripheral vasoconstriction, decreased cardiac output, myocardial ischemia, myocardial infarction, and cardiac arrest. In patients with vascular disease, even relatively modest doses can precipitate peripheral vascular insufficiency, mesenteric ischemia, or myocardial infarction. Given these potential side effects, the minimal amount of vasopressin required should be used to achieve the desired blood pressure goals. In addition, since vasopressin appears to be a pure vasopressor in the context of vasodilatory shock, cardiac output will usually decline. Consideration of placement of an intra-arterial and pulmonary artery catheter (PAC) should be given to all patients receiving vasopressin for shock.
Administration of low- or renal-dose dopamine (1–4 µg/kg per minute) to maintain renal or mesenteric blood flow in sepsis and septic shock is not recommended
Although concurrent infusion of low-dose dopamine during human septic shock does mitigate a decrease in renal perfusion that can occur as a consequence of norepinephrine infusion, the clinical benefit of this therapy is questionable (367,368). Low-dose dopamine infusions can cause a mild transient diuresis in the absence of other vasopressors in nonoliguric critically ill patients (369,370). However, low-dose dopamine does not prevent the development of renal dysfunction in these patients, including those with sepsis and septic shock (371,372).
Invasive and Noninvasive Monitoring
Controversy exists regarding the most appropriate monitoring methods for determining the adequacy of resuscitation in patients with severe sepsis and septic shock. The range of monitoring that must be considered in each patient begins with observation by specially trained nursing personnel, to routine noninvasive devices (e.g., continuous electrocardiographic monitors, intermittent mechanical sphygmomanometry, end-tidal carbon dioxide sensors, percutaneous oximetry), to commonly used invasive techniques (arterial, central venous, and pulmonary artery catheters). Prior to the advent of basic hemodynamic monitoring in the 1950s and early 1960s, clinical examination and manual sphygmomanometry were the only available methods for assessment of cardiovascular status. Clinical judgment correctly predicts the hemodynamic profile (including CO and central venous/pulmonary wedge pressures) of critically ill patients only about half of the time (373,374).
CVP has been considered a useful measure of intravascular volume since the early studies of hypovolemic shock in young men following battlefield trauma (250,251). However, CVP may be much less reliable as a reflection of left ventricular preload in older patients with various cardiopulmonary disorders as are typically found in a modern-day ICU (152,375). Although low filling pressures may reliably indicate hypovolemia in most patients, the presence of a normal or even elevated central venous pressure can be misleading in patients in whom right ventricular afterload is elevated or right ventricular contractility is impaired (376).
The PWP obtained by using a PAC has been considered to reflect intravascular volume more reliably than CVP. In addition, the device allows thermodilution-based derivation of CO (373,374,377). Although the PAC has gained widespread acceptance, significant questions about its use have been raised. Several studies have questioned the relationship of PAC-derived, pressure-based estimates of ventricular preload in specific groups of critically ill patients (375) and, more recently, even in normal subjects (378). In addition, the lack of randomized trials demonstrating benefit and the association of PAC with excess mortality in two observational cohort studies have led to concerns regarding the clinical utility and safety of PACs (379,380). Despite these concerns, the PAC remains the most commonly used modality for hemodynamic monitoring of unstable critically ill patients.
Patients with established septic shock should have continuous monitoring of blood pressure, oxygen saturation, electrocardiogram (ECG), and urine output in a closed ICU staffed with full-time dedicated intensivists and critical care–trained nurses
Several studies have demonstrated that a reduced mortality with decreased length of stay and overall cost for a wide range of individual conditions are obtained when critically ill patients are cared for in closed ICUs staffed with full-time dedicated intensivists and nurses (381,382,383,384,385). Similar improvements in outcome of sepsis and septic shock have been documented with the use of dedicated intensivists in closed ICUs (386). Among the practice differences associated with the use of full-time intensivists is a greater use of invasive monitoring (384).
Patients requiring vasopressor agents for a prolonged period or at high dose should be strongly considered for insertion of an arterial pressure catheter for continuous blood pressure monitoring, as well as to facilitate frequent measurements of arterial blood gases and chemistry
Accurate, continuous monitoring of blood pressure is required for optimal assessment of severity of shock, response to fluid resuscitation, and titration of vasopressors and inotropes. However, intense peripheral vasoconstriction may occur during shock as a consequence of the vascular compensatory response to hypotension or due to administration of vasopressors. Clinical ausculatory and noninvasive mechanical methods can be highly inaccurate in this setting (387,388). Patients with sustained shock, particularly those requiring vasopressor support, should be assessed for placement of an intra-arterial catheter for continuous blood pressure monitoring. However, such catheters should be preferentially placed in peripheral sites in non-end arteries (radial, dorsalis pedis), and should be used with caution in patients at high risk for vascular disease.
If volume resuscitation requirements exceed 2 L, placement of a central venous catheter for monitoring of CVP and for vasopressor/inotrope infusion should be considered. An initial target CVP of ≥8 mm Hg is recommended
Fluid deficits during septic shock in adults typically range from 5 to 10 L (248). In the absence of significant cardiopulmonary dysfunction, central venous pressure should accurately assess intravascular volume status. However, cardiopulmonary dysfunction is not uncommon in patients with septic shock either as an underlying predisposition to critical illness/sepsis or as a consequence of the injury (ARDS/acute lung injury [ALI], myocardial depression). Low central venous pressures remain indicative of hypovolemia; elevated or normal central venous pressures in this patient group may not necessarily indicate euvolemia. CVP monitoring should be entertained if substantial amounts of fluid resuscitation are required to ensure that overt hypovolemia is adequately addressed. The initial target CVP should be ≥8 mm Hg, with additional increases indicated by the effect of fluid boluses on cardiac output and clinical perfusion. The overall goal is to provide adequate cardiac output and tissue perfusion using the lowest necessary cardiac filling pressures.
Initiation of invasive cardiac monitoring using a pulmonary artery catheter should be considered if there has been an inadequate response to fluid resuscitation (3–5 L or CVP 8–12 mm Hg), if there is clinical suspicion of intravascular fluid volume overload, or if the patient has impaired cardiac function. An initial target of PWP of 12–15 mm Hg will ensure that hypovolemia is absent in most patients, but higher pressures may be required in certain subgroups
Although the maintenance of a blood pressure adequate for autoregulation of blood flow to vital organs and tissues is the first objective in the resuscitation of septic shock, support of global perfusion is also critical. Adequacy of global perfusion cannot always be reliably inferred from the clinical examination or CVP/arterial pressure monitoring (373,377,389). Patients who respond poorly to fluid resuscitation or are at high risk for fluid resuscitation–related complications may benefit from pulmonary artery catheterization. A substantial degree of variability in the relationship between PWP and end-diastolic volumes makes it difficult to specify target PWP goals that ensure adequate cardiac output and tissue perfusion (378,390,391). In general, a PWP titrated to at least 12 to 15 cm H2O will optimize cardiac function (152). If hypotension persists, a higher PWP may be beneficial as assessed by measuring the effect of additional fluids on cardiac index. An elevated PWP may risk the development or aggravation of ALI and ARDS (392,393). Specific groups that may require higher PWP include those with congestive heart failure, left ventricular hypertrophy, restrictive or constrictive heart disease, or increased intrathoracic pressures, including those on high levels of positive end-expiratory pressure (PEEP).
In patients with vasopressor-requiring shock who develop progressive organ failure or hypoxemic respiratory failure, pulmonary artery catheterization may be a useful clinical management tool
The information available from a PAC can be used to help determine the cause of shock and provide a guide for interventions to maintain an appropriate cardiac output and intravascular volume to limit the risk of further progression of organ dysfunction/failure. If PACs are beneficial in patients with sepsis, the most likely candidates may be those in whom resuscitation by clinical assessment or CVP fails to reverse the progression of organ failure.
Invasive monitoring using a pulmonary artery catheter is not recommended for routine use in all patients with severe sepsis
At least one major prospective, nonrandomized multicenter study has suggested increased length of stay, costs, and mortality in a cohort of risk-matched patients receiving a PAC in the first 24 hours after ICU admission (394). A recent multicenter randomized controlled trial involving 676 subjects with shock (primarily septic), ARDS, or both has demonstrated no difference in organ failure–free days, renal support needs, vasopressor requirements, mechanical ventilation, ICU/hospital length of stay (14 and 90 day), or mortality between subjects randomized to pulmonary artery catheterization or controls (395). A second, smaller randomized trial of 200 patients (about 100 with sepsis) also demonstrated no mortality difference with or without the use of PAC (396). Other smaller studies, including one randomized trial in high-risk operative patients, failed to demonstrate any difference in mortality with PAC use (397,398). In contrast, one meta-analysis of RCTs demonstrated a reduced mortality risk in surgical ICU patients treated with PAC but no effect on mortality in medical or mixed ICU patients (399). On the basis of the total data available, routine use of PAC in patients with sepsis or other critical illness cannot be recommended.
Specific Therapy
As discussed, patients with severe sepsis and septic shock must first be treated using the following: (i) Appropriate resuscitation, (ii) broad spectrum antimicrobials, (iii) source control, and (iv) physiologic support of organ function in the intensive care unit. Immunomodulatory therapy has been evaluated only in association with adequate treatment based on these four elements.
In the last few decades, the dominant hypotheses regarding the pathogenesis of septic shock and septic organ dysfunction focused on inflammatory mediators including TNF α, IL-1β, interleukin-6, and platelet-activating factor. Several clinical trials have been performed evaluating both nonspecific inhibitors of inflammation such as nonsteroidal anti-inflammatory drugs and high-dose glucocorticoids and specific immunomodulatory agents such as monoclonal antibody against TNF α and IL-1 receptor antagonist (400,401). Despite an expenditure of over 1 billion dollars, these studies have failed to demonstrate a survival benefit. No primary immunomodulatory experimental agent has received regulatory approval.
Recently accepted models of the pathogenesis of sepsis have emphasized a central role for altered hemostatic/coagulant function. Three coagulation modulators have been assessed in large randomized controlled clinical trials: Tissue factor pathway inhibitor, antithrombin III, and drotrecogin alfa (activated) (recombinant human activated protein C). Drotrecogin alfa (activated) is the first and, to date, only specific therapy that has been shown to improve survival in patients with severe sepsis and septic shock.
Recombinant human-activated protein C should be administered in patients with suspected sepsis with organ dysfunction. Acceptable criteria include, but are not necessarily limited to, a minimum of one organ dysfunction with an Acute Physiology and Chronic Health Evaluation (APACHE) II score ≥25; or if an accurate APACHE II score is unavailable, the presence of two or more organ dysfunctions
Although clinical trials of modulation of the coagulation cascade for treatment of sepsis have been performed with several agents (e.g., antithrombin III [27], tissue factor pathway inhibitor [28]), only drotrecogin alfa (activated) has been shown to improve mortality (26). The pivotal study was an international multicenter RCT that compared drotrecogin alfa (activated) to placebo used in conjunction with standard treatment (antibiotics, physiologic support, and surgical source control) (26). Patients were entered into the study if they exhibited acute organ dysfunction due to a suspected infection (severe sepsis) within a 24-hour window. The study was stopped at a planned interim analysis because of definitive statistical evidence that supported a beneficial treatment effect. Using an intention-to-treat analysis, the study demonstrated an absolute mortality reduction of 6.5% from 31.3% in the placebo group to 24.8% in the drotrecogin alfa (activated) group, yielding a highly significant 21% relative risk reduction. Subsequent open-label studies of drotrecogin alfa (activated) using the same criteria as in the pivotal study have demonstrated a consistent mortality rate between 25.1% and 26.1% (402,403). A retrospective analysis of an open-label study suggests that earlier initiation of treatment (<24 hours after diagnosis of severe sepsis) yields superior outcomes (404).
The original study demonstrated a differential treatment effect based on either APACHE II scores or the number of acute organ dysfunctions present at the time of enrollment into the study. The absolute reduction in mortality was 1.7% among patients with a single dysfunctional organ and 7.4% among those with two or more dysfunctional organs (402,405). Similarly, there was no overall reduction in absolute mortality in the first 2 quartiles of APACHE score (score <25), whereas there was a 13% reduction in the last 2 quartiles (score ≥25) (405). A more recent RCT (prematurely terminated for futility) has underlined concerns regarding the utility of drotrecogin alfa (activated) in relatively low-risk (generally APACHE<25 or single organ failure) adult patients with a slight trend toward increased mortality risk in the treatment arm (29). Similarly, a study of drotrecogin alfa (activated) in pediatric septic shock with respiratory failure was also prematurely terminated due to its futility, along with evidence of an increased central nervous system (CNS) bleeding risk in neonates (406).
Drotrecogin alfa (activated) remains approved for management of high-risk patients with severe sepsis/septic shock, but new studies are ongoing to validate the continued use of this agent.
Intravenous immune globulin should be considered for patients suffering from streptococcal toxic shock syndrome
The potential utility of polyclonal immune globulin preparations for severe sepsis and septic shock in general is uncertain at present. One meta-analysis has suggested that sepsis-related mortality is significantly reduced when intravenous immunoglobulin (IVIG) is used in the management of such patients (407). A small randomized controlled trial of trauma patients has also demonstrated a reduced incidence of septic complications including pneumonia and other infections (other than catheter-related infections), although ICU length of stay and mortality were not reduced (408). Evidence favoring the use of polyclonal immunoglobulin for defined invasive streptococcal infections, including streptococcal septic shock, is more definitive. A case-matching study has demonstrated an improved 30-day survival in patients treated with intravenous polyclonal immune globulin, while a randomized controlled trial (aborted prematurely due to low enrollment) has shown decreased early sepsis-related organ failure with a trend toward improved survival (409).
Immunosuppressive doses of corticosteroids are contraindicated in the management of sepsis and septic shock
In the past, high-dose steroids had been advocated for sepsis with organ failure to dampen inflammatory responses and minimize organ dysfunction (410). Several large multicenter randomized controlled trials have definitively demonstrated that administration of high dose (15–30 mg/kg methylprednisolone equivalent) corticosteroids fail to improve outcome in adult septic shock (411,412,413,414). In some of these studies, mortality in specific subgroups appeared to be increased with steroid treatment (412).
Supportive Therapy
Although specific therapies for septic shock continue to be developed, general supportive care, in conjunction with antibiotics, remains the standard of care. Fluid and vasopressor/inotropic support have been addressed in this chapter. In addition, there has been an explosion of data in recent years regarding the efficacy of other elements of supportive care including ventilatory strategies, intensity of dialysis, endocrine support, and glycemic management. In other key areas (e.g., nutritional support), definitive data are lacking. Nonetheless, it is likely that an aggressive approach to optimization of supportive care, in combination with anti-infective therapy and resuscitative efforts, can improve morbidity and mortality. For that reason, application of appropriate support modalities in a timely manner should be the standard of care of septic patients in all ICUs.
Intensive renal replacement therapy (daily intermittent dialysis or continuous renal replacement therapy) is indicated for severe sepsis or septic shock with renal failure
Indications for acute dialysis in the ICU population are not dissimilar to those for other patients. These indications include volume overload, electrolyte imbalance, acid-base disturbances, elevated blood urea nitrogen, uremic pericarditis, or uremic encephalopathy. Unfortunately, ICU patients, especially those with acute renal failure, may have altered hemodialysis kinetics such that standard intermittent dialysis may offer suboptimal urea clearance kinetics despite apparently equivalent doses. Compared to standard intermittent dialysis, daily hemodialysis has been shown to yield higher urea clearance and improved mortality in ICU patients with acute renal failure (415). Similarly, another study has demonstrated that higher urea clearance with continuous venovenous hemodialysis yields reduced mortality (416). Whether these data can be extrapolated to include septic patients with a background of chronic renal failure is unknown. Peritoneal dialysis is not appropriate since even high-frequency exchanges yield relatively low urea clearance kinetics. A recent study of infection-related acute renal failure that included cases of sepsis demonstrated increased mortality among those treated with peritoneal dialysis compared to those treated with hemodialysis (417).
Intensive insulin therapy maintaining a blood glucose of 4.4 to 6.1 mmol/L (80–110 mg/dL) may be beneficial in critically ill ICU patients with severe sepsis
Hyperglycemia is a recognized risk factor for increased mortality in the critically ill independent of the APACHE II score (418). One single-center randomized, controlled, nonblinded trial has indicated that tight glycemic control in surgical ICU patients undergoing mechanical ventilation (mostly post–coronary artery bypass graft or other cardiovascular surgery) reduces the incidence of severe sepsis and decreases mortality, primarily because of a decreased incidence of multiple organ failure with septic foci (419). These data are consistent with other clinical and experimental studies suggesting the presence of granulocyte dysfunction and increased risk of infection in postoperative surgical patients with persistent hyperglycemia (420). However, another RCT by the same group has failed to demonstrate similar improvements in critically ill medical patients (421). A retrospective subgroup analysis, however, suggested mortality improvement in those patients admitted with an ICU length of stay of greater than 3 days. In addition, there was a decreased incidence of renal dysfunction and critical illness polyneuropathy, with fewer days on ventilator support and shorter ICU and hospital length of stay (421,422). No definitive data exist regarding the question of whether a tight control strategy is useful in patients who are already septic. In addition, these data should be interpreted with caution pending replication of these results in other centers.
Stress dose steroids may be administered at presentation to selected patients with septic shock pending the result of an ACTH stimulation test
Several previous large randomized, double-blind, multicenter trials have definitively demonstrated that administration of immunosuppressive (15–30 mg/kg methylprednisolone equivalent) corticosteroids fail to improve outcome in adult septic shock (411,412,413,414). However, some evidence suggests that low “stress-dose” corticosteroids may be beneficial. A relative adrenal insufficiency has been suggested to exist in a substantial subset of patients with septic shock (239,423). Among other deleterious effects, adrenal insufficiency can result in impairment of catecholamine sensitivity (423,424,425). Administration of stress-dose steroids (150–300 mg hydrocortisone daily equivalent) to patients with septic shock can decrease pressor requirements while suppressing inflammatory markers (424,426,427). One recent RCT has demonstrated that 7 days of therapy with hydrocortisone, 50 mg IV every 6 hours, and fludrocortisone, 50 µg orally once daily, generates a significant reduction in mortality in patients with relative adrenal insufficiency (428). Subgroup analysis demonstrated that this improvement was restricted to those who fail to respond to an ACTH challenge (about 75% of septic shock patients), with an increase in serum cortisol of at least 250 nmol/L (9 µg/dL). In the recent past, these data were interpreted as suggesting that patients with pressor-dependent septic shock should undergo ACTH challenge on admission, followed immediately by initiation of stress-dose steroid therapy. If the ACTH stimulation test was within normal limits, corticosteroids were discontinued. If the test results indicated relative adrenal insufficiency, hydrocortisone and fludrocortisone were often continued for 7 days or as otherwise clinically indicated.
The major uncertainty with regard to stress-dose steroid therapy had been the appropriate test and value of serum cortisol to indicate adrenal insufficiency. Various studies supported using random cortisol levels between 275 and 950 nmol/L (10–35 µg/dL) during the acute stress, or increments of cortisol of 250 nmol/L (9 µg/dL) within the first hour following ACTH stimulation (239,423,428). Although no definitive data existed as to which cutoff value was best, many clinicians considered a random value of less than 400 nmol/L (15 µg/dL) to be sufficiently suggestive of relative adrenal insufficiency during the shock state to initiate and continue stress-dose therapy. Similarly, a value greater than 950 nmol/L (35 µg/dL) during shock has been thought to be sufficiently normal to discontinue stress-dose therapy without further assessment. Values between those two extremes were often interpreted to be an indication for ACTH challenge with a response of less than 250 nmol/L (9 µg/dL) supporting the need, for steroid therapy. Unfortunately, a recent study has challenged these accepted cutoffs in the critically ill by questioning the scientific validity of using total as opposed to free serum concentrations of serum cortisol in such patients (429).
Of most concern, a major multicenter, placebo-controlled, double-blind RCT of septic shock has failed to confirm an improvement in survival regardless of ACTH responsiveness (430). The steroid group did exhibit a reduction in pressor days but also had a higher incidence of superinfections and associated sepsis/septic shock events. Confounding these results, the steroid regimen (hydrocortisone alone) differed from the regimen used in the previous positive study and could also be implemented as late as 72 hours following onset of septic shock. Based on these data, stress-dose or low-dose steroid therapy should not be considered part of the routine management of septic shock pending further definitive trials.
Low-volume (6–8 mL/kg ideal body weight), pressure-limited ventilation is indicated in patients with sepsis-associated acute lung injury or acute respiratory distress syndrome
Animal and human studies have suggested that high levels of PEEP and large tidal volumes are associated with increased pulmonary generation of proinflammatory cytokines (431,432) and ventilation-induced lung injury (433). ALI and ARDS represent a manifestation of MODS that may occur in conjunction with severe sepsis and septic shock. Septic patients with bilateral persistent opacities, in association with an acute and persistent defect of oxygenation (PaO2/FiO2 ratio of ≤200 for ARDS and ≤300 for ALI) and no clinical evidence of left atrial hypertension or a pulmonary wedge pressure of ≥18 mm Hg, fit the criteria for this syndrome (434). Small randomized studies have supported the possibility that a lung-protective strategy using low tidal volumes and limited airway pressures may decrease pulmonary injury and decrease mortality (435). A single large multicenter, randomized controlled trial has demonstrated that ventilation of critically ill patients with ARDS with a low tidal volume (tidal volume of 6–8 mL/kg ideal body weight) reduces all-cause absolute mortality by 10% (from 40% to 30%; 25% relative risk reduction) (436). Patients with severe sepsis or septic shock who meet criteria for ALI or ARDS should be ventilated with a low-volume, pressure-limited strategy. Available evidence suggests that ventilation of patients at risk for ALI/ARDS with this strategy does not prevent the development of this pulmonary syndrome (437).
Endotracheal intubation and mechanical ventilation should be considered early in the management of all patients with sepsis and organ failure
Airway intubation is indicated for all patients with impaired airway protection reflexes (e.g., as a consequence of cerebral hypoperfusion or septic encephalopathy), refractory hypoxemia, respiratory acidosis, or respiratory distress associated with ongoing hypotension/hypoperfusion. Though not yet addressed by systematic studies, clinical experience suggests that respiratory arrest is a significant risk in such patients. These observations are consistent with observations of respiratory muscle compromise and respiratory failure in animal models of septic shock (438,439).
Enteral feeding should be considered within 24 hours of admission to the ICU for most patients with sepsis and septic shock. Parenteral feeding should be used only if enteral feeding is not possible despite best efforts
Recent meta-analyses suggest that early enteral feeding lowers the risk of infection and improves survival compared to delayed feeding in the critically ill (440). These findings are consistent with animal studies demonstrating that enteral nutrition maintains gut mucosal integrity, decreases bacterial translocation, and limits the systemic inflammatory response to bacterial toxins (441). Diminished bowel sounds should not prevent a trial of enteral feeding. Most patients will tolerate enteral feeding if a small bowel tube is used. Studies of parenteral feeding in the ICU have, in general, failed to demonstrate an improvement in mortality in critically ill patients (442). Other studies demonstrate the superiority of enteral over parenteral feeding in critically ill patients with respect to costs and complications, including risk of infection (441,443).
Intravenous administration of sodium bicarbonate is not indicated for sepsis-associated metabolic acidosis with a pH ≥7.15
Human investigations demonstrate that intravenous administration of sodium bicarbonate for lactic acidosis (pH ≥7.15) associated with septic shock does not improve cardiac performance or reduce vasopressor requirements compared to administration of an equimolar amount of normal saline (444,445). No human data exist in regard to the effect of intravenous bicarbonate administration for more severe degrees of metabolic acidosis.
|
Table 57.15 Time Line of Implementation of Recommended Diagnostic and Therapeutic Interventions |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
Summary
Severe sepsis and septic shock continue to be a major cause of mortality and morbidity among patients requiring ICU support. In recent years, both basic and clinical research in the field have accelerated substantially. This has led to the publication of several studies with major implications regarding the appropriate management of patients with these conditions. Many of these new studies relate to optimization of supportive care. Although controversial, a single specific therapy, drotrecogin alfa (activated), has been shown to improve mortality in severe sepsis and septic shock. Few major studies in the areas of fluid resuscitation, vasopressors/inotropes, invasive and noninvasive monitoring, or antimicrobial therapy have been published in recent years. Nonetheless, outcome can most likely be improved by taking a systematic approach to therapy as described in Table 57.15. Although significant improvements in outcome have been made possible by new pharmacologic therapies, recent studies focusing on antimicrobial and supportive elements clearly demonstrate that close attention to established therapies can have a substantial impact on survival in severe sepsis and septic shock.
References
1. Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310.
2. Finland M, Jones WF, Barnes MW. Occurence of serious bacterial infections since the introduction of antibacterial agents. JAMA. 1959;84:2188–2197.
3. Hemminki E, Paakkulainen A. Effect of antibiotics on mortality from infectious diseases in Sweden and Finland. Am J Public Health. 1976;66:1180–1184.
4. Kreger BE, Craven DE, McCabe WR. Gram-negative bacteremia. IV. Re-evaluation of clinical features and treatment in 612 patients. Am J Med. 1980;68(3):344–355.
5. Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–1554.
6. Minino AM, Heron MP, Smith BL. Deaths: preliminary data for 2004. Natl Vital Stat Rep. 2006;54(19):1–49.
7. Friedman G, Silva E, Vincent JL. Has the mortality of septic shock changed with time? Crit Care Med. 1998;26(12):2078–2086.
8. Geroulanos S, Douka ET. Historical perspective of the word “sepsis.” Intensive Care Med. 2006;32:2077.
9. Budelmann G. Hugo Schottmuller, 1867–1936. The problem of sepsis [in German]. Internist (Berl). 1969;10(3):92–101.
10. Bone R. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–874.
11. Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31(4):1250–1256.
12. Marshall JC, Cook DJ, Christou NV, et al. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23(10):1638–1652.
13. Ferreira FL, Bota DP, Bross A, et al. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA. 2001;286(14):1754–1758.
14. Bone RC, Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann Intern Med. 1996;125(8):680–687.
15. Padkin A, Goldfrad C, Brady AR, et al. Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med. 2003;31(9):2332–2338.
16. Finfer SBR, Bellomo R, Lipman J, et al. Adult-population incidence of severe sepsis in Australian and New Zealand intensive care units. Intensive Care Med. 2004;30:589–596.
17. Brun-Buisson C, Meshaka P, Pinton P, et al. EPISEPSIS: a reappraisal of the epidemiology and outcome of severe sepsis in French intensive care units. Intensive Care Med. 2004;30(4):580–588.
18. Sundararajan V, Macisaac CM, Presneill JJ, et al. Epidemiology of sepsis in Victoria, Australia. Crit Care Med. 2005;33(1):71–80.
19. Danai P, Martin GS. Epidemiology of sepsis: recent advances. Curr Infect Dis Rep. 2005;7:329–334.
20. Brun-Buisson C, Doyon F, Carlet J, et al. Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA. 1995;274:968–974.
21. Annane D, Aegerter P, Jars-Guincestre MC, et al. Current epidemiology of septic shock: the CUB-Rea Network. Am J Resp Crit Care Med. 2003;168(2):165–172.
22. Sands KE, Bates DW, Lanken PN, et al. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA. 1997;278(3):234–240.
23. Pakhale S, Roberts D, Light B, et al. A geographically and temporally comprehensive analysis of septic shock: impact of age, sex and socioeconomic status. Crit Care Med. 2005;33:A79.
24. Zhan C, Miller MR. Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA. 2003;290(14):1868–1874.
25. Kumar A, Roberts D, Wood KE, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34(6):1589–1596.
26. Bernard GR, Vincent JL, Laterre PF. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709.
27. Warren BL, Eid A, Singer P, et al. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286(15):1869–1878.
28. Abraham E, Reinhart K, Opal S, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290(2):238–247.
29. Abraham E, Laterre PF, Garg R, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353(13):1332–1341.
30. O'Brien JM Jr, Lu B, Ali NA, et al. Alcohol dependence is independently associated with sepsis, septic shock, and hospital mortality among adult intensive care unit patients. Crit Care Med. 2007;35(2):345–350.
31. Rangel-Frausto MS, Pittet D, Costigan M, et al. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study [comment]. JAMA. 1995;273(2):117–123.
32. Loeb HS, Cruz A, Teng CY, et al. Haemodynamic studies in shock associated with infection. Br Heart J. 1967;29(6):883–894.
33. Weil MH, Shubin H, Biddle M. Shock caused by gram-negative microorganisms. Ann Intern Med. 1964;60:384–400.
34. Kasal J, Jovanovic Z, Clermont G, et al. Comparison of Cox and Gray's survival models in severe sepsis. Crit Care Med. 2004;32(3):700–707.
35. Parker MM, Shelhamer JH, Natanson C, et al. Serial cardiovascular variables in survivors and nonsurvivors of human septic shock: heart rate as an early predictor of prognosis. Crit Care Med. 1987;15:923–929.
36. Alberti C, Brun-Buisson C, Burchardi H, et al. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med. 2002;28(2):108–121.
37. Richards MJ, Edwards JR, Culver DH, et al. Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit Care Med. 1999;27(5):887–892.
38. Wisplinghoff H, Bischoff T, Tallent SM, et al. Nosocomial bloodstream infections in US hospitals: analysis of. 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39(3):309–317.
39. Macphail GL, Taylor GD, Buchanan-Chell M, et al. Epidemiology, treatment and outcome of candidemia: a five-year review at three Canadian hospitals. Mycoses. 2002;45(5-6):141–145.
40. Berrouane YF, Herwaldt LA, Pfaller MA, et al. Trends in antifungal use and epidemiology of nosocomial yeast infections in a university hospital. J Clin Microbiol. 1999;37(3):531–537.
41. Abi-Said D, Anaissie E, Uzun O, et al. The epidemiology of hematogenous candidiasis caused by different Candida species. Clin Infect Dis. 1997;24(6):1122–1128.
42. Murray BE, Murray BE. Vancomycin-resistant enterococcal infections. N Engl J Med. 2000;342(10):710–721.
43. Giamarellou H. Multidrug resistance in Gram-negative bacteria that produce extended-spectrum beta-lactamases (ESBLs). Clin Microbiol Infect. 2005;11(Suppl 4):1–16.
44. Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med. 2005;352(14):1445–1453.
45. From the Centers for Disease Control. Staphylococcus aureus resistant to vancomycin–United States, 2002. JAMA. 2002;288(7):824–825.
46. Heumann D, Glauser MP, Calandra T. Molecular basis of host-pathogen interaction in septic shock. Curr Opin Microbiol. 1998;1(1):49–55.
47. Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745.
48. Bauer S, Kirschning CJ, Hacker H, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A. 2001;98(16):9237–9242.
49. Sparwasser T, Miethke T, Lipford G, et al. Bacterial DNA causes septic shock. Nature. 1997;386(6623):336–337.
50. Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738.
51. Dziarski R, Wang Q, Miyake K, et al. MD-2 enables Toll-like receptor 2 (TLR2)-mediated responses to lipopolysaccharide and enhances TLR2-mediated responses to Gram-positive and Gram-negative bacteria and their cell wall components. J Immunol. 2001;166(3):1938–1944.
52. Yang RB, Mark MR, Gray A, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395(6699):284–288.
53. Lien E, Sellati TJ, Yoshimura A, et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274(47):33419–33425.
54. Schwandner R, Dziarski R, Wesche H, et al. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. 1999;274(25):17406–17409.
55. Manocha S, Feinstein D, Kumar A, et al. Novel therapies for sepsis: antiendotoxin therapies. Exp Opin Invest Drugs. 2002;11(12):1795–1812.
56. Martich GD, Boujoukos AJ, Suffredini AF. Response of man to endotoxin. Immunobiology. 1993;187(3–5):403–416.
57. Ing DJ, Zang J, Dzau VJ, et al. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak, and Bcl-x. Circ Res. 1999;84:21–33.
58. Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. FASEB J. 1994;8:504–512.
59. Balligand JL, Ungureanu-Longrois D, Simmons WW, et al. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. J Biol Chem. 1994;269:27580–27588.
60. Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemost. 2001;86(1):51–56.
61. Heumann D. CD14 and LBP in endotoxemia and infections caused by Gram-negative bacteria. J Endotoxin Res. 2001;7(6):439–441.
62. Raetz CR, Ulevitch RJ, Wright SD, et al. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J. 1991;5(12):2652–2660.
63. Rietschel ET, Kirikae T, Schade FU, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8(2):217–225.
64. Hellman J, Warren HS. Antiendotoxin strategies. Infect Dis Clin North Am. 1999;13(2):371–386.
65. Kumar A, Zanotti S, Bunnell G, et al. Interleukin-10 blunts the human inflammatory response to lipopolysaccharide without affecting the cardiovascular response. Crit Care Med. 2005;33(2):331–340.
66. Kumar A, Bunnell E, Lynn M, et al. Experimental human endotoxemia is associated with depression of load-independent contractility indices: prevention by the lipid a analogue E5531. Chest. 2004;126(3):860–867.
67. Suffredini AF, Reda D, Banks SM, et al. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J Immunol. 1995;155(10):5038–5045.
68. Le Roy D, Di Padova F, Tees R, et al. Monoclonal antibodies to murine lipopolysaccharide (LPS)-binding protein (LBP) protect mice from lethal endotoxemia by blocking either the binding of LPS to LBP or the presentation of LPS/LBP complexes to CD14. J Immunol. 1999;162(12):7454–7460.
69. Schumann RR, Leong SR, Flaggs GW, et al. Structure and function of lipopolysaccharide binding protein. Science. 1990;249(4975):1429–1431.
70. Heumann D, Barras C, Severin A, et al. Gram-positive cell walls stimulate synthesis of tumor necrosis factor alpha and interleukin-6 by human monocytes. Infect Immun. 1994;62(7):2715–2721.
71. Mattsson E, Rollof J, Verhoef J, et al. Serum-induced potentiation of tumor necrosis factor alpha production by human monocytes in response to staphylococcal peptidoglycan: involvement of different serum factors. Infect Immun. 1994;62(9):3837–3843.
72. Wright SD, Ramos RA, Tobias PS, et al. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249(4975):1431–1433.
73. Hailman E, Lichenstein HS, Wurfel MM, et al. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179(1):269–277.
74. Ingalls RR, Golenbock DT, Ingalls RR, et al. CD11c/CD18, a transmembrane signaling receptor for lipopolysaccharide. J Exp Med. 1995;181(4):1473–1479.
75. Kusunoki T, Hailman E, Juan TS, et al. Molecules from Staphylococcus aureus that bind CD14 and stimulate innate immune responses. J Exp Med. 1995;182(6):1673–1682.
76. Kirschning CJ, Wesche H, Merrill AT, et al. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med. 1998;188(11):2091–2097.
77. Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749–3752.
78. Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451.
79. Chow JC, Young DW, Golenbock D, et al. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–10692.
80. Tauszig S, Jouanguy E, Hoffmann JA, et al. Toll-related receptors and the control of antimicrobial peptide expression in Drosophila. Proc Natl Acad Sci U S A. 2000;97(19):10520–10525.
81. Lien E, Means TK, Heine H, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105(4):497–504.
82. Muroi M, Ohnishi T, Tanamoto K, et al. MD-2, a novel accessory molecule, is involved in species-specific actions of Salmonella lipid A. Infect Immun. 2002;70(7):3546–3550.
83. Muroi M, Ohnishi T, Tanamoto K, et al. Regions of the mouse CD14 molecule required for toll-like receptor. 2- and 4-mediated activation of NF-kappa B. J Biol Chem. 2002;277(44):42372–42379.
84. Muller-Alouf H, Alouf JE, Gerlach D, et al. Human pro- and anti-inflammatory cytokine patterns induced by Streptococcus pyogenes erythrogenic (pyrogenic) exotoxin A and C superantigens. Infect Immun. 1996;64(4):1450–1453.
85. Cohen J. The immunopathogenesis of sepsis. Review. Nature. 2002;420(6917):885–891.
86. Docke WD, Randow F, Syrbe U, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nature Med. 1997;3(6):678–681.
87. Mira JP, Cariou A, Grall F, et al. Association of TNF2, a TNF-alpha promoter polymorphism, with septic shock susceptibility and mortality: a multicenter study. JAMA. 1999;282(6):561–568.
88. Holmes CL, Russell JA, Walley KR. Genetic polymorphisms in sepsis and septic shock: role in prognosis and potential for therapy. Chest. 2003;124:1103–1115.
89. Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064.
90. Lorente JA, Landin L, Renes E, et al. Role of nitric oxide in the hemodynamic changes of sepsis. Crit Care Med. 1993;21:759–767.
91. Kilbourn RG, Gross SS, Jubran A, et al. N-methyl-L-arginine inhibits tumor necrosis factor-induced hypotension: implications for the involvement of nitric oxide. Proc Natl Acad Sci U S A. 1990;87:3629–3623.
92. Thiemermann C, Szabö C, Mitchell JA, et al. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Natl Acad Sci U S A. 1993;90:267–271.
93. Kubes P. Nitric oxide modulates microvascular permeability. Am J Physiol. 1992;262(2):H611–H615.
94. Kumar A, Thota V, Dee L, et al. Tumor necrosis factor-alpha and interleukin-1 beta are responsible for depression of in vitro myocardial cell contractility induced by serum from humans with septic shock. J Exp Med. 1996;183:949–958.
95. Kumar A, Krieger A, Symeoneides S, et al. Myocardial dysfunction in septic shock, II: role of cytokines and nitric oxide. J Cardiovasc Thorac Anesth. 2001;15(4):485–511.
96. Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–1624.
97. Muta T, Iwanaga S, Muta T, et al. Clotting and immune defense in Limulidae. Progr Molec Subcell Biol. 1996;15:154–189.
98. Nachum R, Nachum R. Antimicrobial defense mechanisms in Limulus polyphemus. Progr Clin Biol Res U S A. 1979;29:513–524.
99. McGilvray ID, Rotstein OD, McGilvray ID, et al. Role of the coagulation system in the local and systemic inflammatory response. World J Surg. 1998;22(2):179–186.
100. Wheeler AP, Bernard GR. Treating patients with severe sepsis. N Engl J Med. 1999;340:207–214.
101. Gando S, Nanzaki S, Sasaki S. Activation of the extrinsic coagulation pathway in patients with severe sepsis and septic shock. Crit Care Med. 1998;26:2005–2009.
102. Mesters R, Helterbrand J, Utterback BG. Prognostic value of protein C concentrations in neutropenic patients at high risk of severe septic complications. Crit Care Med. 2000;28:2209–2216.
103. Rosenberg RD, Aird WC. Vascular-bed-specific hemostasis and hypercoagulable states. N Engl J Med. 1999;340:1555–1564.
104. van der Poll T, Bueller HR, ten Cate H, et al. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med. 1990;322:1622–1627.
105. Esmon NL, Esmon CT. Protein C and the endothelium. Semin Thromb Hemost. 1988;14:210–215.
106. Mesters R, Mannucci PM, Coppola E. Factor VIIA and anti-thrombin III activity during severe sepsis and septic shock in neutropenic patients. Blood. 1996;88:881–886.
107. Abraham E. Tissue factor inhibition and clinical trial results of tissue factor pathway inhibitor in sepsis. Crit Care Med. 2000;28(9 Suppl):S31–S33.
108. Abraham E, Reinhart K, Svoboda P, et al. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: a multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med. 2001;29(11):2081–2089.
109. Sorensen TI, Nielsen GG, Andersen PK, et al. Genetic and environmental influences on premature death in adult adoptees. N Engl J Med. 1988;318(12):727–732.
110. Udhoji VN, Weil MH. Hemodynamic and metabolic studies on shock associated with bacteremia. Ann Intern Med. 1965;62:966–978.
111. Cohn JD, Greenspan M, Goldstein CR, et al. Arteriovenous shunting in high cardiac output shock syndromes. Surg Gynecol Obstet. 1968;127:282–288.
112. Thijs LG, Groenveld ABJ. Peripheral circulation in septic shock. Appl Cardiopul, Pathol. 1988;2:203–214.
113. Wright CJ, Duff JH, McLean APH, et al. Regional capillary blood flow and oxygen uptake in severe sepsis. Surg Gynecol Obstet. 1971;132:637–644.
114. Finley RJ, Duff JH, Holliday RL, et al. Capillary muscle blood flow in human sepsis. Surgery. 1975;78:87–94.
115. Cronenwett JL, Lindenauer SM. Direct measurement of arteriovenous anastomic blood flow in the septic canine hindlimb. Surgery. 1979;85:275–282.
116. Dantzker D. Oxygen delivery and utilization in sepsis. Crit Care Clin. 1989;5:81–98.
117. Wolf YG, Cotev S, Perel A, et al. Dependence of oxygen consumption on cardiac output in sepsis. Crit Care Med. 1987;15:198–203.
118. Shoemaker WC, Chang P, Czer L, et al. Cardiorespiratory monitoring in postoperative patients, I: prediction of outcome and severity of illness. Crit Care Med. 1979;7:237–242.
119. Haupt MT, Gilbert EM, Carlson RW. Fluid loading increases oxygen consumption in septic patients with lactic acidosis. Am Rev Respir Dis. 1985;131:912–916.
120. Samsel RW, Nelson DP, Sanders WM, et al. Effect of endotoxin on systemic and skeletal muscle oxygen extraction. J Appl Physiol. 1988;65:1377–1382.
121. Vincent JL, Roman A, DeBacker D, et al. Oxygen uptake/supply dependency: effects of short-term dobutamine infusion. Am Rev Respir Dis. 1990;142:2–8.
122. Fenwick JC, Dodek PM, Ronco JJ, et al. Increased concentrations of plasma lactate predict pathological dependence of oxygen consumption on oxygen delivery in patients with adult respiratory distress syndrome. J Crit Care. 1990;5:81–87.
123. Gutierrez G, Pohil RJ. Oxygen consumption is linearly related to oxygen supply in critically ill patients. J Crit Care. 1986;1:45–53.
124. Barcroft J. On anoxaemia. Lancet. 1920;2:485–492.
125. Astiz M, Rackow EC, Weil MH, et al. Early impairment of oxidative metabolism and energy production in severe sepsis. Circ Shock. 1988;26:311–320.
126. Mizock B. Septic shock: a metabolic perspective. Arch Intern Med. 1984;144:579–585.
127. Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17(1):219–237.
128. VanderMeer TJ, Wang H, Fink MP. Endotoxemia causes ileal mucosal acidosis in the absence of mucosal hypoxia in a normodynamic porcine model of septic shock. Crit Care Med. 1995;23(7):1217–1226.
129. Hotchkiss RS, Rust RS, Dence CS, et al. Evaluation of the role of cellular hypoxia in sepsis by the hypoxic marker [18F] fluoromisonidazole. Am J Physiol. 1991;261:R965–R972.
130. King CJ, Tytgat S, Delude RL, et al. Ileal mucosal oxygen consumption is decreased in endotoxemic rats but is restored toward normal by treatment with aminoguanidine. Crit Care Med. 1999;27(11):2518–2524.
131. Simonson SG, Welty-Wolf K, Huang YT, et al. Altered mitochondrial redox responses in gram negative septic shock in primates. Circ Shock. 1994;43(1):34–43.
132. Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–223.
133. Griebel JA, Moore FA, Piantadosi CA. In-vivo responses of mitochondrial redox levels to Escherichia coli bacteremia in primates. J Crit Care. 1990;5:1–9.
134. Cairns CB, Moore FA, Haenel JB, et al. Evidence for early supply independent mitochondrial dysfunction in patients developing multiple organ failure after trauma. J Trauma. 1997;42(3):532–536.
135. Mori E, Hasebe M, Kobayashi K, et al. Alterations in metabolite levels in carbohydrate and energy metabolism of rat in hemororrhagic shock and sepsis. Metabolism. 1987;36:14–20.
136. Tanaka J, Sato T, Kamiyama Y, et al. Bacteremic shock: aspects of high-energy metabolism of rat liver following Escherichia coli injection. J Surg Res. 1982;33:49–57.
137. Yassen KA, Galley HF, Lee A, et al. Mitochondrial redox state in the critically ill. Br J Anaesth. 1999;83(2):325–327.
138. Grum CM, Simon RH, Dantzker D, et al. Evidence for adenosine triphosphate degradation in critically ill patients. Chest. 1985;88:763–767.
139. Ozawa K, Aoyama H, Yasuda K, et al. Metabolic abnormalities associated with postoperative organ failure. A redox theory. Arch Surg. 1983;118(11):1245–1251.
140. Bergstrom J, Bostrom H, Furst P, et al. Preliminary studies of energy-rich phosphagens in muscle from severely ill patients. Crit Care Med. 1976;4(4):197–204.
141. Liaw KY, Askanazi J, Michelson CB, et al. Effect of injury and sepsis on high-energy phosphates in muscle and red cells. J Trauma. 1980;20(9):755–759.
142. Song SK, Hotchkiss RS, Karl IE, et al. Concurrent quantification of tissue metabolism and blood flow via 2H/31P NMR in vivo, III: alterations of muscle blood flow and metabolism during sepsis. Magn Reson Med. 1992;25:67–77.
143. Hotchkiss RS, Karl IE. Reevaluation of the role of cellular hypoxia and bioenergetic failure in sepsis. JAMA. 1992;267:1503–1510.
144. Solomon MA, Correa R, Alexander HR, et al. Myocardial energy metabolism and morphology in a canine model of sepsis. Am J Physiol. 1994;266:H757–H768.
145. Hotchkiss RS, Song SK, Neil JJ, et al. Sepsis does not impair tricarboxylic acid cycle in the heart. Am J Physiol. 1991;260:C50–C57.
146. Chaudry IH, Wichterman KA, Baue AE. Effect of sepsis on tissue adenine nucleotide levels. Surgery. 1979;85:205–211.
147. Geller ER, Tankauskas S, Kirpatrick JR. Mitochondrial death in sepsis: a failed concept. J Surg Res. 1986;40:514–517.
148. MacLean LD, Mulligan WG, McLean APH, et al. Patterns of septic shock in man: a detailed study of 56 patients. Ann Surg. 1967;166:543–562.
149. Nishijima H, Weil MH, Shubin H, et al. Hemodynamic and metabolic studies on shock associated with gram-negative bacteremia. Medicine (Baltimore). 1973;52:287–294.
150. Weil MH, Nishijima H. Cardiac output in bacterial shock. Am J Med. 1978;64:920–922.
151. Blain CM, Anderson TO, Pietras RJ, et al. Immediate hemodynamic effects of gram-negative vs gram-positive bacteremia in man. Arch Intern Med. 1970;126:260–265.
152. Packman MI, Rackow EC. Optimum left heart filling pressure during fluid resuscitation of patients with hypovolemic and septic shock. Crit Care Med. 1983;11:165–169.
153. Winslow EJ, Loeb HS, Rahimtoola SH, et al. Hemodynamic studies and results of therapy in 50 patients with bacteremic shock. Am J Med. 1973;54:421–432.
154. Krausz MM, Perel A, Eimerl D, et al. Cardiopulmonary effects of volume loading in patients with septic shock. Ann Surg. 1977;185:429–434.
155. Parker MM, Suffredini AF, Natanson C, et al. Responses of left ventricular function in surviviors and non-survivors of septic shock. J Crit Care. 1989;4:19–25.
156. Teule GJJ, Van Lingen A, Verweij-van Vught MA, et al. Role of peripheral pooling in porcine Escherichia coli sepsis. Circ Shock. 1984;12:115–123.
157. Natanson C, Fink MP, Ballantyne HK, et al. Gram-negative bacteremia produces both severe systolic and diastolic cardiac dysfunction in a canine model that simulates human septic shock. J Clin Invest. 1986;78:259–270.
158. Carroll GC, Snyder JV. Hyperdynamic severe intravascular sepsis depends on fluid administration in cynomolgus monkey. Am J Physiol. 1982;243:131–141.
159. Teule GJJ, Den Hollander W, Bronsveld W, et al. Effect of volume loading and dopamine on hemodynamics and red cell distribution in canine endotoxic shock. Circ Shock. 1983;10:41–50.
160. Magder S, Vanelli G. Circuit factors in the high cardiac output of sepsis. J Crit Care. 1996;11(4):155–166.
161. Donnino M, Nguyen HB, Rivers EP. A hemodynamic comparison of early and late phase severe sepsis and septic shock. Chest. 2002;122:4S.
162. Kumar A, Haery C, Parrillo JE. Myocardial dysfunction in septic shock, I: clinical manifestation of cardiovascular dysfunction. J Cardiothor Vasc Anesth. 2001;15(3):364–376.
163. Ognibene FP, Parker MM, Natanson C, et al. Depressed left ventricular performance. Response to volume infusion in patients with sepsis and septic shock. Chest. 1988;93:903–910.
164. Parker MM, Shelhamer JH, Bacharach SL, et al. Profound but reversible myocardial depression in patients with septic shock. Ann Intern Med. 1984;100:483–490.
165. Garner LB, Willis MS, Carlson DL, et al. Macrophage migration inhibitory factor is a cardiac-derived myocardial depressant factor. Am J Physiol. 2003;285(6):H2500–H2509.
166. Eichenholz PW, Eichacker PQ, Hoffman WD, et al. Tumor necrosis factor challenges in canines: patterns of cardiovascular dysfunction. Am J Physiol. 1992;263:H668–H675.
167. Vincent JL, Bakker J, Marecaux G, et al. Administration of anti-TNF antibody improves left ventricular function in septic shock patients: results of a pilot study. Chest. 1992;101:810–815.
168. Hosenpud JD, Campbell SM, Mendelson DJ. Interleukin-1-induced myocardial depression in an isolated beating heart preparation. J Heart Transplant. 1989;8:460–464.
169. Massey CV, Kohout TR, Gaa ST, et al. Molecular and cellular actions of platelet-activating factor in rat heart cells. J Clin Invest. 1991;88:2106–2116.
170. Schutzer KM, Haglund U, Falk A. Cardiopulmonary dysfunction in a feline septic model: possible role of leukotrienes. Circ Shock. 1989;29:13–25.
171. Werdan K, Muller U, Reithmann C. “Negative inotropic cascades” in cardiomyocytes triggered by substances relevant to sepsis. In: Schlag G, Redl H, eds. Pathophysiology of Shock, Sepsis and Organ Failure. Berlin, Germany: Springer-Verlag; 1993:787–834.
172. Pathan N, Hemingway CA, Alizadeh AA, et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet. 2004;363(9404):203–209.
173. Papadopoulos MC, Davies DC, Moss R, et al. Pathophysiology of septic encephalopathy: a review. Crit Care Med. 2000;28(8):3019–3024.
174. Sprung CL, Peduzzi PN, Shatney CH, et al. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med. 1990;18(8):801–806.
175. Pine RW, Wertz MJ, Lennard ES, et al. Determinants of organ malfunction or death in patients with intra-abdominal sepsis. A discriminant analysis. Arch Surg. 1983;118(2):242–249.
176. Young GB, Bolton CF, Austin TW, et al. The encephalopathy associated with septic illness. Clin Invest Med. 1990;13(6):297–304.
177. Young GB, Bolton CF, Archibald Y, et al. The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol. 1992;9(1):145–152.
178. Kollef MH, Sherman G. Acquired organ system derangements and hospital mortality: are all organ systems created equally? Am J Crit Care. 1999;8(3):180–188.
179. De Jonghe B, Lacherade JC, Durand MC, et al. Critical illness neuromuscular syndromes. Crit Care Clin. 2006;22(4):805–818.
180. Leijten FS, Harinck-de Weerd JE, Poortvliet DC, et al. The role of polyneuropathy in motor convalescence after prolonged mechanical ventilation. JAMA. 1995;274(15):1221–1225.
181. Bercker S, Weber-Carstens S, Deja M, et al. Critical illness polyneuropathy and myopathy in patients with acute respiratory distress syndrome. Crit Care Med. 2005;33(4):711–715.
182. Lorin S, Sivak M, Nierman DM. Critical illness polyneuropathy: what to look for in at-risk patients. J Crit Illness. 1998;13(10):608–612.
183. Tennila A, Salmi T, Pettila V, et al. Early signs of critical illness polyneuropathy in ICU patients with systemic inflammatory response syndrome or sepsis [see comment]. Intensive Care Med. 2000;26(9):1360–1363.
184. Tepper M, Rakic S, Haas JA, et al. Incidence and onset of critical illness polyneuropathy in patients with septic shock. Neth J Med. 2000;56(6):211–214.
185. De Jonghe B, Sharshar T, Lefaucheur JP, et al. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA. 2002;288(22):2859–2867.
186. Cunnion RE, Schaer GL, Parker MM, et al. The coronary circulation in human septic shock. Circulation. 1986;73:637–644.
187. Dhainaut JF, Huyghebaert MF, Monsallier JF, et al. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation. 1987;75:533–541.
188. Reilly JM, Cunnion RE, Burch-Whitman C, et al. A circulating myocardial depressant substance is associated with cardiac dysfunction and peripheral hypoperfusion (lactic acidemia) in patients with septic shock. Chest. 1989;95:1072–1080.
189. Parrillo JE, Burch C, Shelhamer JH, et al. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. J Clin Invest. 1985;76:1539–1553.
190. Jones SB, Romano RD. Myocardial beta adrenergic receptor coupling to adenylate cyclase during developing septic shock. Circ Shock. 1990;30:51–61.
191. Bristow MR, Ginsburg R, Minobe W, et al. Decreased catecholamine sensitivity and beta adrenergic receptor density in failing human hearts. N Engl J Med. 1982;307:205–210.
192. Silverman HJ, Penaranda R, Orens JB, et al. Impaired beta-adrenergic receptor stimulation of cyclic adenosine monophosphate in human septic shock: association with myocardial hyporesponsiveness to catecholamines. Crit Care Med. 1993;21:31–39.
193. Kumar A, Brar R, Wang P, et al. The role of nitric oxide and cyclic GMP in human septic serum-induced depression of cardiac myocyte contractility. Am J Physiol. 1999;276:R265–R276.
194. Kumar A, Paladugu B, Mensing J, et al., Nitric oxide-dependent and -independent mechanisms are involved in TNFa-induced depression of cardiac myocyte contractility. Am J Physiol. 2007;292:R1900–R1906.
195. Anel R, Paladugu B, Makkena R, et al. TNFa induces a proximal defect of beta-adrenoreceptor signal transduction in cardiac myocytes. Crit Care Med. 1999;27:A95.
196. Gulick T, Chung MK, Pieper SJ, et al. Interleukin-1 and tumor necrosis factor inhibit cardiac myocyte adrenergic responsiveness. Proc Natl Acad Sci.U S A. 1989;86:6753–6757.
197. Chung MK, Gulick TS, Rotondo R, et al. Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic AMP in rat cardiac myoctyes: impairment of signal transduction. Circ Res. 1990;67:753–763.
198. Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Crit Care Med. 1999;27(9):1775–1780.
199. ver Elst KM, Spapen HD, Nguyen DN, et al. Cardiac troponins I and T are biological markers of left ventricular dysfunction in septic shock. Clin Chem. 2000;46(5):650–657.
200. Mehta NJ, Khan IA, Gupta V, et al. Cardiac troponin I predicts myocardial dysfunction and adverse outcome in septic shock. Intl J Cardiol. 2004;95(1):13–17.
201. Schrier RW, Wang W. Acute renal failure and sepsis [see comment]. N Engl J Med. 2004;351(2):159–169.
202. Ware LB, Matthay MA, Ware LB, et al. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349.
203. Sevransky JE, Levy MM, Marini JJ, et al. Mechanical ventilation in sepsis-induced acute lung injury/acute respiratory distress syndrome: an evidence-based review. Crit Care Med. 2004;32(11 Suppl):S548–S553.
204. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1301–1308.
205. Piantadosi CA, Schwartz DA, Piantadosi CA, et al. The acute respiratory distress syndrome. Ann Intern Med. 2004;141(6):460–470.
206. Hoste EA, Lameire NH, Vanholder RC, et al. Acute renal failure in patients with sepsis in a surgical ICU: predictive factors, incidence, comorbidity, and outcome. J Am Soc Nephrol. 2003;14(4):1022–1030.
207. Rasmussen HH, Ibels LS. Acute renal failure. Multivariate analysis of causes and risk factors. Am J Med. 1982;73:211–218.
208. Neveu H, Kleinknecht D, Brivet F, et al. Prognostic factors in acute renal failure due to sepsis. Results of a prospective multicentre study. The French Study Group on Acute Renal Failure. Nephrol Dial Transplant. 1996;11(2):293–299.
209. Brivet FG, Kleinknecht DJ, Loirat P, et al. Acute renal failure in intensive care units–causes, outcome, and prognostic factors of hospital mortality; a prospective, multicenter study. French Study Group on Acute Renal Failure. Crit Care Med. 1996;24(2):192–198.
210. Astiz ME, Rackow EC, Weil MH. Pathophysiology and treatment of circulatory shock. Crit Care Clin. 1993;9:183–203.
211. Deitch E, Bridges W, Baker J, et al. Hemorrhagic shock-induced bacterial translocation is reduced by xanthine oxidase inhibition or inactivation. Surgery. 1988;104:191–198.
212. Mainous MR, Deitch EA. Bacterial translocation. In: Schlag G, Redl H, eds. Pathophysiology of Shock, Sepsis and Organ Failure. Berlin, Germany: Springer-Verlag; 1993:265–278.
213. Lillehei RC, MacLean LD. The intestinal factor in irreversible endotoxin shock. Ann Surg. 1958;148:513–519.
214. Meakins JL, Marshall JC. The gut as the motor of multiple organ failure. In: Marston A, Bulkley GB, Fiddian-Green RG, et al., eds. Splanchnic Ischemia and Multiple Organ Failure. London, England: Edward Arnold; 1989:339.
215. Marrero J, Martinez FJ, Hyzy R, et al. Advances in critical care hepatology. Am J Resp Crit Care Med. 2003;168(12):1421–1426.
216. Moseley RH. Sepsis and cholestasis. Clin Liver Disease. 1999;3:465–475.
217. Champion HR, Jones RT, Trump BF, et al. A clinicopathologic study of hepatic dysfunction following shock. Surg Gynecol Obstet. 1976;142:657–663.
218. Levi M, ten Cate H, Levi M, et al. Disseminated intravascular coagulation.[see comment]. N Engl J Med. 1999;341(8):586–592.
219. Bone RC. Gram positive organisms and sepsis. Arch Intern Med. 1994;154:26–34.
220. Zeerleder S, Hack CE, Wuillemin WA, et al. Disseminated intravascular coagulation in sepsis. Chest. 2005;128(4):2864–2875.
221. ten Cate H. Pathophysiology of disseminated intravascular coagulation in sepsis. Crit Care Med. 2000;28(9 Suppl):S9–S11.
222. Woolf PD. Endocrinology of shock. Ann Emerg Med. 1986;15:1401–1405.
223. Arnold J, Leinhardt D, Little RA. Metabolic response to trauma. In: Schlag G, Redl H, eds. Pathophysiology of Shock Sepsis and Organ Failure. Berlin, Germany: Springer-Verlag; 1993:145–160.
224. Bessey PQ, Brooks DC, Black PR, et al. Epinephrine acutely mediates skeletal muscle insulin resistance. Surgery. 1983;94:172–179.
225. Brierre S, Kumari R, Deboisblanc BP, et al. The endocrine system during sepsis. Am J Med Sci. 2004;328(4):238–247.
226. Mizock B. Metabolic derangements in sepsis and septic shock. Crit Care Clin. 2000;16(2):319–336.
227. Naylor JM, Kronfeld DS. In-vivo studies of hypoglycemia and lactic acidosis in endotoxic shock. Am J Physiol. 1985;248:E309–E316.
228. Daniel AM, Pierce CH, Shizgal HM, et al. Protein and fat utilization in shock. Surgery. 1978;84:588–594.
229. Bagby GJ, Spitzer JA. Decreased myocardial extracellular and muscle lipoprotein lipase activities in endotoxin-treated rats. Proc Soc Exp Biol Med. 1981;168:395–398.
230. Ho HC, Chapital AD, Yu M, et al. Hypothyroidism and adrenal insufficiency in sepsis and hemorrhagic shock. Arch Surg. 2004;139(11):1199–1203.
231. Gardelis JG, Hatzis TD, Stamogiannou L, et al. Activity of the growth hormone/insulin-like growth factor-I axis in critically ill children. J Pediatr Endocrinol. 2005;18(4):363–372.
232. Beishuizen A, Thijs LG, Beishuizen A, et al. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J Endotoxin Res. 2003;9(1):3–24.
233. Heemskerk VH, Daemen MA, Buurman WA, et al. Insulin-like growth factor-1 (IGF-1) and growth hormone (GH) in immunity and inflammation. Cytokine Growth Factor Rev. 1999;10(1):5–14.
234. Woods RJ, David J, Baigent S, et al. Elevated levels of corticotrophin-releasing factor binding protein in the blood of patients suffering from arthritis and septicaemia and the presence of novel ligands in synovial fluid. Br J Rheumatol. 1996;35(2):120–124.
235. Holmes CL, Patel BM, Russell J, et al. Physiology of vasopressin relevant to management of septic shock. Chest. 2001;120(3):989–1002.
236. Landry DW, Levin HR, Gallant EM, et al. Vasopressin deficiency contributes to the vasodilatation of septic shock. Circulation. 1997;95:1122–1125.
237. Marik PE, Zaloga GP. Adrenal insufficiency during septic shock. Crit Care Med. 2003;31(1):141–145.
238. Zaloga GP, Marik P. Hypothalamic-pituitary-adrenal insufficiency. Crit Care Clin. 2001;17(1):25–42.
239. Annane D, Sebille V, Troche G, et al. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283(8):1038–1045.
240. Manglik S, Flores E, Lubarsky L, et al. Glucocorticoid insufficiency in patients who present to the hospital with severe sepsis: a prospective clinical trial [see comment]. Crit Care Med. 2003;31(6):1668–1675.
241. Marik P, Rotello L, Zaloga G. Secondary adrenal insufficiency is common in critically ill patients. Crit Care Med. 2001;29:A163.
242. de Groof F, Joosten KF, Janssen JA, et al. Acute stress response in children with meningococcal sepsis: important differences in the growth hormone/insulin-like growth factor I axis between nonsurvivors and survivors. J Clin Endocrinol Metab. 2002;87(7):3118–3124.
243. Felmet KA, Hall MW, Clark RS, et al. Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and multiple organ failure. J Immunol. 2005;174(6):3765–3772.
244. Medina P, Noguera I, Aldasoro M, et al. Enhancement by vasopressin of adrenergic responses in human mesenteric arteries. Am J Physiol. 1997;272(3 Pt 2):H1087–H1093.
245. Peres BD, Lopes FF, Melot C, et al. Body temperature alterations in the critically ill. Intensive Care Med. 2004;30(5):811–816.
246. Gomez-Jimenez J, Salgado A, Mourelle M, et al. L-arginine: nitric oxide pathway in endotoxemia and human septic shock. Crit Care Med. 1995;23:253–258.
247. Kubes P. Nitric oxide affects microvascular permeability in the intact inflamed vasculature. Microcirculation. 1995;2(3):235–244.
248. Rackow EC, Kaufman BS, Falk JL, et al. Hemodynamic response to fluid repletion in patients with septic shock: evidence for early depression of cardiac performance. Circ Shock. 1987;22:11–22.
249. Parrillo JE, Parker MM, Natanson C, et al. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–242.
250. Cannon WB. Traumatic Shock. New York, NY: Appleton; 1923.
251. Cournand A, Riley RL, Bradley SE, et al. Studies of the circulation in clinical shock. Surgery. 1943;13:964–995.
252. Carcillo JA, Davis AL, Zaritsky A. Role of early fluid resuscitation in pediatric septic shock. JAMA. 1991;266(9):1242–1245.
253. Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–1377.
254. Levraut J, Ichai C, Petit I, et al. Low exogenous lactate clearance as an early predictor of mortality in normolactatemic critically ill septic patients. Crit Care Med. 2003;31(3):705–710.
255. Abramson D, Scalea TM, Hitchcock R, et al. Lactate clearance and survival following injury. J Trauma. 1993;35(4):584–588.
256. Davis JW, Shackford SR, Mackersie RC, et al. Base deficit as a guide to volume resuscitation. J Trauma. 1988;28(10):1464–1467.
257. Oud L, Haupt MT. Persistent gastric intramucosal ischemia in patients with sepsis following resuscitation from shock. Chest. 1999;115(5):1390–1396.
258. Wo CC, Shoemaker WC, Appel PL, et al. Unreliability of blood pressure and heart rate to evaluate cardiac output in emergency resuscitation and critical illness. Crit Care Med. 1993;21(2):218–223.
259. Ward KR, Ivatury RR, Barbee WR. Endpoints of resuscitation for the victim of trauma. Intensive Care Med. 2001;16(2):55–75.
260. Bakker J, Coffemils M, Leon M, et al. Blood lactate levels are superior to oxygen-derived variables in predicting outcome in human septic shock. Chest. 1992;99:956–962.
261. Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med. 2004;32(8):1637–1642.
262. James JH, Luchette FA, McCarter FD, et al. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 1999;354(9177):505–508.
263. Ernest D, Belzberg AS, Dodek PM. Distribution of normal saline and 5% albumin infusions in septic patients. Crit Care Med. 1999;27(1):46–50.
264. Rackow EC, Falk JL, Fein IA, et al. Fluid resuscitation in circulatory shock: a comparison of the cardiorespiratory effects of albumin, hetastarch, and saline solutions in patients with hypovolemic and septic shock. Crit Care Med. 1983;11:839–850.
265. Haupt MT, Teerapong P, Green D, et al. Increased pulmonary edema with crystalloid compared to colloid resuscitation of shock associated with increased vascular permeability. Circ Shock. 1984;12:213–224.
266. Haupt MT, Rackow EC. Colloid osmotic pressure and fluid resuscitation with hetastarch, albumin, and saline solutions. Crit Care Med. 1982;10:159–162.
267. Wilkes MM, Navickis RJ. Patient survival after human albumin administration. A meta-analysis of randomized, controlled trials. Ann Intern Med. 2001;135(3):149–164.
268. Alderson P, Bunn F, Lefebvre C, et al. Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database Syst Rev. 2002;(1):CD001208.
269. Finfer S, Bellomo R, Boyce N, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350(22):2247–2256.
270. Schierhout G, Roberts I. Fluid resuscitation with colloid or crystalloid solutions in critically ill patients: a systematic review of randomised trials [see comment]. BMJ. 1998;316(7136):961–964.
271. Meehan TP, Fine MJ, Krumholz HM, et al. Quality of care, process, and outcomes in elderly patients with pneumonia. JAMA. 1997;278(23):2080–2084.
272. Ibrahim EH, Sherman G, Ward S, et al. The influence of inadequate antimicrobial treatment of bloodstream infections on patient outcomes in the ICU setting [see comment]. Chest. 2000;118(1):146–155.
273. Kollef MH, Sherman G, Ward S, et al. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest. 1999;115(2):462–474.
274. Kumar A, Suppes R, Gulati H, et al. The impact of initiation of inadequate antimicrobial therapy on survival in human septic shock. Antimicrob Agents Chemother. 2007;111:271.
275. Young LS, Martin WJ, Meyer RD, et al. Gram-negative rod bacteremia: Microbiologic, immunologic, and therapeutic considerations. Ann Intern Med. 1977;86:456–471.
276. Romero-Vivas J, Rubio M, Fernandez C, et al. Mortality associated with nosocomial bacteremia due to methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 1995;21(6):1417–1423.
277. Nguyen MH, Peacock JE Jr, Tanner DC, et al. Therapeutic approaches in patients with candidemia. Evaluation in a multicenter, prospective, observational study. Arch Intern Med. 1995;155(22):2429–2435.
278. Vergis EN, Hayden MK, Chow JW, et al. Determinants of vancomycin resistance and mortality rates in enterococcal bacteremia. a prospective multicenter study. Ann Intern Med. 2001;135(7):484–492.
279. Byl B, Clevenbergh P, Jacobs F, et al. Impact of infectious diseases specialists and microbiological data on the appropriateness of antimicrobial therapy for bacteremia [see comment]. Clin Infect Dis. 1999;29(1):60–66.
280. Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med. 1998;129(11):862–869.
281. Miner JR, Heegaard W, Mapes A, et al. Presentation, time to antibiotics, and mortality of patients with bacterial meningitis at an urban county medical center. J Emerg Med. 2001;21(4):387–392.
282. Proulx N, Frechette D, Toye B, et al. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM. 2005;98(4):291–298.
283. Houck PM, Bratzler DW, Nsa W, et al. Timing of antibiotic administration and outcomes for Medicare patients hospitalized with community-acquired pneumonia. Arch Intern Med. 2004;164(6):637–644.
284. Natsch S, Kullberg BJ, Van der Meer JW, et al. Delay in administering the first dose of antibiotics in patients admitted to hospital with serious infections. Eur J Clin Microbiol Infect Dis. 1998;17(10):681–684.
285. Pinder M, Bellomo R, Lipman, et al. Pharmacological principles of antibiotic prescription in the critically ill. Anaesth Intensive Care. 2002;30(2):134–144.
286. Pimentel FL, Abelha F, Trigo MA, et al. Determination of plasma concentrations of amikacin in patients of an intensive care unit. J Chemother. 1995;7(1):45–49.
287. Whipple JK, Ausman RK, Franson T, et al. Effect of individualized pharmacokinetic dosing on patient outcome. Crit Care Med. 1991;19(12):1480–1485.
288. Joukhadar C, Frossard M, Mayer BX, et al. Impaired target site penetration of beta-lactams may account for therapeutic failure in patients with septic shock. Crit Care Med. 2001;29(2):385–391.
289. Franson TR, Quebbeman EJ, Whipple J, et al. Prospective comparison of traditional and pharmacokinetic aminoglycoside dosing methods. Crit Care Med. 1988;16(9):840–843.
290. Chelluri L, Jastremski MS. Inadequacy of standard aminoglycoside loading doses in acutely ill patients. Crit Care Med. 1987;15(12):1143–1145.
291. Tegeder I, Schmidtko A, Brautigam L, et al. Tissue distribution of imipenem in critically ill patients. Clin Pharmacol Ther. 2002;71(5):325–333.
292. Moore RD, Smith CR, Lietman PS. The association of aminoglycoside plasma levels with mortality in patients with gram-negative bacteremia. J Infect Dis. 1984;149(3):443–448.
293. Moore RD, Smith CR, Lietman PS. Association of aminoglycoside plasma levels with therapeutic outcome in gram-negative pneumonia. Am J Med. 1984;77(4):657–662.
294. Forrest A, Nix DE, Ballow CH, et al. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother. 1993;37(5):1073–1081.
295. Preston SL, Drusano GL, Berman AL, et al. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA. 1998;279(2):125–129.
296. Drusano GL, Preston SL, Fowler C, et al. Relationship between fluoroquinolone area under the curve: minimum inhibitory concentration ratio and the probability of eradication of the infecting pathogen, in patients with nosocomial pneumonia. J Infect Dis. 2004;189(9):1590–1597.
297. Moore RD, Lietman PS, Smith CR. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J Infect Dis. 1987;155(1):93–99.
298. Kashuba AD, Nafziger AN, Drusano GL, et al. Optimizing aminoglycoside therapy for nosocomial pneumonia caused by gram-negative bacteria. Antimicrob Agents Chemother. 1999;43(3):623–629.
299. Schentag JJ, Smith IL, Swanson DJ, et al. Role for dual individualization with cefmenoxime. Am J Med. 1984;77(6A):43–50.
300. Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26(1):1–10.
301. Lodise TP, Lomaestro BM, Drusano GL. Piperacillin-tazobactam for Pseudomonas aeruginosa infection: Clinical implications of an extended-infusion dosing strategy. Clin Infect Dis. 2007;44:357–363.
302. Craig WA, Ebert SC. Continuous infusion of beta-lactam antibiotics. Antimicrob Agents Chemother. 1992;36(12):2577–2583.
303. Craig WA. Once-daily versus multiple-daily dosing of aminoglycosides. J Chemother. 1995;7(Suppl 2):47–52.
304. Kashuba AD, Bertino JS Jr, Nafziger AN. Dosing of aminoglycosides to rapidly attain pharmacodynamic goals and hasten therapeutic response by using individualized pharmacokinetic monitoring of patients with pneumonia caused by gram-negative organisms. Antimicrob Agents Chemother. 1998;42(7):1842–1844.
305. Bodey GP, Ketchel SJ, Rodriguez V. A randomized study of carbenicillin plus cefamandole or tobramycin in the treatment of febrile episodes in cancer patients. Am J Med. 1979;67(4):608–616.
306. Daenen S, Vries-Hospers H. Cure of Pseudomonas aeruginosa infection in neutropenic patients by continuous infusion of ceftazidime. Lancet. 1988;1(8591):937.
307. Egerer G, Goldschmidt H, Hensel M, et al. Continuous infusion of ceftazidime for patients with breast cancer and multiple myeloma receiving high-dose chemotherapy and peripheral blood stem cell transplantation. Bone Marrow Transplant. 2002;30(7):427–431.
308. Benko AS, Cappelletty DM, Kruse JA, et al. Continuous infusion versus intermittent administration of ceftazidime in critically ill patients with suspected gram-negative infections. Antimicrob Agents Chemother. 1996;40(3):691–695.
309. Thalhammer F, Traunmuller F, El Manyawi I, et al. Continuous infusion versus intermittent administration of meropenem in critically ill patients. J Antimicrob Chemother. 1999;43(4):523–527.
310. Sfeir T, Saha DC, Astiz M, et al. Role of interleukin-10 in monocyte hyporesponsiveness associated with septic shock. Crit Care Med. 2001;29(1):129–133.
311. Haupt W, Riese J, Mehler C, et al. Monocyte function before and after surgical trauma. Dig Surg. 1998;15(2):102–104.
312. Brandtzaeg P, Osnes L, Ovstebo R, et al. Net inflammatory capacity of human septic shock plasma evaluated by a monocyte-based target cell assay: identification of interleukin-10 as a major functional deactivator of human monocytes. [Erratum in J Exp Med. 1996;184(5):2075.]. J Exp Med. 1996;184(1):51–60.
313. Williams MA, Withington S, Newland AC, et al. Monocyte anergy in septic shock is associated with a predilection to apoptosis and is reversed by granulocyte-macrophage colony-stimulating factor ex vivo. J Infect Dis. 1998;178(5):1421–1433.
314. Tavares-Murta BM, Zaparoli M, Ferreira RB, et al. Failure of neutrophil chemotactic function in septic patients. Crit Care Med. 2002;30(5):1056–1061.
315. Holzer K, Konietzny P, Wilhelm K, et al. Phagocytosis by emigrated, intra-abdominal neutrophils is depressed during human secondary peritonitis. Eur Surg Res. 2002;34(4):275–284.
316. Benjamim CF, Ferreira SH, Cunha FQ. Role of nitric oxide in the failure of neutrophil migration in sepsis. J Infect Dis. 2000;182(1):214–223.
317. Barriere SL. Monotherapy versus combination antimicrobial therapy: a review. Pharmacotherapy. 1991;11(2 Pt 2):64S–71S.
318. Hughes WT, Armstrong D, Bodey GP, et al. 2002 guidelines for the use of antimicrobial agents in neutropenic patients with cancer. Clin Infect Dis. 2002;34(6):730–751.
319. Bochud PY, Glauser MP, Calandra T, et al. Antibiotics in sepsis. Intensive Care Med. 2001;27(Suppl 1):S33–S48.
320. Safdar N, Handelsman J, Maki DG, et al. Does combination antimicrobial therapy reduce mortality in Gram-negative bacteraemia? A meta-analysis [see comment]. Lancet Infect Dis. 2004;4(8):519–527.
321. Hilf M, Yu VL, Sharp J, et al. Antibiotic therapy for Pseudomonas aeruginosa bacteremia: outcome correlations in a prospective study of. 200 patients. Am J Med. 1989;87(5):540–546.
322. Chow JW, Fine MJ, Shlaes DM, et al. Enterobacter bacteremia: clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991;115(8):585–590.
323. Korvick JA, Bryan CS, Farber B, et al. Prospective observational study of Klebsiella bacteremia in 230 patients: outcome for antibiotic combinations versus monotherapy. Antimicrob Agents Chemother. 1992;36(12):2639–2644.
324. Anderson ET, Young LS, Hewitt WL. Antimicrobial synergism in the therapy of gram-negative rod bacteremia. Chemotherapy. 1978;24(1):45–54.
325. Baddour LM, Yu VL, Klugman KP, et al. Combination antibiotic therapy lowers mortality among severely ill patients with pneumococcal bacteremia. Am J Resp Crit Care Med. 2004;170(4):440–4.
326. Waterer GW, Somes GW, Wunderink RG. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med. 2001;161(15):1837–1842.
327. Martinez JA, Horcajada JP, Almela M, et al. Addition of a macrolide to a beta-lactam-based empirical antibiotic regimen is associated with lower in-hospital mortality for patients with bacteremic pneumococcal pneumonia [see comment]. Clin Infect Dis. 2003;36(4):389–395.
328. Rodriguez A, Mendia A, Sirvent JM, et al. Combination antibiotic therapy improves survival in patients with community-acquired pneumonia and shock. Crit Care Med. 2007;35(6):1493–1498.
329. Sprung CL, Finch RG, Thijs LG, et al. International sepsis trial (INTERSEPT): role and impact of a clinical evaluation committee. Crit Care Med. 1996;24(9):1441–1447.
330. Sudarsky LA, Laschinger JC, Coppa GF, et al. Improved results from a standardized approach in treating patients with necrotizing fasciitis. Ann Surg. 1987;206(5):661–665.
331. Moss RL, Musemeche CA, Kosloske AM. Necrotizing fasciitis in children: prompt recognition and aggressive therapy improve survival. J Pediatr Surg. 1996;31(8):1142–1146.
332. Kumar A, Wood K, Gurka D, et al. Outcome of septic shock correlates with duration of hypotension prior to source control implementation. ICAAC Proceedings. 2004;350:K–1222.
333. Kumar A, Parrillo JE. Shock: pathophysiology, classification and approach to management. In: Parrillo JE, Dellinger RP, eds. Critical Care Medicine: Principles of Diagnosis and Management in the Adult. 3rd ed. St. Louis, MO: Mosby; 2007:379–422.
334. Bond RF. Peripheral macro- and microcirculation. In: Schlag G, Redl H, eds. Pathophysiology of Shock, Sepsis and Organ Failure. Berlin, Germany: Springer-Verlag; 1993:893–907.
335. Desjars P, Pinaud M, Potel G, et al. A reappraisal of norepinephrine therapy in human septic shock. Crit Care Med. 1987;15(2):134–137.
336. Desjars P, Pinaud M, Bugnon D, et al. Norepinephrine therapy has no deleterious renal effects in human septic shock. Crit Care Med. 1989;17(5):426–429.
337. Fukuoka T, Nishimura M, Imanaka H, et al. Effects of norepinephrine on renal function in septic patients with normal and elevated serum lactate levels. Crit Care Med. 1989;17(11):1104–1107.
338. Hesselvik JF, Brodin B. Low dose norepinephrine in patients with septic shock and oliguria: effects on afterload, urine flow, and oxygen transport. Crit Care Med. 1989;17(2):179–180.
339. Meadows D, Edwards JD, Wilkins RG, et al. Reversal of intractable septic shock with norepinephrine therapy. Crit Care Med. 1988;16(7):663–666.
340. Redl-Wenzl EM, Armbruster C, Edelmann G, et al. The effects of norepinephrine on hemodynamics and renal function in severe septic shock states. Intensive Care Med. 1993;19(3):151–154.
341. Neviere R, Mathieu D, Chagnon JL, et al. The contrasting effects of dobutamine and dopamine on gastric mucosal perfusion in septic patients. Am J Resp Crit Care Med. 1996;154(6 Pt 1):1684–1688.
342. Marin C, Eon B, Saux P, et al. Renal effects of norepinephrine used to treat septic shock patients. Crit Care Med. 1990;18(3):282–285.
343. Marik PE, Mohedin M. The contrasting effects of dopamine and norepinephrine on systemic and splanchnic oxygen utilization in hyperdynamic sepsis. JAMA. 1994;272(17):1354–1357.
344. Ruokonen E, Takala J, Kari A, et al. Regional blood flow and oxygen transport in septic shock. Crit Care Med. 1993;21(9):1296–1303.
345. Meier-Hellmann A, Specht M, Hannemann L, et al. Splanchnic blood flow is greater in septic shock treated with norepinephrine than in severe sepsis. Intensive Care Med. 1996;22(12):1354–1359.
346. Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study [see comment]. Intensive Care Med. 1997;23(3):282–287.
347. De Backer D, Creteur J, Silva E, et al. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med. 2003;31(6):1659–1667.
348. Martin C, Papazian L, Perrin G, et al. Norepinephrine or dopamine for the treatment of hyperdynamic septic shock? Chest. 1993;103(6):1826–1831.
349. Devins SS, Miller A, Herndon BL, et al. The effects of dopamine on T-cell proliferative response and serum prolactin in critically ill patients. Crit Care Med. 1992;20:1644–1649.
350. Kumar A, Schupp E, Bunnell E, et al. The cardiovascular response to dobutamine in septic shock. Clin Invest Med. 1994;17, B18 Abstract #107.
351. Rhodes A, Lamb FJ, Malagon R, et al. A prospective study of the use of a dobutamine stress test to identify outcome in patients with sepsis, severe sepsis or septic shock. Crit Care Med. 1999;27(11):2361–2366.
352. Barton P, Garcia J, Kouatli A, et al. Hemodynamic effects of i.v. milrinone lactate in pediatric patients with septic shock. A prospective, double-blinded, randomized, placebo-controlled, interventional study. Chest. 1996;109:1302–1312.
353. Gattinoni L, Brazzi L, Pelosi P, et al. A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 Collaborative Group. N Engl J Med. 1995;333:1025–1032.
354. Hayes MA, Timmins AC, Yau EHS, et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717–1722.
355. Shoemaker WC, Appel PL, Kram HB, et al. Temporal hemodynamic and oxygen transport patterns in medical patients. Septic shock. Chest. 1993;104(5):1529–1536.
356. Shoemaker WC, Appel PL, Kram HB, et al. Sequence of physiologic patterns in surgical septic shock. Crit Care Med. 1993;21(12):1876–1889.
357. Ivanov R, Allen J, Calvin JE. The incidence of major morbidity in critically ill patients managed with pulmonary artery catheters: a meta-analysis. Crit Care Med. 2000;28(3):615–619.
358. Tuchschmidt J, Fried J, Astiz M, et al. Elevation of cardiac output and oxygen delivery improves outcome in septic shock. Chest. 1992;102:216–220.
359. Yu M, Levy MM, Smith P, et al. Effect of maximizing oxygen delivery on morbidity and mortality rates in critically ill patients: a prospective, randomized, controlled study. Crit Care Med. 1993;21(6):830–838.
360. Yu M, Burchell S, Hasaniya NW, et al. Relationship of mortality to increasing oxygen delivery in patients > or = 50 years of age: a prospective, randomized trial. Crit Care Med. 1998;26(6):1011–1019.
361. Alia I, Esteban A, Gordo F, et al. A randomized and controlled trial of the effect of treatment aimed at maximizing oxygen delivery in patients with severe sepsis or septic shock. Chest. 1999;115(2):453–461.
362. Heyland DK, Cook DJ, King D, et al. Maximizing oxygen delivery in critically ill patients: a methodologic appraisal of the evidence. Crit Care Med. 1996;24(3):517–524.
363. Bussolino F, Camussi G, Baglioni C. Synthesis and release of platelet activating factor by human vascular endothelial cells treated with tumor necrosis factor or interleukin-1. J Biol Chem. 1988;263:11856–11861.
364. Landry DW, Levin HR, Gallant EM, et al. Vasopressin pressor sensitivity in vasodilatory septic shock. Crit Care Med. 1997;25:1279–1282.
365. Patel BM, Chittock DR, Russell JA, et al. Beneficial effects of short-term vasopressin infusion during severe septic shock. Anesthesiology. 2002;96(3):576–582.
366. Russell JA, Walley KR, Singer J, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358:877–887.
367. Schaer GL, Fink MP, Parrillo JE. Norepinephrine alone versus norepinephrine plus low-dose dopamine: enhanced renal blood flow with combination pressor therapy. Crit Care Med. 1985;13:492–496.
368. Hoogenberg K, Smit AJ, Girbes AR. Effects of low-dose dopamine on renal and systemic hemodynamics during incremental norepinephrine infusion in healthy volunteers. Crit Care Med. 1998;26(2):260–265.
369. Olson D, Pohlman A, Hall JB. Administration of low-dose dopamine to nonoliguric patients with sepsis syndrome does not raise intramucosal gastric pH nor improve creatinine clearance. Am J Resp Crit Care Med. 1996;154(6 Pt 1):1664–1670.
370. Ichai C, Passeron C, Carles M, et al. Prolonged low-dose dopamine infusion induces a transient improvement in renal function in hemodynamically stable, critically ill patients: a single-blind, prospective, controlled study. Crit Care Med. 2000;28(5):1329–1335.
371. Bellomo R, Chapman M, Finfer S, et al. Low-dose dopamine in patients with early renal dysfunction: a placebo-controlled randomised trial. Australia New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000;356(9248):2139–2143.
372. Marik PE, Iglesias J. Low-dose dopamine does not prevent acute renal failure in patients with septic shock and oliguria. NORASEPT II Study Investigators. Am J Med. 1999;107(4):387–390.
373. Mimoz O, Rauss A, Rekik N, et al. Pulmonary artery catheterization in critically ill patients: a prospective analysis of outcome changes associated with catheter-prompted changes in therapy. Crit Care Med. 1994;22(4):573–579.
374. Connors AF Jr, McCaffree DR, Gray BA. Evaluation of right-heart catheterization in the critically ill patient without acute myocardial infarction. N Engl J Med. 1983;308:263–267.
375. Weisel RD, Vito L, Dennis RC, et al. Myocardial depression during sepsis. Am J Surg. 1977;133:512–521.
376. Cohn JN. Central venous pressure as a guide to volume expansion. Ann Intern Med. 1967;66(6):1283–1287.
377. Connors AF Jr, Dawson NV, Shaw PK, et al. Hemodynamic status in critically ill patients with and without acute heart disease. Chest. 1990;98(5):1200–1206.
378. Kumar A, Anel R, Bunnell E, et al. Pulmonary artery occlusion pressure and central venous pressure fail to predict ventricular filling volume, cardiac performance, or the response to volume infusion in normal subjects. Crit Care Med. 2004;32(3):691–699.
379. Robin ED. The cult of the Swan-Ganz catheter. Overuse and abuse of pulmonary flow catheters. Ann Intern Med. 1985;103(3):445–449.
380. Dalen JE, Bone RC. Is it time to pull the pulmonary artery catheter [see comment]? JAMA. 1996;276(11):916–918.
381. Pronovost PJ, Jenckes MW, Dorman T, et al. Organizational characteristics of intensive care units related to outcomes of abdominal aortic surgery. JAMA. 1999;281:1310–1317.
382. Carson SS, Stocking C, Podsadecki T, et al. Effects of organizational change in the medical intensive care unit of a teaching hospital: a comparison of ‘open’ and ‘closed’ formats. JAMA. 1996;276:322–328.
383. Pronovost PJ, Angus DC, Dorman T, et al. Physician staffing patterns and clinical outcomes in critically ill patients: a systematic review. JAMA. 2002;288(17):2151–2162.
384. Li TC, Phillips MC, Shaw L, et al. On-site physician staffing in a community hospital intensive care unit. Impact on test and procedure use and on patient outcome. JAMA. 1984;252(15):2023–2027.
385. Pollack MM, Katz RW, Ruttimann UE, et al. Improving the outcome and efficiency of intensive care: the impact of an intensivist. Crit Care Med. 1988;16(1):11–17.
386. Reynolds HN, Haupt MT, Thill-Baharozian MC, et al. Impact of critical care physician staffing on patients with septic shock in a university hospital medical intensive care unit. JAMA. 1988;260:3446–3450.
387. Hutton P, Dye J, Prys-Roberts C. An assessment of the Dinamap. Anaesthesia. 1984;39:261–267.
388. Cohn JN. Blood pressure measurement in shock. Mechanism of inaccuracy in ausculatory and palpatory methods. JAMA. 1967;199(13):118–122.
389. Connors AF Jr, Dawson NV, McCaffree DR, et al. Assessing hemodynamic status in critically ill patients: do physicians use clinical information optimally? J Crit Care. 1987;2:174–180.
390. Michard F, Teboul JL. Predicting fluid responsiveness in ICU patients: a critical analysis of the evidence. Chest. 2002;121(6):2000–2008.
391. Marik PE. Pulmonary artery catheterization and esophageal Doppler monitoring in the ICU. Chest. 1999;116(4):1085–1091.
392. Humphrey H, Hall J, Sznajder I, et al. Improved survival in ARDS patients associated with a reduction in pulmonary capillary wedge pressure. Chest. 1990;97(5):1176–1180.
393. Mitchell JP, Schuller D, Calandrino FS, et al. Improved outcome based on fluid management in critically ill patients requiring pulmonary artery catheterization. Am Rev Resp Dis. 1992;145(5):990–998.
394. Connors AF Jr, Speroff T, Dawson NV, et al. The effectiveness of right heart catheterization in the initial care of critically ill patients. JAMA. 1996;276:889–897.
395. Richard C, Warszawski J, Anguel N, et al. Early use of the pulmonary artery catheter and outcomes in patients with shock and acute respiratory distress syndrome: a randomized controlled trial. JAMA. 2003;290(20):2713–2720.
396. Rhodes A, Cusack RJ, Newman PJ, et al. A randomised, controlled trial of the pulmonary artery catheter in critically ill patients. Intensive Care Med. 2002;28(3):256–264.
397. Polanczyk CA, Rohde LE, Goldman L, et al. Right heart catheterization and cardiac complications in patients undergoing noncardiac surgery: an observational study. JAMA. 2001;286(3):309–314.
398. Sandham JD, Hull RD, Brant RF, et al. A randomized, controlled trial of the use of pulmonary-artery catheters in high-risk surgical patients. N Engl J Med. 2003;348(1):5–14.
399. Ivanov RI, Allen J, Sandham JD, et al. Pulmonary artery catheterization: a narrative and systematic critique of randomized controlled trials and recommendations for the future. New Horiz. 1997;5(3):268–276.
400. Zanotti S, Kumar A, Kumar A. Cytokine modulation in sepsis and septic shock. Expert Opin Investig Drugs. 2002;11(8):1061–1075.
401. Anel RL, Kumar A. Experimental and emerging therapies for sepsis and septic shock. Expert Opin Investig Drugs. 2001;10(8):1471–1485.
402. Ely EW, Bernard GR, Vincent JL. Activated protein C for severe sepsis. N Engl J Med. 2002;347(13):1035–1036.
403. Beale R, Wright TJ, Wong K, et al. Safety of drotrecogin alfa (activated) in adult patients with severe sepsis: comparison of global ENHANCE to PROWESS. Chest. 2003;124:102S.
404. Vincent JL, Bernard GR, Beale R, et al. Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE: further evidence for survival and safety and implications for early treatment. Crit Care Med. 2005;33(10):2266–2277.
405. Siegel JP. Assessing the use of activated protein C in the treatment of severe sepsis. N Engl J Med. 2002;347(13):1030–1034.
406. Nadel S, Goldstein B, Williams MD, et al. Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial [see comment]. Lancet. 2007;369(9564):836–843.
407. Alejandria MM, Lansang MA, Dans LF, et al. Intravenous immunoglobulin for treating sepsis and septic shock. Cochrane Database Syst Rev. 2002;(1):CD001090.
408. Douzinas EE, Pitaridis MT, Louris G, et al. Prevention of infection in multiple trauma patients by high-dose intravenous immunoglobulins. Crit Care Med. 2000;28(1):8–15.
409. Darenberg J, Ihendyane N, Sjolin J, et al. Intravenous immunoglobulin G therapy in streptococcal toxic shock syndrome: a European randomized, double-blind, placebo-controlled trial. Clin Infect Dis. 2003;37(3):333–340.
410. Pitcairn M, Schuler J, Erve PR, et al. Glucocorticoid and antibiotic effect on experimental gram-negative bacteremic shock. Arch Surg. 1975;110(8):1012–1015.
411. Sprung CL, Caralis PV, Marcial EH, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984;311(18):1137–1143.
412. Bone RC, Fisher CJ Jr, Clemmer TP, et al. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317(11):653–658.
413. Luce JM, Montgomery AB, Marks JD, et al. Ineffectiveness of high-dose methylprednisolone in preventing parenchymal lung injury and improving mortality in patients with septic shock. Am Rev Resp Dis. 1988;138(1):62–68.
414. The Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. N Engl J Med. 1987;317(11):659–665.
415. Schiffl H, Lang SM, Fischer R. Daily hemodialysis and the outcome of acute renal failure. N Engl J Med. 2002;346(5):305–310.
416. Ronco C, Bellomo R, Homel P, et al. Effects of different doses in continuous veno-venous haemofiltration on outcomes of acute renal failure: a prospective randomised trial. Lancet. 2000;356(9223):26–30.
417. Phu NH, Hien TT, Mai NT, et al. Hemofiltration and peritoneal dialysis in infection-associated acute renal failure in Vietnam. N Engl J Med. 2002;347(12):895–902.
418. Krinsley JS. Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients. Mayo Clin Proc. 2003;78(12):1471–1478.
419. Vanden Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345(19):1359–1367.
420. Latham R, Lancaster AD, Covington JF, et al. The association of diabetes and glucose control with surgical-site infections among cardiothoracic surgery patients. Infect Control Hosp Epidemiol. 2001;22(10):607–612.
421. Vanden Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354(5):449–461.
422. Hermans G, Wilmer A, Meersseman W, et al. Impact of intensive insulin therapy on neuromuscular complications and ventilator dependency in the medical intensive care unit. Am J Resp Crit Care Med. 2007;175(5):480–489.
423. Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348(8):727–734.
424. Annane D, Bellissant E, Sebille V, et al. Impaired pressor sensitivity to noradrenaline in septic shock patients with and without impaired adrenal function reserve. Br J Clin Pharmacol. 1998;46:589–597.
425. Briegel J, Forst H, Haller M, et al. Stress doses of hydrocortisone reverse byperdynamic septic shock: a prospective randomized, double-blind, single-center study. Crit Care Med. 1999;27(4):723–732.
426. Bollaert PE, Charpentier C, Levy B, et al. Reversal of late septic shock with supraphysiologic doses of hydrocortisone. Crit Care Med. 1998;26(4):645–650.
427. Keh D, Boehnke T, Weber-Cartens S, et al. Immunologic and hemodynamic effects of “low-dose” hydrocortisone in septic shock: a double-blind, randomized, placebo-controlled, crossover study. Am J Resp Crit Care Med. 2003;167(4):512–520.
428. Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288(7):862–871.
429. Hamrahian AH, Oseni TS, Arafah BM. Measurements of serum free cortisol in critically ill patients. N Engl J Med. 2004;350(16):1629–1638.
430. Sprung C, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–124.
431. Ranieri VM, Suter PM, Tortorella C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282(1):54–61.
432. Tremblay L, Valenza F, Ribeiro SP, et al. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997;99(5):944–952.
433. International consensus conferences in intensive care medicine: Ventilator-associated Lung Injury in ARDS. This official conference report was cosponsored by the American Thoracic Society, The European Society of Intensive Care Medicine, and The Societe de Reanimation de Langue Francaise, and was approved by the ATS Board of Directors, July, 1999. Am J Resp Crit Care Med. 1999;160(6):2118–2124.
434. Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Resp Crit Care Med. 1994;149(3 Pt 1):818–824.
435. Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338(6):347–354.
436. The Acute Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1301–1308.
437. Stewart TE, Meade MO, Cook DJ, et al. Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N Engl J Med. 1998;338(6):355–361.
438. Hussain SN, Graham R, Rutledge F, et al. Respiratory muscle energetics during endotoxic shock in dogs. J Appl Physiol. 1986;60(2):486–493.
439. Leon A, Boczkowski J, Dureuil B, et al. Effects of endotoxic shock on diaphragmatic function in mechanically ventilated rats. J Appl Physiol. 1992;72(4):1466–1472.
440. Marik PE, Zaloga GP. Early enteral nutrition in acutely ill patients: a systematic review. Crit Care Med. 2001;29(12):2264–2270.
441. Heyland DK. Nutritional support in the critically ill patients. A critical review of the evidence. Crit Care Clin. 1998;14(3):423–440.
442. Heyland DK, MacDonald S, Keefe L, et al. Total parenteral nutrition in the critically ill patient: a meta-analysis. JAMA. 1998;280(23):2013–2019.
443. Moore FA, Feliciano DV, Andrassy RJ, et al. Early enteral feeding, compared with parenteral, reduces postoperative septic complications. The results of a meta-analysis. Ann Surg. 1992;216(2):172–183.
444. Cooper DJ, Walley KR, Wiggs BR, et al. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study Ann Intern Med. 1990;112:492–498.
445. Mathieu D, Neviere R, Billard V, et al. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med. 1991;19(11):1352–1356.
446. van der Poll T, van Deventer SJ. Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am. 1999;13(2):413–426.
447. Leaver SK, Finney SJ, Burke-Gaffney A, et al. Sepsis since the discovery of Toll-like receptors: disease concepts and therapeutic opportunities. Crit Care Med. 2007;35(5):1404–1410.
448. Van Amersfoort ES, Van Berkel TJ, Kuiper J, et al. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin Micro Rev. 2003;16(3):379–414.
449. Arcaroli J, Fessler MB, Abraham E, et al. Genetic polymorphisms and sepsis. Shock. 2005;24(4):300–312.
450. Lin MT, Albertson TE, Lin MT, et al. Genomic polymorphisms in sepsis. Crit Care Med. 2004;32(2):569–579.
451. Texereau J, Pene F, Chiche JD, et al. Importance of hemostatic gene polymorphisms for susceptibility to and outcome of severe sepsis. Crit Care Med. 2004;32(5 Suppl):S313–S319.
452. Papathanassoglou ED, Giannakopoulou MD, Bozas E, et al. Genomic variations and susceptibility to sepsis. AACN Advanced Critical Care. 2006;17(4):394–422.
453. Sharma S, Mink S. Septic shock. http://www.emedicine.com/MED/topic2101.htm. 2007. Accessed Dec. 1, 2007.